
Transplantace hematopoetických buněk u pěti pacientů s chronickou granulomatózní nemocí v České republice
Transplantation of hematopoietic cells in five patients with chronic granulomatous disease in the Czech Republic
Chronic granulomatous disease (CGD) is a primary immunodeficiency caused by defective phagocytosis. Conservative treatment is applied in most of the patients. Currently, the only available curative treatment is allogeneic hematopoietic stem cell transplantation (HSCT).
Thirty-seven patients were diagnosed with CGD in the Czech and Slovak Republic between 1957 and 2011. Calculated incidence of the disease in the Czech Republic is 1 : 280,000 live births. Five patients with serious course of the disease underwent allogeneic HSCT in 2007–2010. Median age of CGD diagnosis in these patients was 6 months (6–17 months), median age at transplantation was 21 months (1–17 years). One patient was transplanted in myeloablative regimen, 4 patients in regimen with reduced intensity. All of the grafts were from HLA-matched unrelated donors. The procedure was complicated with graft versus host disease – GvHD (3 patients), infections (4 patients), autoimmunity (3 patients) and toxic side effects (3 patients). Median follow-up after the transplantation was 12 months (6–44 months). In all of the patients normalization of granulocyte function was documented within 2 months after the transplantation. Mixed chimerism was present in all of them. The oldest patient (transplanted at the age of 17 years, 4 months) died 6 months after transplantation due to intracranial haemorrhage after operational revision of healed brain abscess. All the other patients (4) are in a good clinical condition, without signs of CGD or GvHD.
In concordance with other transplantation centres we show that early allogeneic HSCT is a suitable treatment of patients with severe CGD.
Key words:
chronic granulomatous disease, primary immunodeficiency, hematopoietic stem cell transplantation
Authors:
A. Janda 1; P. Keslová 1; R. Formánková 1; J. Litzman 2; A. Šedivá 3; H. Houšťková 4; P. Rozsíval 5; E. Pařízková 5; J. Starý 1; P. Sedláček 1
Authors‘ workplace:
Klinika dětské hematologie a onkologie UK 2. LF a FN Motol, Praha, přednosta prof. MUDr. J. Starý, DrSc., Ústav klinické imunologie a alergologie LF Masarykovy univerzity, FN U sv. Anny, Brno , přednosta prof. MUDr. J. Litzman, CSc., Ústav imunolog
1
Published in:
Čes-slov Pediat 2012; 67 (2): 81-88.
Category:
Original Papers
Overview
Chronická granulomatózní nemoc (CGD) patří mezi vrozené imunodeficience způsobené poruchou fagocytózy. Většina pacientů je léčena konzervativně. V současné době je jedinou dostupnou kurativní terapií alogenní transplantace hematopoetických buněk (HSCT).
V letech 1957 až 2011 bylo v České a Slovenské republice diagnostikováno 37 pacientů s CGD. Odhadovaná incidence onemocnění v České republice je 1 : 280 000 živě narozených dětí. Pět pacientů se závažným průběhem onemocnění podstoupilo v letech 2007–2010 alogenní HSCT. Medián stanovení diagnózy byl u těchto pacientů 6 měsíců (6–17 měsíců), medián věku při transplantaci 21 měsíců (1 rok až 17 let). Jeden pacient byl transplantován v myeloablativním režimu, 4 pacienti s použitím redukovaného režimu, vždy štěpy od HLA-shodných nepříbuzných dárců. Výkon byl komplikován GvHD II. stupně (3 pacienti), infekcí (4 pacienti), autoimmunitou (3 pacienti) a vedlejšími účinky léčby (3 pacienti). Medián sledování po transplantaci byl 12 měsíců (6–44 měsíců). U všech pacientů došlo v průběhu 2 měsíců po transplantaci k normalizaci funkce granulocytů, u všech byl přítomen smíšený chimerismus. Nejstarší pacient (transplantován ve věku 17 let, 4 měsíce) zemřel 6 měsíců po transplantaci na intrakraniální krvácení po operační revizi zhojeného mozkového abscesu. Ostatní pacienti (4) jsou v dobrém klinickém stavu, bez známek CGD a GvHD.
Ve shodě se zahraničními pracovišti ukazují naše výsledky, že časná alogenní transplantace hematopoetických buněk představuje vhodnou alternativu léčby pacientů se závažnou formou chronické granulomatózní nemoci.
Klíčová slova:
chronická granulomatózní nemoc, primární imunodeficience, transplantace buněk krvetvorby
ÚVOD
Chronická granulomatózní nemoc (Chronic Granulomatous Disease, CGD) patří mezi vrozené imunodeficience způsobené poruchou fagocytózy s incidencí 1/100–250 tisíc živě narozených dětí. Onemocnění je podmíněno defektem v genech kódujících součásti NADPH oxidázy, což vede k poruše produkce reaktivních metabolitů kyslíku ve fagozomu fagocytujících buněk (neutrofily, monocyty, makrofágy a eozinofily). Absence těchto metabolitů vede ke snížení mikrobicidie vůči některým mikroorganismům a k poruše regulace zánětu. Nejčastější (70 %) a také nejzávažnější je X-vázaná (XL) forma onemocnění (MIM #306400), kdy je poškozen gen CYBB kódující membránový glykoprotein 91phox (gp91phox, NOX2). Autozomálně recesivní (AR) formy způsobené mutacemi v genech pro jiné součásti NADPH komplexu, tj. p22phox, p47phox, p67phox a p40phox (MIM #233690, #233700, #233710 a #601488) jsou vzácnější a klinické obtíže jsou většinou méně výrazné [1, 2].
Onemocnění se projevuje vyšší náchylností k bakteriálním a mykotickým infekcím měkkých tkání. Většina infekcí je způsobena těmito 5 organismy: Staphylococcus aureus, Burkholderia (Pseudomonas) cepacia, Serratia marcescens, Nocardia a Aspergillus spp. Infekce většinou pramení z nevyhnutelného environmentálního kontaktu. Častý je výskyt abscesů a granulomů postihujících kůži, lymfatické uzliny, plíce, játra a střevo, relativně častěji se lze u pacientů setkat s osteomyelitidou, perianálními abscesy a gingivitidou. Nejčastějším vyvolavatelem abscesů je Staphylococcus aureus. Granulomy jsou většinou sterilní a mohou způsobovat obstrukční obtíže. Intraluminální granulomy v genitoureterálním traktu např. ztěžují odtok moči a přispívají k vyšší incidenci infekcí močových cest. Granulomatózní postižení trávicí trubice je obtížně odlišitelné od Crohnovy choroby [1–4].
Chronická granulomatózní choroba je pomalu progredující onemocnění s akutními infekčními i neinfekčními komplikacemi. Většina pacientů dříve umírala v první dekádě života. Se zlepšováním péče a především s profylaktickým používáním antimikrobiálních léků se jejich výhled významně zlepšil. Při dobré spolupráci pacienta sledovaného ve specializovaném centru je možno dosáhnout dobré kontroly infekcí, ovlivnění ostatních projevů nemoci (např. Crohn-like enterokolitida, granulomy, fibrotizace plicního parenychmu) už tak úspěšné není. Mortalitu podmíněnou infekcemi lze snížit, morbidita zůstává však stále vysoká, což přispívá k významnému snížení dlouhodobé kvality života těchto pacientů. CGD zůstává smrtelnou chorobou a dlouhodobá prognóza pacientů je nejistá. V současnosti dochází k většině úmrtí v dospělosti. Tento fakt souvisí zřejmě s kolonizací pacientů, s orgánovými komplikacemi onemocnění, s neinfekčními projevy a také s obecně horší dostupností specialistů pro imunodeficity mimo pediatrii. Udává se, že 40. roku věku se dožije méně než polovina pacientů [1, 5–8].
Průběh onemocnění je variabilní. Poškození genu pro gp91phox na X chromozomu (X-CGD) je obecně spojeno s těžším průběhem a s horší prognózou. Kromě poškozeného genu je také důležitý typ poškození (např. missense versus nonsense mutace) a jeho vliv na funkci NADPH systému. U pacientů se zbytkovou aktivitou oxidázy je dlouhodobé přežití významně lepší. Tento vztah však neplatí u komplikací onemocnění (autoimunita, granulomy) [9]. Z funkčních a genetických vyšetření tedy nelze vyvozovat závěry týkající se budoucí kvality života pacientů.
K funkčnímu vyšetření fagocytujících buněk se nyní nejčastěji používá cytometrický test oxidativního vzplanutí, nazývaný „burst test“. Vyšetření je založeno na detekci zeleně fluoreskujícího rhodaminu 123, který vzniká oxidací dihydrorhodaminu 123 působením reaktivních metabolitů kyslíku v granulocytech. Výhodou testu je kromě rychlosti provedení a relativní technické nenáročnosti i možnost odhalení nosiček onemocnění u X-vázané formy, u kterých jsou typicky přítomny normální i defektní granulocyty. Je-li dostupné molekulárně genetické vyšetření, mělo by být provedeno u všech pacientů.
V rámci diferenciální diagnostiky abnormálně nízkého respiračního vzplanutí je třeba zvážit deficit glukózo-6-fosfát dehydrogenázy, která bývá spojena s určitým stupněm hemolýzy; infekci některými patogeny, které snižují expresi glykoproteinu 91phox a tak negativně ovlivňují produkci reaktivních metabolitů kyslíku (Legionella pneumophila, Toxoplasma gondii, Chlamydia spp., Entamoeba histolytica, Ehrlichia risticii) [10]; a hypozinkémii [11].
Péče o pacienty s chronickou granulomatózou je založena především na prevenci infekcí. Zásadní jsou režimová opatření (zákaz koupání ve stojatých vodách, vyhýbání se prašnému prostředí a místům se zvýšeným výskytem plísní, prevence zranění) a celoživotní antibiotická (trimetoprim-sulfometoxazol, 5 mg/kg/den, maximálně 160 mg trimetoprimu denně, v 1–2 dávkách) a antimykotická profylaxe (itrakonazol 100 mg denně u dětí mladších 13 let, 200 mg u starších). V případě akutních infekcí je třeba postupovat razantně a vždy se snažit o mikrobiologické stanovení vyvolávajícího agens. Preventivní protiinfekční strategie snižuje rizika infekce, ale riziko závažné infekce (bakteriální i mykotické) zcela neeliminuje. Neúspěch může být v poddávkování léků (rostoucí organismus, neuzpůsobení dávky, nespolupráce pacienta) a v trvalém stavu imunodeficience, dlouhodobém podávání kortikoidů a postupném orgánovém postižení (játra, plíce, trávicí trakt). V léčbě granulomů jsou používány kortikoidy (prednison, iniciální dávka 1 mg/kg/den), eventuálně chirurgická léčba. Spornou je aplikace interferonu gama (0,05 mg/kg, třikrát týdně). Infuze dárcovských granulocytů může vyvolat vznik autoprotilátek proti součástem NADPH komplexu a zkomplikovat tak případnou alogenní transplantaci hematopoetických buněk; tato léčba by měla být indikována výjimečně. Genová terapie CGD přináší v současné době pouze přechodný efekt srovnatelný s transfuzí dárcovských granulocytů při vysokém nebezpečí vzniku malignity díky náhodné inzerci virového vektoru [5, 12, 13].
Jedinou dostupnou kurativní metodou je alogenní transplantace hematopoetických buněk (HSCT). Na základě empiricky stanovených klinických kritérií, zpracovaných v rámci skupiny IE-EBMT (Working Party Inborn Errors of the European Blood and Marrow Transplantation), je indikací k jejímu provedení přítomnost minimálně jedné závažné život ohrožující infekce nebo těžké granulomatózní poškození životně důležitých orgánů, případně non-compliance při užívání antibiotické profylaxe, eventuálně dlouhodobě nedostupná specializovaná péče. Pacienti s chronickými, resp. těžce probíhajícími a chronickými infekcemi, případně s refrakterní granulomatózou, jsou považováni za vysoce rizikové. Na základě jejich stavu je nutné upravit přípravný režim, jejich prognóza je výrazně horší než u pacientů vstupujících do transplantace bez aktivně probíhající nemoci [2, 14].
Myeloablativní přípravný režim (busulfan, cyklofosfamid) v kombinaci s deplecí T buněk je obecně úspěšnější v dosahování plného dárcovského chimerismu s dobrým dlouhodobým přežitím pacientů. Nevýhodou je možné zhoršení probíhající infekce či aktivního zánětu ve spojení s těžkou reakcí štěpu proti hostiteli (GvHD) u vysoce rizikových a starších pacientů. U těchto pacientů je nutné použít méně toxické režimy s redukovanou intenzitou používající nižší dávky busulfanu a fludarabinu, či kombinace fludarabinu s melfalanem či treosulfanem. Vzhledem k tomu, že cílem transplantace je náhrada myeloidní řady, není dosažení plného chimerismu nutné. Přípravný režim s redukovanou intenzitou je tedy nyní obecně preferovaným přístupem i u pacientů ve standardním riziku.
Riziko úmrtí při transplantaci vyplývající z několika studií je přibližně 5% při převodu štěpu od HLA-identického sourozence a 15% při použití štěpů od HLA-shodných nepříbuzných dárců [2, 14]. Čím starší je pacient v době transplantace, tím vyšší je riziko komplikací i úmrtí. Některá transplantační centra proto doporučují časnou HSCT u všech pacientů s diagnózou CGD bez ohledu na klinický průběh onemocnění, tj. manifestaci život ohrožující infekce, resp. refrakterní granulomatózy. Některá pracoviště takto postupují jen v případě, že je k dispozici jako dárce zdravý HLA-shodný sourozenec a riziko úmrtí při transplantaci je tak nízké [2, 14], jiná centra preferují transplantaci u všech pacientů s geneticky prokázanou X-vázanou formou onemocnění do 5 let věku i s pomocí štěpu od nepříbuzného dárce (osobní sdělení: A. Gennery, A. H. Filipovich).
V letech 1957 až 2011 bylo v České a Slovenské republice diagnostikováno 37 pacientů s CGD [15]. Pět pacientů se závažným průběhem onemocnění podstoupilo v letech 2007–2010 alogenní HSCT.
PACIENTI
Přehled všech transplantovaných pacientů je uveden v tabulce 1.
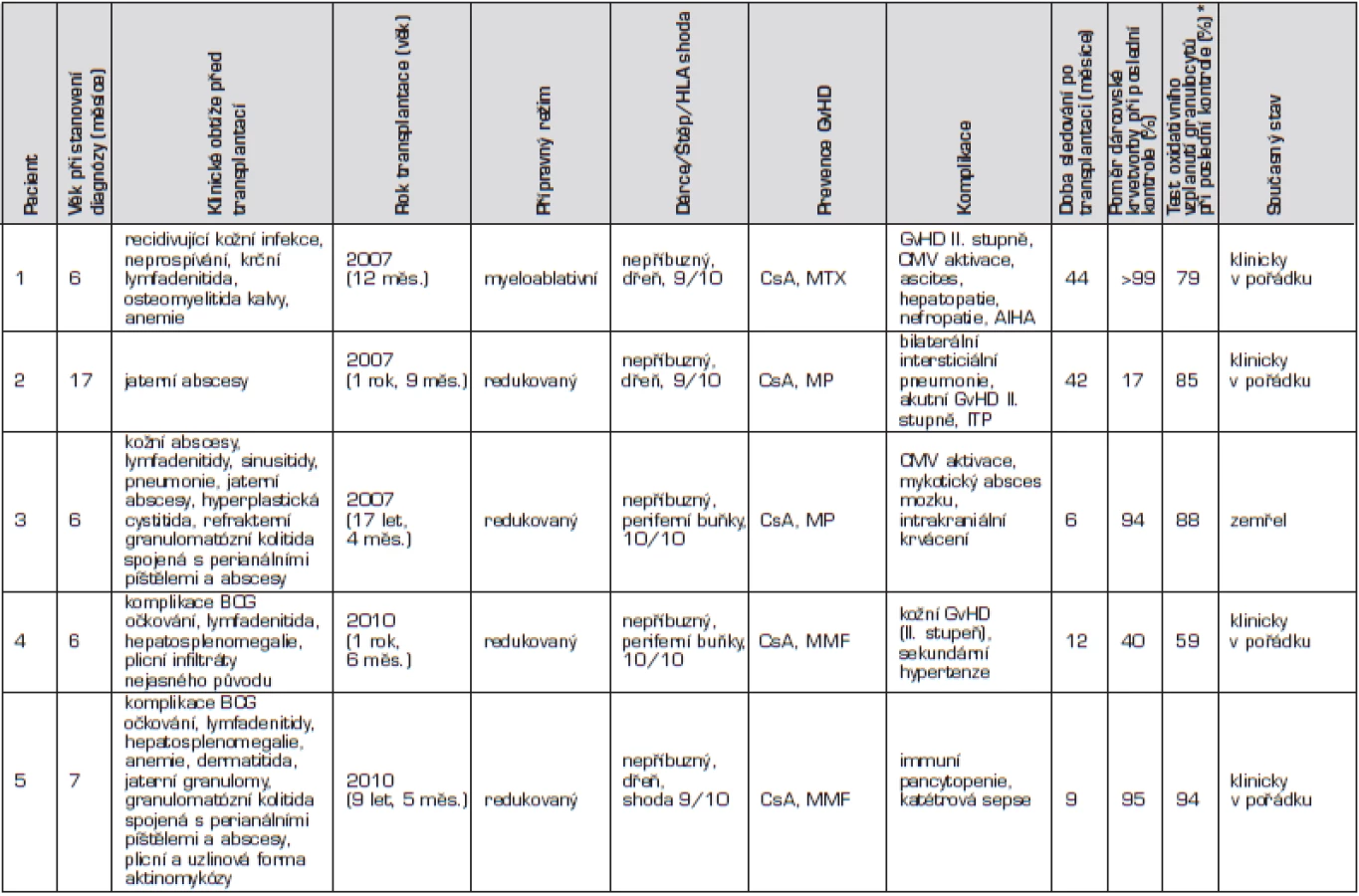
U prvního chlapce se onemocnění manifestovalo v prvním měsíci života, a to recidivující kožní infekcí a neprospíváním. Diagnóza byla stanovena v 6 měsících věku, kdy se objevila krční lymfadenitida a osteomyelitida kalvy (Serratia marcescens) s intrakraniální penetrací (obr. 1).
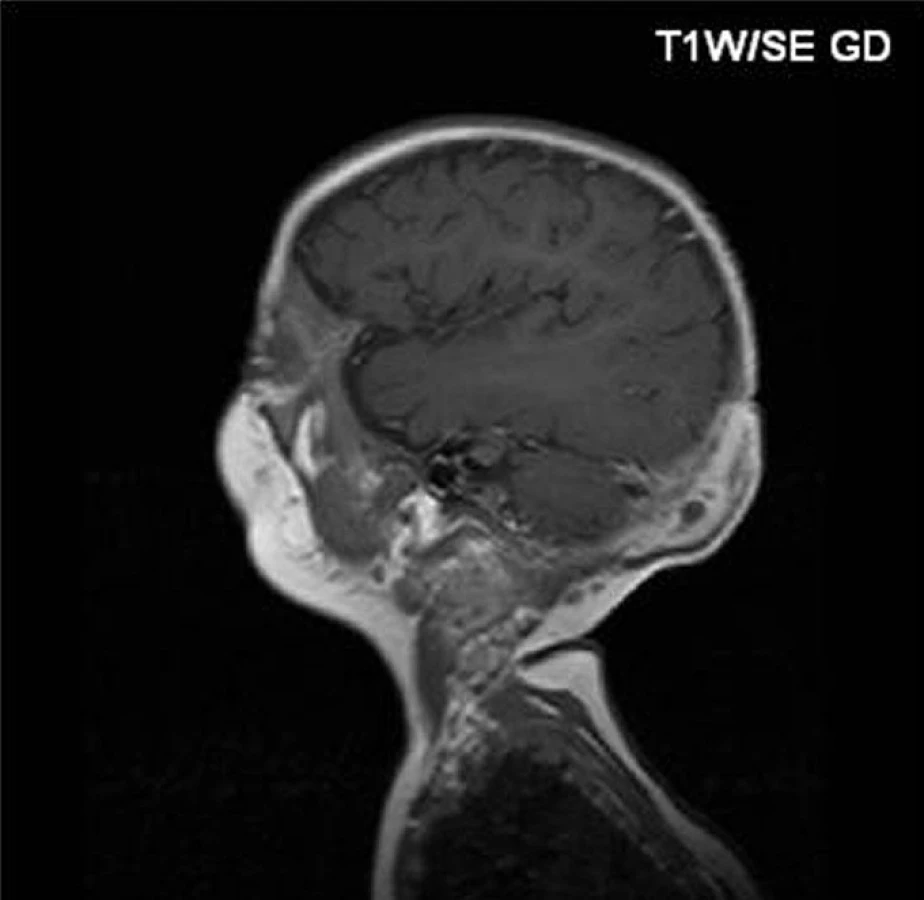
Infekce byla doprovázena výraznou hepatosplenomegalií a anemií. Vzhledem k závažnosti stavu, věku pacienta a dostupnosti vhodného dárce bylo rozhodnuto o HSCT. Po stabilizaci infekce (léčen amikacinem, ciprofloxacinem, linezolidem, itrakonazolem, trimetoprim-sulfometoxazolem, kortikosteroidy a interferonem gama) byl chlapec ve věku 12 měsíců transplantován od nepříbuzného dárce. Preventivně byla aplikována antituberkulotika. Granulocyty se přihojily 19 dní po transplantaci. Kompletní dárcovská krvetvorba byla detekována 21 dní po transplantaci, normální funkce granulocytů byla dokumentována 2 měsíce po převodu štěpu. Přechodně se objevil smíšený chimerismus, od 22. měsíce po transplantaci byla opakovaně nalezena prakticky pouze dárcovská krvetvorba. Výkon byl komplikován akutní střevní GvHD II. stupně (léčen mykofenolátem) a opakovanou CMV aktivací (léčen ganciklovirem); přechodně se manifestovaly ascites, pericholecystitida, hepatopatie a nefropatie, nejspíše na autoimunitním podkladě. Do ambulantního sledování byl propuštěn 110 dní po transplantaci. V průběhu sledování po transplantaci byly nutné tři chirurgické intervence nesouvisející s vlastní transplantací: operace inguinální hernie a následná revize pro krvácení, a operační korekce hydrokély. Potransplantační období bylo dále komplikováno rozvojem akutní imunní hemolytické anemie (AIHA) 10 měsíců po převodu štěpu (léčen vysokodávkovanými intravenózními imunoglobuliny a rituximabem) a katétrovou sepsí (rezistentní Klebsiella oxytoca). Nyní, ve věku 4 roky a 8 měsíců, tj. 44 měsíců po transplantaci, má chlapec normální funkci granulocytů a kompletní dárcovskou krvetvorbu. Je bez klinických obtíží, frekvence infekcí je běžná. Zatím užívá běžnou potransplantační antimikrobiální chemoprofylaxi (její ukončení je plánováno za rok), je bez imunosuprese, tvorba vlastních imunoglobulinů je nižší, nevyžaduje však substituční terapii.
U druhého chlapce byla diagnóza stanovena v 17 měsících věku, kdy byly při laparotomii zjištěny mnohočetné jaterní abscesy (Staphylococcus aureus, Enterococcus faecalis, Hafnia, Acinetobacter). Chlapec byl léčen intenzivní antibiotickou terapií (cefotaxim, oxacilin, amikacin, meropenem, teicoplanin, ciprofloxacin), interferonem gama, kortikosteroidy a granulocytárními náplavy. Na této terapii došlo postupně ke zřetelné regresi jaterních ložisek. Transplantace HSCT byla provedena ve věku 21 měsíců od nepříbuzného dárce. K přihojení granulocytů došlo 12 dní po transplantaci, normalizace funkce granulocytů byla dokumentována od 60. dne po převodu štěpu. Kompletní dárcovské krvetvorby se nepodařilo dosáhnout, autologní krvetvorba postupně narůstala až na současných 83 %. Počet funkčních dárcovských granulocytů kolísal, při poslední kontrole však navzdory významné autologní krvetvorbě dosahoval normálních hodnot, což odráží převažující dárcovský původ granulocytů.
Ultrazvukovým vyšetřením břicha byl již 16 dní po transplantaci dokumentován normální jaterní parenchym bez ložiskových lézí. Výkon byl komplikován bilaterální intersticiální pneumonií a akutní izolovanou gastrointestinální GvHD II. stupně. Do ambulantního sledování byl chlapec propuštěn 83. den po transplantaci. Imunosuprese byla ukončena 5 měsíců po transplantaci. Následně prodělal několik mírných respiračních infektů, nekomplikované plané neštovice (léčen herpesinem) a virovou parotitidu. Tři roky po transplantaci došlo k manifestaci imunní trombocytopenie (léčen vysokodávkovanými intravenózními imunoglobuliny, IVIG), byla zjištěna pozitivita antiglobulinového testu, ale bez známek klinické hemolýzy. Nyní, ve věku 5 let a 3 měsíců, tj. 42 měsíců po transplantaci, má chlapec normální počet funkčních granulocytů a je zcela bez klinických obtíží v domácí péči a bez imunosuprese. Trvá pozitivita antitrombocytárních protilátek, antiglobulinový test je negativní. Pokračuje přechodná standardní antimikrobiální chemoprofylaxe.
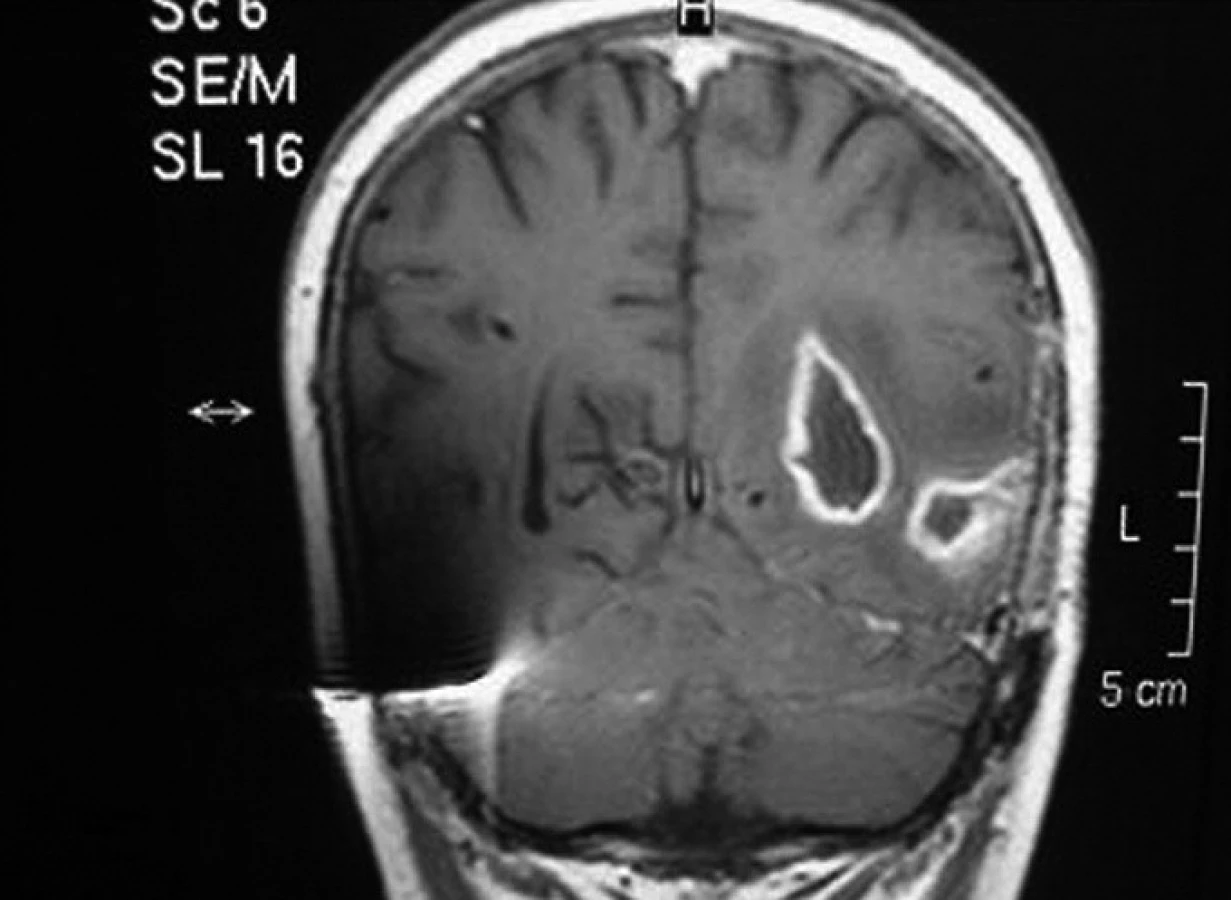
U pacienta č. 3, nejstaršího chlapce z našeho souboru, transplantovaného ve věku 17 let, se onemocnění manifestovalo drobnými intradermálními abscesy v obličeji bezprostředně po porodu a rozsáhlými abscesy po vpiších při očkování. Diagnóza byla stanovena v 6 měsících věku, kdy byl hospitalizován pro krční lymfadenitidu a jaterní abscesy. Chlapec byl trvale na chemoprofylaxi s výbornou compliance. Do 10 let věku měl obtíže především charakteru lymfadenitid, později došlo k rozvoji respiračních infektů – opakované sinusitidy a pneumonie. Ve 14 letech byla zjištěna granulomatózní kolitida, v témže roce bylo nutno chirurgicky řešit jaterní absces, zároveň chlapec prodělal hyperplastickou cystitidu. Postupně došlo k rozvoji perianálních píštělí a abscesů, kolitida se stala refrakterní na konzervativní léčbu. Pro nepříznivý průběh střevního onemocnění byl chlapec indikován k HSCT štěpem z periferních buněk od nepříbuzného dárce. Granulocyty se přihojily 11 dní po transplantaci, od 60. dne dokumentována normální funkce granulocytů, došlo ke zhojení kolitidy i komunikujících píštělí. Bezprostřední potransplantační období bylo komplikováno aktivací CMV. Čtyřicet devět dní po převodu štěpu byl zjištěn mykotický absces mozku (Aspergillus calidoustus, viz obr. 2). Původ patogena není jasný, jednalo se o solitární postižení, do CNS pronikl zřejmě již před transplantací. V důsledku rekonstituce normální funkce dárcovských granulocytů došlo k rozpuštění pseudoabscesu a k manifestaci infekce. Infekce byla vyléčena kombinací antimykotik (lipozomální amphotericin B, Ambisome) s chirurgickým odstraněním ložisek (2 operace). Chlapec následně absolvoval více operací s cílem zavedení a úpravy komorové drenáže sekundárního hydrocefalu. Při poslední operaci došlo krátce po výkonu k závažnému krvácení do CNS s projevy mozkové smrti (šest měsíců po transplantaci).
Diagnóza chronické granulomatózní nemoci u čtvrtého pacienta byla stanovena v 7 měsících věku při hospitalizaci pro axilární lymfadenitidu po BCG vakcinaci. Postižená abscedující uzlina byla exstirpována a chlapec byl léčen co-trimoxazolem a izoniazidem. Krátce nato byla rentgenovým vyšetřením plic zjištěna infiltrace v oblasti pravého plicního křídla a zvětšení mediastinálních uzlin. Nález byl potvrzen vyšetřením HRCT. Opakovaná mikrobiologická vyšetření (krev, sputum, žaludeční aspirát) neprokázala infekční agens. Klinické obtíže byly minimální, chlapec byl afebrilní, zajištěn antituberkulotiky, nález na plicích byl podle zobrazovacích metod několik měsíců stacionární. Přibližně v jednom roce věku se objevily teploty, opakovaně došlo k významné elevaci zánětlivých parametrů. Přes trvající nález plicního infiltrátu byl chlapec bez respirační symptomatologie, infekční agens se kultivačně ani metodou PCR nepodařilo prokázat (krev, bronchoalveolární laváž). Scintigraficky byla vyloučena osteomyelitida. Chlapec byl léčen antibiotiky, antituberkulotiky, antimykotiky a IVIG. Postupně se rozvíjela hepatosplenomegalie, chlapec neprospíval. Po stabilizaci stavu byl chlapec v roce a půl věku transplantován štěpem z periferních buněk od nepříbuzného dárce. Výkon byl komplikován sekundární hypertenzí a akutní kožní GvHD. Čtyři týdny po transplantaci byl propuštěn do domácího ošetřování. Kompletní dárcovská krvetvorba byla přítomna od 14. dne po převodu štěpu, později se objevila autologní složka, která stoupla v průběhu sledování na 60 %. Rok po transplantaci je chlapec klinicky v pořádku s uspokojivou funkcí granulocytů. Stále užívá imunosupresi a standardní antimikrobiální profylaxi. Sekundární hypertenze je kompenzována.
Poslední transplantovaný pacient č. 5 byl od 5. týdne věku sledován pro nehojící se absces po BCG vakcinaci. V průběhu prvního roku věku se manifestovala hepatopatie, histologicky byla potvrzena přítomnost jaterního granulomu, byla stanovena diagnóza CGD. Chlapec byl zajištěn co-trimoxazolem a itrakonazolem v preventivních dávkách, jeho stav byl dlouhodobě uspokojivý, nemocnost odpovídala věku. V šesti letech se objevily bolesti břicha a časté průjmy, v sedmi letech byl kolonoskopickým a bioptickým vyšetřením zjištěn nespecifický chronický zánět tenkého i tlustého střeva, který přechodně zareagoval na imunosupresivní léčbu. O rok později se objevily abscesy na bradě a hýždích, následně rozsáhlá krční lymfadenitida, která si vyžádala opakovanou chirurgickou intervenci. V exstirpovaných uzlinách byla metodou PCR opakovaně prokázána aktinomykóza (Actinomyces naeslundii). Doplňující celotělové PET/CT vyšetření odhalilo vícečetná ložiska infekce v plicní tkáni. Byla zahájena léčba penicilinem a pacient byl indikován k alogenní transplantaci kostní dřeně od nepříbuzného dárce. Za 14 dní po transplantaci byla dokumentována téměř kompletní dárcovská krvetvorba. Výkon byl bez výrazných komplikací, do domácího ošetřování byl chlapec propuštěn měsíc po převodu štěpu. Normální funkce granulocytů byla přítomna od 60. dne po transplantaci. Kontrolní PET/CT (100 dní po transplantaci) prokázalo vymizení všech infekčních ložisek. V dalším sledování došlo postupně k rozvoji přechodné pancytopenie vyžadující aplikaci růstových faktorů (G-CSF) a steroidů. Pět měsíců po transplantaci došlo k rozvoji katétrové sepse (Burkholderia cepacia), která si vyžádala odstranění centrálního žilního katétru. Po celou dobu léčby byl chlapec bez známek GvHD. Devět měsíců po transplantaci je pacient v pořádku, bez klinických obtíží, s normální funkcí granulocytů. Zatím užívá standardní antimikrobiální profylaxi a imunosupresiva.
Podle cytometrického burst testu provedeného před transplantací byla u všech pacientů zjištěna nulová produkce reaktivních metabolitů kyslíku, u čtyř matek postižených chlapců výsledek testu ukazoval heterozygotní stav. Poškození genu CYBB bylo potvrzeno molekulárně genetickým vyšetřením u 4 rodin (2krát nonsense, 1krát missense, 1krát nejasný efekt mutace). U jedné rodiny vyšetření probíhá.

DISKUSE
Alogenní transplantace krvetvorných buněk je v současnosti jedinou standardně používanou kauzální léčbou CGD [2, 4, 14]. Na rozdíl od pacientů s těžkými kombinovanými imunodeficiencemi, kdy je HSCT bezprostředně život zachraňujícím výkonem, vstupuje do diskuse o léčbě pacientů s CGD mírnější průběh onemocnění a stále významná časná a pozdní toxicita transplantace.
U všech našich pacientů byla transplantace indikována z důvodu život ohrožujícího stavu (infekce, refrakterní granulomatóza) a u všech byl použit štěp od nepříbuzného dárce. Zásadní rozdíl mezi pacienty byl v jejich věku v době transplantace a v předtransplantační morbiditě. Nejmladší pacienti byli transplantováni časně po diagnóze, záhy po první závažné infekci. Nejstarší chlapec vstupoval do transplantace jako vysoce rizikový, dlouhodobě léčený pro opakované infekce, aktuálně s floridní granulomatózou střeva. Mykotický absces mozku, který se mu stal osudným, zřejmě souvisel s klinicky němou kolonizací pacienta danou jeho věkem a délkou expozice infekčním agens.
Frekvence komplikací byla v našem souboru pacientů poměrně vysoká. U 3 pacientů se objevila akutní GvHD II. stupně, infekční komplikace se vyskytly u 4 pacientů (2krát aktivace CMV, 1krát katétrová sepse, 1krát mykotický absces mozku). Další vedlejší účinky (hepatopatie, nefropatie, intersticiální pneumonie, hypertenze) byly přítomny u 3 pacientů. Nápadná byla vyšší incidence autoimunitních projevů (1krát AIHA, 1krát ITP, 1krát imunní pancytopenie) související zřejmě se smíšeným lymfocytárním chimerismem po přípravném režimu s redukovanou intenzitou.
U všech pacientů došlo k reparaci funkce granulocytů a tedy k vyléčení CGD. Ve shodě s literaturou vedla HSCT k vyhojení probíhající střevní granulomatózy [14], aktinomykózy [16] i zhojení jaterních abscesů. I pacienty, u kterých je proporce funkčních granulocytů menší než u zdravých kontrol, lze považovat za vyléčené, neboť podle publikovaných údajů je minimální počet funkčních granulocytů nutných k ochraně před infekčními komplikacemi přibližně 10 % [1]. Otázkou zůstává, zda se v delší perspektivě u těchto pacientů s dvěma populacemi granulocytů nevyskytnou autoimunitní komplikace, tak jako u přenašeček X-vázané formy onemocnění [17].
Indikace k HSCT u pacientů s CGD by měla být zvážena u každého nově diagnostikovaného případu. Rozhodujícími kritérii jsou závažnost klinického průběhu onemocnění, tíže genetického defektu a jeho funkční dopad a samozřejmě také dostupnost vhodného dárce a souhlas rodičů po podání komplexní informace o prognóze pacienta (přežití i kvalita života). Při časování transplantace je třeba mít na mysli, že výsledky jsou příznivější u dětí transplantovaných v kojeneckém a batolecím věku. Na druhou stranou jsou tito pacienti více ohroženi pozdní toxicitou léčby.
Délka sledování našich pacientů je zatím příliš krátká na vyvozování rozsáhlých závěrů, ale dosavadní průběh a zkušenosti z jiných transplantačních center [2, 14] naznačují, že HSCT by mohla přispět ke zvýšení kvality i k prodloužení jejich života.
Práce byla podpořena granty MZO FNM2005 a MSM 00216812.
Dr. Janda je stipendistou fellowship programu Evropské společnosti pro imunodeficience (ESID) financované firmou Baxter. Rádi bychom poděkovali prof. Roosovi z Holandska a dr. Avčinovi ze Slovinska za ochotné provedení molekulárně genetických vyšetření u našich pacientů a kolegům z Kliniky zobrazovacích metod UK 2. LF za zapůjčení fotodokumentace.
Došlo: 13. 12. 2011
Přijato: 6. 2. 2012
MUDr. Aleš Janda, MSc., Ph.D.
Centrum für Chronische Immundefizienz (CCI) Universitätsklinikum Freiburg
Breisacher Str. 117
79106 Freiburg im Breisgau
Německo
e-mail: ales.janda@uniklinik-freiburg.de
Sources
1. Roos D, Kuijpers TW, Curnutte J. Chronic granulomatous disease, a molecular and cellular approach. In: Ochs HD, Smith E, Puck JM. Primary Immunodeficiency Diseases. Oxford: Oxford University Press, 2006 : 525–549.
2. Kang EM, et al. Chronic granulomatous disease: overview and hematopoietic stem cell transplantation. J Allergy Clin Immunol 2011; 127 (6): 1319–1326; quiz 1327–1328.
3. Marciano BE, et al. Gastrointestinal involvement in chronic granulomatous disease. Pediatrics 2004; 114 (2): 462–468.
4. Seger RA. Chronic granulomatous disease: recent advances in pathophysiology and treatment. Neth J Med 2010; 68 (11): 334–340.
5. Seger RA. Modern management of chronic granulomatous disease. Br J Haematol 2008; 140 (3): 255–266.
6. Winkelstein JA, et al. Chronic granulomatous disease. Report on a national registry of 368 patients. Medicine (Baltimore) 2000; 79 (3): 155–169.
7. Jones LB, et al. Special article: chronic granulomatous disease in the United Kingdom and Ireland: a comprehensive national patient-based registry. Clin Exp Immunol 2008; 152 (2): 211–218.
8. van den Berg JM, et al. Chronic granulomatous disease: the European experience. PLoS One 2009; 4 (4): e5234.
9. Kuhns DB, et al. Residual NADPH oxidase and survival in chronic granulomatous disease. N Engl J Med 2010; 363 (27): 2600–2610.
10. Banerjee R, et al. Cutting edge: infection by the agent of human granulocytic ehrlichiosis prevents the respiratory burst by down-regulating gp91phox. J Immunol 2000; 164 (8): 3946–3949.
11. Honzík T, et al. Prolonged impairment of polymorphonuclear cells functions in one infant with transient zinc deficiency: A case report. Prague Med Rep 2008; 109 (2–3): 184–193.
12. Seger RA. Advances in the diagnosis and treatment of chronic granulomatous disease. Curr Opin Hematol 2011; 18 (1): 36–41.
13. Stein S, et al. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat Med 2010; 16 (2): 198–204.
14. Seger RA. Hematopoietic stem cell transplantation for chronic granulomatous disease. Immunol Allergy Clin North Am 2010; 30 (2): 195–208.
15. Janda A. Pacienti s chronickou granulomatózní nemocí v České a Slovenské republice. Alergie 2010; 12 (2): 112–120.
16. Reichenbach J, et al. Actinomyces in chronic granulomatous disease: an emerging and unanticipated pathogen. Clin Infect Dis 2009; 49 (11): 1703–1710.
17. Cale CM, Mortonand L, Goldblatt D. Cutaneous and other lupus-like symptoms in carriers of X-linked chronic granulomatous disease: incidence and autoimmune serology. Clin Exp Immunol 2007; 148 (1): 79–84.
Labels
Neonatology Paediatrics General practitioner for children and adolescentsArticle was published in
Czech-Slovak Pediatrics

2012 Issue 2
- The Importance of Limosilactobacillus reuteri in Administration to Diabetics with Gingivitis
- What Effect Can Be Expected from Limosilactobacillus reuteri in Mucositis and Peri-Implantitis?
Most read in this issue
- Transplantace hematopoetických buněk u pěti pacientů s chronickou granulomatózní nemocí v České republice
- Postižení kardiovaskulárního systému u Turnerova syndromu
- Případ akcidentální otravy chlapců veterinárním anestetikem
- Růst a pubertální vývoj u dětí s intrauterinní růstovou retardací v moravské větvi studie ELSPAC