
Hirschsprungova choroba a její genetické příčiny
Hirschsprung´s disease and its genetic cause
Hirschsprung´s disease (HSCR) is a congenital developmental disease of enteric nervous system. The cause is the congenital primary loss of intramural innervation of certain bowel segment. The affected part of bowel is permanently contracted and the healthy bowel above is dilatated and it leads to the development of megacolon. Several variants of HSCR are distinguished by different length of affected colon. It is occurred alone or syndromic, sporadic (80%) or familial form (20%) of HSCR. Genetic cause of HSCR is complex, 12 causative genes are known to be involved in different signal cascades. The major gene is the RET proto-oncogene, where causative inactivating mutations or predisposing variants in coding or non-coding part of gene are found. The other genes are involved in RET/GDNF or EDNRB/ENT3 cascades and transcription factors modulating activities of these cascades. These cascades and factors are in connection and influence themselves. Beside causative genes several modifying genes are known that can influence phenotype of HSCR. Due to the recognition of genetic cause of HSCR it is possible to find the cause of development of HSCR, distinguish the inheritance and the penetrance of each mutation. Because mutations in the RET proto-oncogene are known to be the cause of development of medullary thyroid carcinoma (MTC), patients with HSCR are in higher risk of MTC and all patients should be recommended to genetic screening.
Key words:
Hirschsprung´s disease, neurocristopathy, genetics, RET proto-oncogene, medullary thyroid carcinoma, signal cascades
:
Š. Dvořáková 1; E. Václavíková 1,2; R. Škába 3; L. Kavalcová 3; B. Bendlová 1
![]()
:
Oddělení molekulární endokrinologie, Endokrinologický ústav, Prahavedoucí Mgr. J. Včelák
1; Katedra biochemie, Přírodovědecká fakulta, Univerzita Karlova, Prahavedoucí doc. RNDr. M. Šulc, Ph. D.
2; Klinika dětské chirurgie, UK 2. LF a FN Motol, Prahapřednosta prof. MUDr. J. Šnajdauf, DrSc.
3
:
Čes-slov Pediat 2013; 68 (3): 167-176.
:
Review
Hirschsprungova choroba (HSCR) je vrozené vývojové onemocnění enterického nervového systému. Příčinou je chybějící intramurální inervace určitého úseku střeva. Postižený střevní úsek je trvale stažen a zdravé střevo nad ním se postupně dilatuje a vytváří megakolon. Rozlišuje se několik variant HSCR podle délky postiženého úseku střeva. HSCR se vyskytuje izolovaně nebo syndromicky, sporadicky (80 %) nebo familiárně (20 %). Genetická podstata HSCR je komplexní, je známo 12 příčinných genů zapojených v různých signalizačních kaskádách. Majoritním genem je RET proto-onkogen, kde se nalézají příčinné inaktivující mutace nebo predispoziční varianty v kódující i nekódující části genu. Dalšími geny jsou geny zapojené do RET/GDNF kaskády nebo EDNRB/ENT3 kaskády a s nimi spojené transkripční faktory modulující aktivitu těchto drah. Tyto dráhy a faktory spolu souvisejí a vzájemně se ovlivňují. Kromě příčinných genů je známa řada modifikujících genů, které mohou ovlivňovat fenotyp HSCR. Poznáním genetické podstaty HSCR je možné odhalit příčinu vzniku HSCR, zjistit dědičnost a penetranci jednotlivých mutací. Protože RET proto-onkogen je také příčinou vzniku medulárního karcinomu štítné žlázy (MTC), jsou pacienti s HSCR ve zvýšeném riziku MTC a všichni pacienti by měli být doporučeni ke genetickému screeningu.
Klíčová slova:
Hirschsprungova choroba, neurokristopatie, genetika, RET proto-onkogen, medulární karcinom štítné žlázy, signalizační dráhy
ÚVOD
Hirschsprungova choroba (HSCR), nebo-li kongenitální (vrozená) střevní aganglionóza, historicky megacolon congenitum, je charakterizována absencí parasympatických gangliových buněk v submukózním a myenterickém plexu gastrointestinálního traktu. Postižený střevní úsek je trvale stažen a zdravé střevo nad ním se postupně dilatuje a vytváří megakolon. Přechod mezi postiženým a zdravým střevem má typický nálevkovitý tvar. Příčinou HSCR je předčasné zastavení migrace, proliferace nebo diferenciace buněk neurální lišty při tvorbě střevního nervového systému mezi 5. a 12. týdnem embryonálního vývoje [1]. HSCR má své jméno po dánském pediatrovi Haraldu Hirschsprungovi, který v roce 1888 publikoval práci o dvou chlapcích, kteří zemřeli a až při pitvě se nalezlo neobyčejné rozšíření tlustého střeva (megakolon), které Hirschsprung považoval za vrozené. V roce 1940 bylo popsáno chybění nervových buněk v Auerbachově myenterické nervové pleteni ve střevě u HSCR. Orvar Swenson v roce 1948 navrhl chirurgický postup pro léčbu pacientů s HSCR, který se v různých modifikacích používá dodnes. V roce 1974 Robert P. Bolande použil termín neurokristopatie pro onemocnění, syndromy nebo nádory pocházející z neurální lišty a HSCR byla zařazena do skupiny jednoduchých neurokristopatií [2].
Střevní nebo-li enterický nervový systém je velmi komplexní, koordinuje střevní motilitu, imunitní reakce, změny krevního průtoku a sekreci vody a elektrolytů. Během vývoje vzniká z vysoce migrující populace buněk neurální lišty. Kolonizace gastrointestinálního traktu a diferenciace buněk neurální lišty do neuronů je velmi složitý proces. Tvorba funkčního střevního nervového systému vyžaduje koordinaci mnoha procesů, včetně přežití, migrace, proliferace a diferenciace prekurzorových buněk v gastrointestinálním traktu. Načasování a harmonizace transkripčních faktorů, růstových faktorů, jejich receptorů a jimi aktivovaných signálů jsou nepostradatelné pro všechna stadia vývoje enterického nervového systému. Každý defekt v tomto procesu může vést k vývoji HSCR [3]. Selhání buněk neurální lišty při kolonizaci gastrointestinálního traktu vede k tomu, že v různě dlouhých úsecích chybí enterické neurony (aganglionární část střeva). Aganglionóza začíná vždy v análním kanálu a plynule postupuje orálně. Protože jsou enterické neurony nepostradatelné pro motilitu, aganglionární část brání průchodu materiálu nacházejícího se v lumen střeva. Podle délky postiženého úseku rozeznáváme z chirurgického pohledu ultrakrátkou nízkou formu (UKS – do 10 %) postihující pouze anální kanál, klasickou rektosigmoideální formu (70–80 %), formu s aganglionózou celého tlustého střeva a části terminálního ilea (do 10 %) a diseminované formy s postižením téměř celého gastrointestinálního traktu (NTBA – 4 %). Z genetického pohledu se rozeznává krátká forma (S-HSCR – 80 %), dlouhá forma (L-HSCR – 15 %) a totální střevní aganglionóza (TIA – 5 %). Zajímavé je, že S-HSCR, chirurgicky klasická forma HSCR, je 4x častější u mužů než u žen, L-HSCR a TIA se vyskytují se stejnou frekvencí u mužů i u žen [4].
HSCR je vrozené onemocnění, jehož incidence je 1 : 5000 narozených dětí. Choroba je nejčastěji zjištěna již po narození. U novorozenců dochází k pozdržení odchodu smolky za hranici 48 hodin, postupně nastupuje zvracení a rozvíjí se ileózní stav. Někteří pacienti mohou být výjimečně diagnostikováni až v předškolním věku. Případy HSCR diagnostikované v dospělosti jsou velmi vzácné a projeví se většinou krytou perforací dilatovaného, skybalem naplněného střeva.
Jedinou léčbou je chirurgické odstranění aganglionárního úseku střeva, jinak hrozí obávaný zánět zbytku tlustého i tenkého střeva (enterokolitida spojená s HSCR) a sepse u malých dětí. U adolescentů a dospělých je to výše zmíněná perforace tlustého střeva [5, 6].
Genetické příčiny HSCR
HSCR je spojena zhruba u 12 % pacientů s chromozomálními abnormalitami (v 90 % doprovází Downův syndrom) a u 18 % pacientů s dalšími vrozenými anomáliemi (tab. 1). V 70 % se vyskytuje jako izolovaná nesyndromická forma, a to buď familiárně (20 %), nebo sporadicky (80 %) [7].

Izolovaná familiární forma HSCR má komplexní dědičnost s pohlavně dependentní, většinou nekompletní penetrancí a fenotypovou variabilitou. U dlouhé formy HSCR (L-formy) je navrhován dominantní či aditivní model dědičnosti, zatímco pro krátkou formu (S-formu) spíše recesivní či multifaktoriální model dědičnosti. HSCR je multigenní onemocnění, kde stejný fenotyp může být spojen s mutacemi v různých příčinných genech a různou variabilitou tzv. modifikujících genů. V současné době je známo 12 příčinných genů (RET, EDNRB, EDN3, GDNF, NTN, SOX10, PHOX2B, ECE1, KIAA1279/KBP, ZFHX1B, TTF-1, NRG1) a 5 chromozomálních lokusů zapojených do rozvoje HSCR [1]. Všechny tyto geny jsou zapojeny do časného embryonálního vývoje střevní nervové soustavy. Geny kódují hlavně komponenty signalizačních drah RET/GDNF a EDN3/EDNRB a transkripční faktory, jako SOX10, PHOX2B nebo ZFHX1B, které se v těchto drahách uplatňují (obr. 1).

U některých pacientů může být fenotyp výsledkem kumulativního efektu nejméně dvou mutací v různých genech. Genetický screening však odhaluje mutace v těchto genech jen u 50 % pacientů, zhruba u poloviny pacientů tak zatím zůstává genetická příčina onemocnění neznámá. Avšak důležitou roli v projevu a variabilitě HSCR hrají také epigenetické faktory. Některé varianty genů v nekódujících úsecích DNA, studované především u RET proto-onkogenu, mohou ovlivňovat fenotyp HSCR. Důležité funkční varianty jsou nalézány v promotorech a enhancerech genů, mohou se také tvořit nová sestřihová místa v intronech genů. Tím se může regulovat exprese genů, které jsou zodpovědné za diferenciaci a migraci buněk neurální lišty [8].
1. Signalizační dráha RET/GDNF
RET proto-onkogen (REarranged during Transfection) je majoritním genem u HSCR, kde je v kódující části genu nalézáno přes 100 různých typů mutací včetně velkých delecí, mikrodelecí, inzercí, nesmyslných, záměnných a nestřihových mutací. Byly definovány i predisponující haplotypy (specifická kombinace alel na jednom chromozomovém vlákně). Mutace jsou nalézány v rámci celého genu, nejsou zde žádné „hot-spot“ mutace, což ztěžuje molekulárně genetickou analýzu [9]. RET proto-onkogen se skládá z 21 exonů, jeho velikost je 55 kB a je lokalizován na 10q11.2. Gen kóduje RET protein, což je transmembránový receptor s extracelulární částí zahrnující dvě funkčně důležité domény (doména podobná kadherinu, doména bohatá na cysteiny), které jsou důležité pro vazbu ligandu a dimerizaci receptoru. RET protein dále sestává z transmembránové domény a intracelulární části se dvěma tyrozinkinázovými doménami, které jsou po vazbě ligandu autofosforylovány a tím dochází k přenosu signálu dále do buňky. RET protein reguluje diferenciaci, proliferaci a migraci buněk, buněčné přežívání a má také proapoptotickou aktivitu závislou na kaspázách. K aktivaci RET signalizace je potřeba multiproteinový komplex tvořený z některého z rozpustných ligandů (GDNF (neurotrofický faktor), neurturin, persefin nebo artemin), koreceptoru GFRα1-4 (membránově ukotvený glykosyl-fosfatidylinozitol koreceptor GDNF) a RETu. Vytvoření tohoto komplexu vede k homodimerizaci RETu a autofosforylaci tyrozinových zbytků v tyrozinkinázové doméně. Tím dochází k aktivaci dvou signalizačních kaskád vedoucích k aktivaci MAPK (mitogenem aktivovaná proteinová kináza) a PI3K (fosfatidylinozitol-3-kináza). Funkční studie ukazují různé vlastnosti mutací u pacientů s HSCR. Všechny tyto mutace RET způsobují ztrátu funkce (haploinsuficienci) [10, 11]. Zárodečné mutace v RET genu byly identifikovány u 50 % familiárních a 3–15 % sporadických HSCR. Pokud rozdělíme pacienty podle formy HSCR, záchyt mutací v RET genu je mnohem vyšší u dlouhé formy HSCR oproti formě krátké (u L-HSCR je 70–80 % a u S-HSCR 17–38 %).
Klinicky významné je možné spojení HSCR s medulárním karcinomem štítné žlázy (MTC) či syndromy mnohočetné endokrinní neoplazie typu 2 (MEN2). K tomuto souběhu obou onemocnění dochází v rodinách s mutacemi v 10. exonu RET proto-onkogenu (kodony 609, 611, 618 a 620). Jsou to tzv. duální mutace, kde ve štítné žláze dochází k nekontrolovatelné proliferaci C-buněk a vzniku medulárního karcinomu štítné žlázy, naproti tomu ve střevě dochází k apoptóze enterických ganglií [12]. Všichni pacienti s HSCR jsou proto v riziku MTC a měli by být geneticky vyšetřeni na rizikové exony RET proto-onkogenu. Přestože se nenalezly mutace v RET proto-onkogenu u všech pacientů, vazebné analýzy odhalily RET gen jako hlavní rizikový faktor HSCR téměř u všech rodin HSCR (90 %), kde je silná vazba mezi určitým haplotypem a HSCR [13, 14]. Tento haplotyp začíná v promotoru RETu asi 4 kb před startem transkripce genu a pokračuje na začátek 2. exonu. Další důležité nekódující místo je na 3´konci genu, záměna sekvence v tomto úseku snižuje stabilitu RET mRNA. Mutace RET proto-onkogenu jsou sice hlavním rizikovým faktorem, ale samotné mutace nemusí být pro vznik HSCR dostatečným impulzem. Svědčí pro to nekompletní penetrance mutovaných alel, tzn. že i prokazatelně patogenní mutace můžeme nacházet u klinicky asymptomatických jedinců. U většiny pacientů se tak na patogenezi HSCR pravděpodobně podílejí ještě další mutace v neznámých genech. Byly objeveny další lokusy 9q31, 19q12, 3p21, 16q23.3, 4q31-32, které segregují s onemocněním [15]. Pro vysvětlení poměrně nízkého záchytu mutací RET genu existuje několik hypotéz:
- k vyjádření fenotypu HSCR je třeba interakce několika genů;
- je zvažován možný vliv neutrálních genových variant působících jako nízko penetrující alely (specifické jednonukleotidové záměny nebo haplotypy);
- tyto varianty RET genu mohou být ve vazebné nerovnováze s dosud neznámým příčinným lokusem, který je v blízkosti RET genu;
- možná je i přítomnost stále nedetekovaných mutací v nekódující části RET genu (regulační funkce nebo role v procesování transkriptu či maturaci proteinu).
Vzácněji jsou u pacientů s HSCR nalézány mutace v genech kódujících ligandy či koreceptory RETu. Mutace v neurotrofickém faktoru (GDNF) jsou detekovány méně než u 5 % pacientů s HSCR. GDNF je secernovaný protein (211 aminokyselin), jehož proteolytickým štěpením vzniká zralý protein (134 aminokyselin). Tento majoritní ligand RETu se váže na koreceptor GFRα1 a společně aktivují RET protein. GDNF je znám jako hlavní přežívací faktor pro mnoho typů neuronů. Absence GDNF ve střevě může způsobit ztrátu nebo snížení aktivace RET receptoru, způsobující zastavení migrace neuroblastů. Mutace GDNF možná nejsou dostačující pro vznik HSCR, protože u několika pacientů byla nalezena kromě mutace GDNF ještě mutace RET, nebo trizomie 21 [16].
Mutace v genu dalšího známého ligandu RETu – neurturinu (NTN) byla identifikována u 1 rodiny spolu s mutací RET.
U pacientů s HSCR zatím nebyla identifikována žádná mutace v genu pro koreceptor GFRα1, kromě delece s nekompletní penetrancí u 1 rodiny [17].
2. Signalizační dráha EDN3/EDNRB
Tato endotelinová dráha reguluje správnou migraci nervových buněk podél střeva, zachovává proliferační fázi, ale zejména diferenciace buněk je prostřednictvím této kaskády inhibována. Detekční záchyt mutací v těchto genech je asi u 5 % pacientů s HSCR. Mutace v genech pro endotelinový receptor B (EDNRB) a pro endotelin 3 (EDN3) způsobují Shah-Waardenburgův syndrom (HSCR, vrozená ztráta sluchu, pigmentační anomálie vlasů, kůže a očí).
EDN3 je secernovaný peptid (21 aminokyselin), který je exprimovaný v nezralé formě a musí být přeměněn na aktivní peptid endotelin konvertujícím enzymem 1 (ECE1). Preproendoteliny jsou proteolyticky štěpeny pomocí dvou souvisejících membránově vázaných metaloproteáz, které dávají vznik zralému endotelinu. Mutace v EDN3 jsou zřídka nalézány u izolovaných i syndromických forem HSCR.
EDNRB je G-protein-vázající receptor důležitý pro vývoj střevních neuronů. EDNRB váže všechny tři ligandy EDN1, 2, 3, ale hlavním ligandem je EDN3. Gen je lokalizován na chromozomu 13q22 a mutace v genu pro EDNRB byly nalezeny u asi 5 % pacientů s HSCR, přičemž penetrance mutací je nekompletní [18].
ECE1 je enzym přeměňující inaktivní prekurzor EDN3 do aktivního EDN3 peptidu. ECE1 procesuje jen EDN1 a EDN3. Mutace v genu kódujícím ECE1 byla objevena jen u jediného pacienta s HSCR, který měl navíc vrozené vady obličeje a srdce [19].
3. Transkripční faktory
V patogenezi HSCR se uplatňují i trans-kripční faktory (SOX10, PHOX2B, ZFHX1B a KIAA1279), které bezprostředně ovlivňují výše zmíněné signalizační dráhy.
SOX10 (SRY (sex determining factor)-like region Y-box10) patří do rodiny DNA vazebných proteinů. SOX10 je exprimován migrujícími buňkami neurální lišty. SOX10 reguluje expresi RETu i EDNRB. Mutace tohoto transkripčního faktoru zvyšují DNA vazebnou afinitu a inhibují diferenciaci nervových buněk [20]. Mutace jsou nalézány u 5 % syndromických (Waardenburgův syndrom) i izolovaných forem HSCR.
PHOX2B (paired-like homeobox 2b) je další transkripční faktor exprimovaný migrujícími buňkami neurální lišty. Gen je lokalizován na chromozomu 4p12. Reguluje expresi RETu. Mutace tohoto genu jsou nalézány u 2/3 pacientů s Haddadovým syndromem (HSCR a vrozený centrální hypoventilační syndrom). Nacházeny jsou hlavně polyalaninové expanze, záměnné mutace či delece [21].
ZFHX1B (zink finger homeodomain trans-kripční faktor, nebo také SMAD interagující protein 1 (SMADIP1/SIP1)) je transkripčním korepresorem cílových genů a gen je lokalizován na chromozomu 2q22. Je esenciální pro vývoj migrujících buněk neurálních prekurzorů. Mutace v tomto genu jsou spojeny s Mowatovým-Wilsonovým syndromem (mentální retardace, epilepsie, opožděný motorický vývoj, HSCR, fenotyp obličeje, srdeční vada, urogenitální anomálie). Nalézány jsou delece nebo záměnné mutace [22].
KIAA1279/KBP (kinesin-binding protein) je protein se stále neznámou funkcí, je však důležitý pro periferní i centrální nervový systém. Gen je lokalizován na chromozomu 10q21.3-q22.1. Mutace se nacházejí u Goldbergové-Shprintzenova syndromu (HSCR, mikrocefalie, mentální retardace, rozštěp patra, hypotonie, malý vzrůst, silné řasy a řídké vlasy) [23].
4. Interakce mezi signalizačními kaskádami a modifikující geny
RET/GDNF a EDN3/EDNRB signalizační cesty byly dlouho považovány za biochemicky nezávislé, avšak nyní se ukazuje, že jsou vzájemně nepřímo propojeny (obr. 2). Mohou interagovat modulací aktivity stejné signální molekuly [24, 25], kterou je nejspíše proteinkináza A (PKA). EDN3/EDNRB a RET/GDNF dráhy působí synergicky na proliferaci, ale působí antagonisticky na migraci buněk. Ačkoliv EDN3/EDNRB cesta je pro normální migraci buněk nezbytná, EDN3/EDNRB signalizace inhibuje migrační odpověď aktivovanou cestou RET/GDNF. GDNF zahajuje proliferaci migrujících buněk a diferenciaci, EDN3 inhibuje diferenciaci způsobenou GDNF. EDN3 je také inhibitorem proteinkinázy A. EDN3 tak vyvažuje vliv GDNF, chrání buňky před předčasnou diferenciací a zachovává tyto buňky v proliferačním stavu, aby kolonizovaly střevo po celé délce.

Interakce mezi RET a EDNRB signalizačními cestami na molekulární a biochemické úrovni mohou tvořit základ pro vznik HSCR (obr. 3) a ovlivnit výskyt a závažnost agan-glionózy. Např. byl popsán pacient s HSCR, který měl slabou hypomorfní mutaci ve dvou genech RET i EDNRB, každou mutaci zdědil po zdravém rodiči. Je také popsána genetická vazba mezi EDNRB a RET, kdy se dědí mutace EDNRB s určitými alelami RET genu.
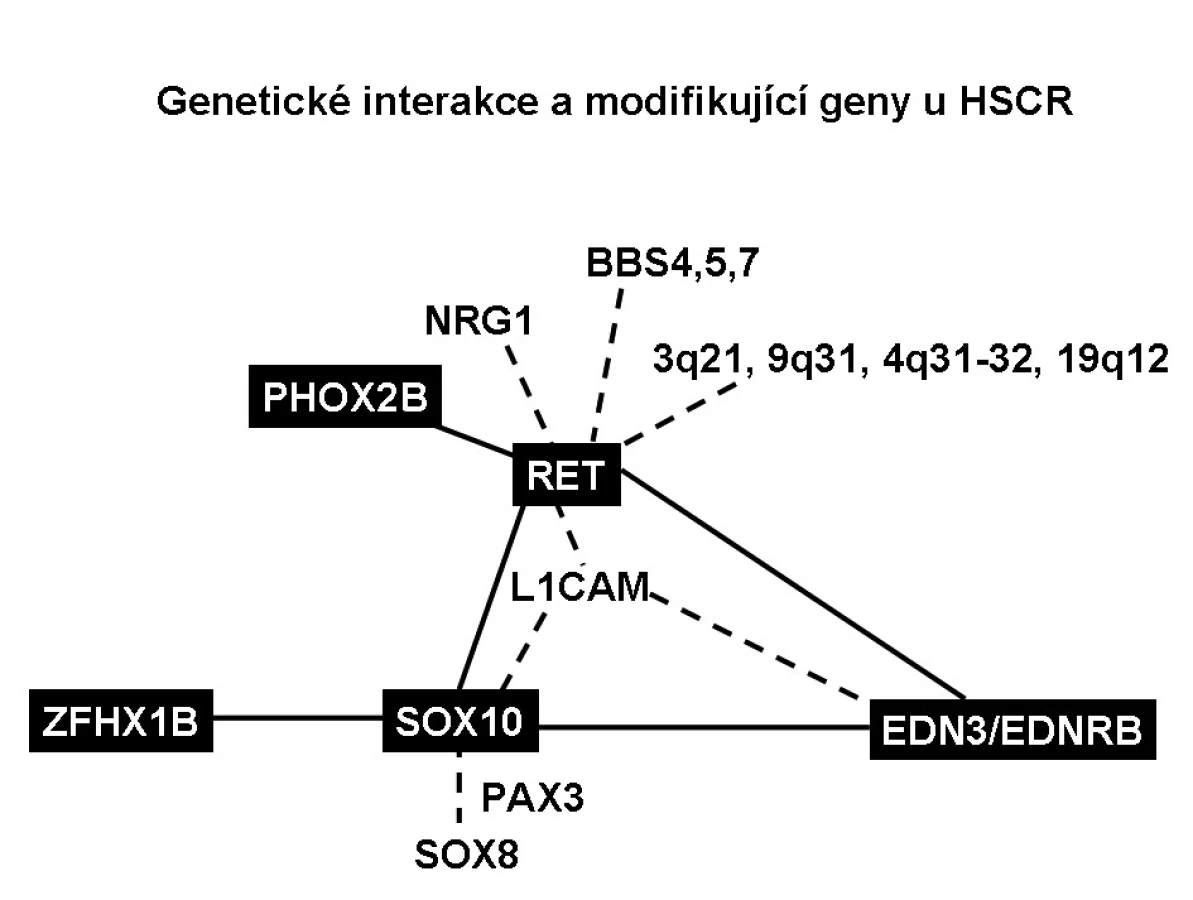
Byla nalezena genetická vazba SOX10 s EDNRB a SOX10 s EDN3, které jsou schopny způsobit těžkou aganglionózu. Ačkoliv celogenomová vazebná studie nenašla genetickou interakci mezi SOX10 a RET, SOX10 tvoří komplex s transkripčním faktorem PAX3, který přímo reguluje expresi RETu [26]. SOX10 také ovlivňuje expresi EDNRB tím, že v enhancerové oblasti EDNRB promotoru je vazebné místo pro SOX10. Snížení exprese genu SOX10 tak vede ke snížení exprese EDNRB. Mutace v EDN3 či EDNRB a SOX10 spolupůsobí oproti mutacím v genech samotných a zhoršují aganglionózu. Transkripční faktory SOX10 a ZFHX1B spolu také interagují, mutace v obou genech dohromady zhoršují aganglionózu, ale přesný mechanismus jejich působení není znám. Interakce genových variant RET, PHOX2B a BBS genů je zodpovědná za syndrom Bardet-Biedlův a centrální hypoventilační syndrom. Funkční spojení spočívá v tom, že transkripční faktor PHOX2B se váže na promotor RETu a reguluje jeho transkripci.
Existují i další signální dráhy zapojené ve vývoji HSCR – NOTCH, WNT a TGFβ/BMP. NOTCH dráha je zapojena v různých aspektech neurogeneze. Může snižovat buněčnou proliferaci, působí jako negativní regulátor diferenciace neuronů. WNT signalizace indukuje buněčnou proliferaci. TGFβ/BMP dráha je důležitá v buněčné agregaci a tvorbě gangliových neuronů. Je známa i interakce mezi RET a TGFβ/BMP.
Vzhledem k nekompletní penetranci onemocnění a velmi rozdílné expresivitě mutací i v rámci jednotlivých rodin je v patogenezi HSCR zvažována i role modifikujících genů [8]. Modifikující gen je takový gen, jehož mutace samy o sobě nepostačují k projevení patologického fenotypu, ale ve spojení s genetickými mutacemi jiných genů už efekt projevují nebo potencují efekt příčinných mutací. Zatím bylo identifikováno jen málo modifikujících genů. Např. je známo, že fenotypické projevy některých mutací RET genu modifikují určité chromozomální oblasti – 3q21, 4q31-32, 8p12, 9q31 a 19q12, ale geny, které se zde vyskytují, nebyly ještě identifikovány. Díky studiu genetického pozadí Bardet-Biedelova syndromu, jehož jedním ze symptomů bývá HSCR, byly nalezeny tři další modifikující geny RETu BBS4, BBS5 a BBS7. Dalším genem, který může modifikovat expresi mutací RET, ale i EDN3/EDNRB signalizační kaskádu a zhoršovat tak fenotyp HSCR, je L1 nervová buněčná adhezivní molekula (L1CAM) lokalizovaná na chromozomu X [27]. Funkčně důležité je ovlivnění transkripční aktivity SOX10, která je modifikována nejen L1CAM, ale také EDNRB, PHOX2B a SOX8. Posledně zmíněný transkripční faktor SOX8 je exprimován též v buňkách neurální lišty a je schopen aktivovat velmi podobné geny jako SOX10. Jsou známy genetické varianty SOX8, které zhoršují aganglionózu. Také např. specifický haplotyp genu neuregulinu 1 (NRG1) zvyšuje riziko HSCR [28]. NRG1 je zapojen v regulaci vývoje neurální lišty a je také regulován SOX10.
Závěr
HSCR je geneticky velmi heterogenní a mutace se vyskytují v různých signalizačních cestách. Je nezpochybnitelné, že klíčovým genem u HSCR je RET proto-onkogen a kromě různorodých mutací zde hrají velkou roli i rizikové haplotypy, což dokazují vazebné analýzy i studium polymorfismů. Bylo identifikováno mnoho dalších klíčových genů, které jsou schopny indukovat HSCR. Nyní je třeba studovat interakce mezi nimi, zjistit, proč je fenotyp onemocnění tak variabilní a penetrance mutací tak rozdílná. Jsou známé raritní případy, kdy pacienti mají mutace ve více než jednom rizikovém genu (RET + GDNF, RET + NTN, RET + EDNRB). Zdá se, že čím více mutací je u pacienta detekováno, tím horší průběh onemocnění má. Intenzivně se studuje i vliv modifikujících genů. Je potřeba syntetizovat výsledky vazebných a asociačních studií s profily genové exprese a s výsledky studií modelových organismů a pečlivě korelovat nalezený genotyp s fenotypovými projevy. Co se týče léčby, výzkum se zaměřuje jednak na vývoj kmenových buněk s cílem obnovy aganglionárního úseku bez nutnosti operačního zákroku, či na různé transplantační studie, kde by se injekcí do postižené střevní stěny zaváděly pacientovy vlastní buňky obdržené z biopsie normálně kolonizovaného úseku střeva [3].
Pro klinickou praxi a genetické poradenství je důležité u pacienta najít genetickou příčinu vzniku HSCR. I když je to značně komplikované, jednotlivým rodinám zjištění příčinné mutace velmi pomůže. U pacientů s HSCR je ale zejména velmi důležité zjištění rizika vzniku medulárního karcinomu štítné žlázy. V našem souboru pacientů s HSCR jsme klasické duální mutace RET genu spojené se vznikem MTC detekovali ve dvou rodinách. Při záchytu známé rizikové mutace je pacientovi doporučena profylaktická totální tyreoidektomie, která by jej měla, pokud je provedena do 5 let věku, celoživotně uchránit před rozvojem tohoto nádorového onemocnění. Při záchytu atypických mutací v MTC rizikových exonech RETu jsou tito naši pacienti i jejich rodinní příslušníci, kteří sice nemají HSCR, ale jsou nositeli téže mutace, dispenzarizováni a v pravidelných intervalech je u nich vyšetřována štítná žláza a je sledována hladina kalcitoninu. Doporučujeme, aby se všichni pacienti s HSCR podrobovali genetickému screeningu, zaměřenému především na mutace v 10. a 11. exonu RET proto-onkogenu, který provádíme na našem pracovišti [29].
Poděkování
Tato práce byla finančně podpořena granty IGA MZ ČR NT 13901-4, GAUK 411611 a projektem Ministerstva zdravotnictví ČR koncepčního rozvoje výzkumné organizace 00023761 (Endokrinologický ústav, Praha).
Došlo: 16. 1. 2013
Přijato: 7. 4. 2013
RNDr. Šárka Dvořáková, Ph.D.
Oddělení molekulární endokrinologie
Endokrinologický ústav
Národní 8
116 94 Praha 1
e-mail: sarka@obloha.cz
sdvorakova@endo.cz
Sources
1. Amiel J, Sproat-Emison E, Garcia-Barcelo M, et al. Hirschsprung disease, associated syndromes and genetics: a review. J Med Genet 2008; 45 : 1–14.
2. Martucciello G, Ceccherini I, Lerome M, et al. Pathogenesis of Hirschsprung’s disease. J Pediatr Surg 2000; 35 : 1017–1025.
3. Heanue TA, Pachnis V. Enteric nervous system development and Hirschsprung‘s disease: advances in genetic and stem cell studies. Nat Rev Neurosci 2007; 8 : 466–479.
4. Amiel J, Lyonnet S. Hirschsprung disease, associated syndromes, and genetics: a review. J Med Genet 2001; 38 : 729–739.
5. Kessmann J. Hirschsprung‘s disease: diagnosis and management. Am Fam Physician 2006; 74 : 1319–1322.
6. de Lorijn F, Kremer LC, Reitsma JB, et al. Diagnostic tests in Hirschsprung disease: a systematic review. J Pediatr Gastroenterol Nutr 2006; 42 : 496–505.
7. Godbole K. Many faces of Hirschsprung‘s disease. Indian Pediatr 2004; 41 : 1115–1123.
8. Wallace AS, Anderson RB. Genetic interactions and modifier genes in Hirschsprung‘s disease. World J Gastroenterol 2011; 17 : 4937–4944.
9. Emison ES, Garcia-Barcelo M, Grice EA, et al. Differential contributions of rare and common, coding and noncoding Ret mutations to multifactorial Hirschsprung disease liability. Am J Hum Genet 2010; 87 : 60–74.
10. Lantieri F, Griseri P, Ceccherini I. Molecular mechanisms of RET-induced Hirschsprung pathogenesis. Ann Med 2006; 38 : 11–19.
11. Asai N, Jijiwa M, Enomoto A, et al. RET receptor signaling: dysfunction in thyroid cancer and Hirschsprung‘s disease. Pathol Int 2006; 56 : 164–172.
12. Arighi E, Popsueva A, Degl’Innocenti D, et al. Biological effects of the dual phenotypic Janus mutation of ret co-segregating with both multiple endocrine neoplasia type 2 and Hirschsprung’s disease. Mol Endocrinol 2004; 18 : 1004–1017.
13. Crockett DK, Piccolo SR, Ridge PG, et al. Predicting phenotypic severity of uncertain gene variants in the RET proto-oncogene. PLoS One 2011; 6: e18380.
14. Burzynski GM, Nolte IM, Bronda A, et al. Identifying candidate Hirschsprung disease-associated RET variants. Am J Hum Genet 2005; 76 : 850–858.
15. Bolk S, Pelet A, Hofstra RM, et al. A human model for multigenic inheritance: phenotypic expression in Hirschsprung disease requires both the RET gene and a new 9q31 locus. Proc Natl Acad Sci U S A 2000; 97 : 268–273.
16. Angrist M, Bolk S, Halushka M, et al. Germline mutations in glial cell line-derived neurotrophic factor (GDNF) and RET in a Hirschsprung disease patient. Nat Genet 1996; 14 : 341–344.
17. Doray B, Salomon R, Amiel J, et al. Mutation of the RET ligand, neurturin, supports multigenic inheritance in Hirschsprung disease. Hum Mol Genet 1998; 7 : 1449–1452.
18. Puffenberger EG, Hosoda K, Washington SS, et al. A missense mutation of the endothelin-B receptor gene in multigenic Hirschsprung’s disease. Cell 1994; 79 : 1257–1266.
19. Hofstra RM, Valdenaire O, Arch E, et al. A loss-of-function mutation in the endothelin-converting enzyme 1 (ECE-1) associated with Hirschsprung disease, cardiac defects, and autonomic dysfunction. Am J Hum Genet 1999; 64 : 304–308.
20. Pingault V, Bondurand N, Kuhlbrodt K, et al. SOX10 mutations in patients with Waardenburg-Hirschsprung disease. Nat Genet 1998; 18 : 171–173.
21. Amiel J, Laudier B, Attié-Bitach T, et al. Trang polyalanine expansion and frameshift mutations of the paired-like homeobox gene PHOX2B in congenital central hypoventilation syndrome. Nat Genet 2003; 33 : 459–461.
22. Zweier C, Albrecht B, Mitulla B, et al. Mowat-Wilson syndrome with and without Hirsch-sprung disease is a distinct, recognizable multiple congenital anomalies-mental retardation syndrome caused by mutations in the zinc finger homeo box 1B gene. Am J Med Genet 2002; 108 : 177–181.
23. Brooks AS, Bertoli-Avella AM, Burzynski GM, et al. Homozygous nonsense mutations in KIAA1279 are associated with malformations of the central and enteric nervous systems. Am J Hum Genet 2005; 77 : 120–126.
24. Carrasquillo MM, McCallion AS, Puffen-berger EG, et al. Genome wide association study and mouse model identify interaction between RET and EDNRB pathways in Hirschsprung’s disease. Nat Genet 2002; 32 : 237–244.
25. Barlow A, de Graaff E, Pachnis V. Enteric nervous system progenitors are coordinately controlled by the G protein-coupled receptor EDNRB and the receptor tyrosine kinase RET. Neuron 2003; 40 : 905–916.
26. Lang D, Epstein JA. Sox10 and Pax3 physically interact to mediate activation of a conserved c-RET enhancer. Hum Mol Genet 2003; 12 : 937–945.
27. Parisi MA, Kapur RP, Neilson I, et al. Hydrocephalus and intestinal aganglionosis: is L1CAM a modifier gene in Hirschsprung’s disease? Am J Med Genet 2002; 108 : 51–56.
28. Garcia-Barcelo MM, Tang CS, Ngan ES, et al. Genome-wide association study identifies NRG1 as a susceptibility locus for Hirschsprung‘s disease. Proc Natl Acad Sci U S A 2009; 106 : 2694–2699.
29. Vaclavikova E, Kavalcova L, Skaba R, et al. Hirschsprung‘s disease and medullary thyroid carcinoma: 15-year experience with molecular genetic screening of the RET proto-oncogene. Pediatr Surg Int 2012; 28 : 12312–12318.
Labels
Neonatology Paediatrics General practitioner for children and adolescentsArticle was published in
Czech-Slovak Pediatrics

2013 Issue 3
- What Effect Can Be Expected from Limosilactobacillus reuteri in Mucositis and Peri-Implantitis?
- The Importance of Limosilactobacillus reuteri in Administration to Diabetics with Gingivitis
Most read in this issue
- Hirschsprung´s disease and its genetic cause
- Nutritional screening upon admission to hospital – NutriAction
- Congenital surfactant deficiency due to ABCA3 mutations leading to fatal respiratory failure in a newborn
-
Incidence of idiopathic enteric inflammations in children and adolescents in the Plzeň (Pilsen) Region in 2001–2011.
A Prospective study