
Osteogenesis imperfecta asociovaná s izolovaným deficitom rastového hormónu
Osteogenesis imperfecta associated with isolated growth hormone deficiency
We report a case of 9.5-year-old boy with isolated GH deficiency and osteogenesis imperfecta type IV. The basis for prevention of pathological fractures in patients with this „brittle bone disease“ is the therapy with antiresorptive drugs – bisphosphonates. Experimental therapy of osteogenesis imperfecta is based on treatment with growth hormone because of its osteoanabolic effect. Due to combination of his two major diagnoses, we start simultaneous treatment with intravenous pamidronate and rHuGH (recombinant growth hormone).
Key words:
osteogenesis imperfecta, growth hormone deficiency, bisphosphonates, recombinant growth hormone
:
K. Kubejová; Ľ. Podracká
:
Klinika detí a dorastu LF UPJŠ a DFN, Košice
:
Čes-slov Pediat 2014; 69 (2): 114-120.
:
Case Report
Autori prezentujú kazuistiku 9,5-ročného chlapca s izolovaným deficitom rastového hormónu a s geneticky potvrdenou osteogenesis imperfecta (typ IV). Základom prevencie vzniku patologických zlomenín u pacientov s touto „chorobou krehkých kostí“ je antiresorbčná liečba bisfosfonátmi. Súčasťou experimentálnej terapie osteogenesis imperfecta je osteoanabolická liečba rastovým hormónom. Práve kombinácia dvoch závažných diagnóz nám umožnila začať nielen liečbu intravenóznym pamidronátom, ale aj substitučnú liečbu rHuGH (rekombinantný rastový hormón).
Kľúčové slová:
osteogenesis imperfecta, deficit rastového hormónu, bisfosfonáty, rekombinantný rastový hormón
ÚVOD
Osteogenesis imperfecta (OI) je zriedkavá dedičná porucha spojivového tkaniva charakterizovaná zvýšenou krehkosťou kostí, čo vhodne vystihuje v anglosaskej literatúre zaužívaný názov „brittle bone disease“ – „choroba krehkých kostí“. Výskyt ochorenia sa odhaduje na 1 prípad na 20 000 živonarodených detí [3, 4]. Vek v čase stanovenia diagnózy závisí od závažnosti klinických prejavov. Príčinou OI je mutácia génov kódujúcich alfa-1 a alfa-2 reťazce kolagénu I (90 % všetkých prípadov, tzv. kolagénové formy OI typ I – IV) a/alebo bielkoviny posttranslačnej modifikácie kolagénu (10 % prípadov, tzv. nekolagénové formy OI) [10]. Genetické mutácie spôsobujú kvantitatívnu alebo kvalitatívnu zmenu kolagénu, ktorý je hlavný proteín kostnej hmoty a tiež dôležitá stavebná bielkovina pre šľachy, väzy, kožu a skléry.
Fenotypické prejavy ochorenia sú pestré, závisia od stupňa kostnej fragility (v závažných prípadoch vznikajú progresívne skeletálne abnormality spôsobené početnými a opakovanými zlomeninami) a telesného rastu (normálna telesná výška až výrazne porušený rast). Na opozdenom raste postihnutých jedincov sa podpisujú najmä kostné deformity dlhých kostí, kompresívne fraktúry stavcov a kyfoskolióza. Ku klinickým znakom ochorenia patria tiež modré skléry (vyskytujú sa približne u polovice pacientov), rôzne závažné prejavy dentinogenesis imperfecta, môže byť pridružená aj hypermobilita kĺbov. Prenatálna diagnostika ochorenia sa opiera o ultrasonografické vyšetrenie. Pri náleze intrauterinných deformít a fraktúr, resp. ak už v rodine boli identifikované príčinné mutácie, sa diagnóza OI môže potvrdiť molekulovou analýzou DNA z amniocytov alebo z buniek choriových klkov [12]. Treba však uviesť, že negatívny výsledok genetického vyšetrenia túto diagnózu nevylučuje. Liečba osteogenesis imperfecta vyžaduje koordinovaný multidisciplinárny prístup a okrem farmakologickej terapie zahŕňa komplexnú ortopedickú, rehabilitačnú a fyzioterapeutickú starostlivosť. Hlavným cieľom je znížiť výskyt zlomenín a následných kostných deformít, a tým zlepšiť funkčnosť a mobilitu pacienta.
V kazuistike prezentujeme vzácny prípad 9,5–ročného chlapca s geneticky potvrdenou osteogenesis imperfecta (typ IV) a s izolovaným deficitom rastového hormónu.
KAZUISTIKA
Pacient, 9,5-ročný Marek, bol prijatý na našu kliniku pre dva týždne trvajúce intenzívne bolesti chrbtice, ktoré sa zhoršovali najmä pri chôdzi. RTG snímka preukázala klinovito deformované telá hrudných a driekových stavcov, preto ortopéd s podozrením na kompresívnu zlomeninu stavcov odoslal pacienta na hospitalizáciu.
V rodinnej anamnéze chlapca je pozoruhodný údaj o úmrtí otca na infarkt myokardu vo veku 45 rokov a výskyt opakovaných zlomenín u strýka a starého otca z matkinej strany. Marek sa narodil v 39. gestačnom týždni s pôrodnou hmotnosťou 3000 g a dĺžkou 50 cm. Priebeh gravidity, pôrod a perinatálna adaptácia boli fyziologické. Prvé dva roky bol jeho telesný vývoj primeraný, ale od troch rokov mal opakovane zlomené horné aj dolné končatiny (celkovo až 5 fraktúr), ktoré si v dvoch prípadoch vyžiadali operačné riešenie. Pri cielených otázkach matke na mechanizmy a okolnosti úrazu vysvitlo, že išlo o „nízko“ traumatické zlomeniny. V osobnej anamnéze bol pozoruhodný aj údaj o endokrinologickej dispenzarizácii pre zaostávanie v raste, ktoré bolo zjavné už v predškolskom období, avšak podrobnejšie sa začalo prešetrovať až v školskom veku. Z endokrinologickej dokumentácie sme zistili, že Marek vo veku7 rokov meral 113 cm (<3. percentil, Z-skóre -2,2 SD), pričom rastová rýchlosť bola pod 2 cm za rok. RTG snímka ľavej ruky a distálnej časti predlaktia preukázala zaostávanie kostného veku takmer o 1 rok. Sérová koncentrácia IGF-1 bola na dolnej hranici normy s prihliadnutím na jeho vek a pohlavie (51 ng/ml; N 49–351 ng/ml). Inzulínový, spánkový, záťažový a klonidínový test potvrdili izolovaný deficit rastového hormónu. Testy sa realizovali podľa štandardných protokolov, sérová koncentrácia rastového hormónu sa pohybovala v rozmedzí 0,11–5,85 ng/ml (priemer 1,7 ng/ml). Keďže pacient spĺňal indikačné kritéria na terapiu rekombinantným rastovým hormónom (rHuGH), vo veku 8 rokov začal substitučnú liečbu rHuGH a v priebehu nasledujúceho roka vyrástol až o 7 cm. Avšak po roku matka liečbu svojvoľne prerušila.
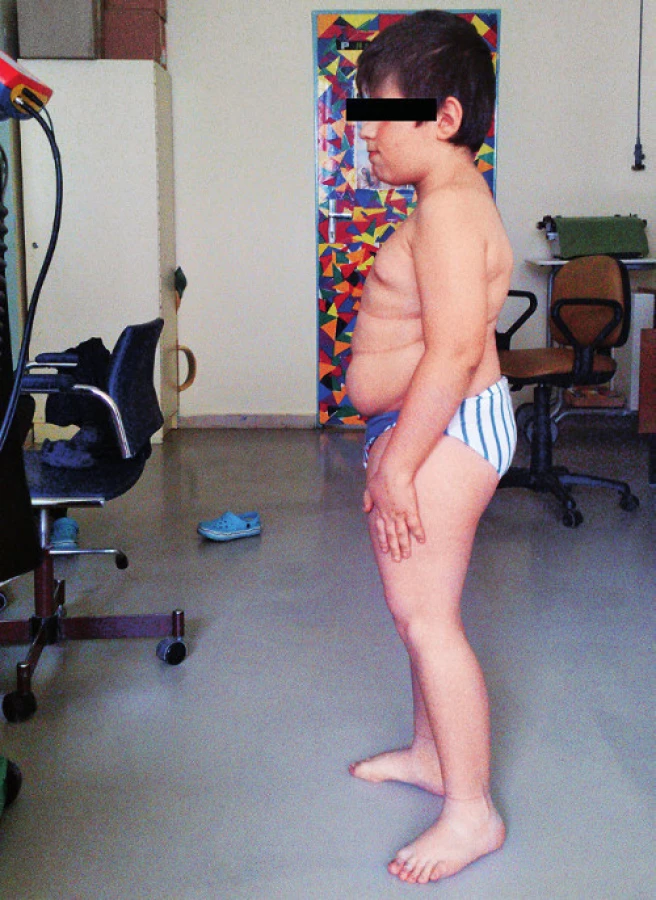
Pri prijatí na našu kliniku Marek udával bolesti chrbtice s maximom v oblasti Th11–L2. Bolesť sa stupňovala najmä pri pohybe, chôdza bola antalgická, kačacia. Vo fyzikálnom náleze dominovali šedo sfarbené skléry, nízky vzrast (129 cm, 3.–10. percentil), disproporcionálna postava s veľkým trupom a kratšími končatinami a mierna kyfoskolióza (obr. 1). Genitál bol chlapčenský, s náznakom inci-pientnej puberty (Tanner P2).
Vzhľadom na opakované zlomeniny ako aj súčasné ťažkosti, sme u Mareka suponovali diagnózu osteogenesis imperfecta a urgentne sme realizovali CT vyšetrenie. Nález preukázal difúznu a pomerne výraznú osteoporózu stavcov hrudnej i driekovej chrbtice a multietážovú bikonkávnu deformáciu tiel stavcov. Výška stavcov bola znížená na 7–8 mm v dôsledku intrakorporálneho preliačenia krycích doštičiek a medzistavcových platničiek do spongiózy tiel stavcov (obr. 2). Čerstvé traumatické zmeny stavcov či porušenie kostnej integrity sa nedokázali.

Denzitometrické vyšetrenie potvrdilo osteoporózu (Z-skóre TBLH -2,5 SD a L2–L4 -3,2 SD) [5]. Sérové koncentrácie vápnika, fosforu, alkalickej fosfatázy a jej kostného izoenzýmu, kalcidiolu, kalcitriolu a parathormónu boli v referenčnom rozmedzí. Index kalcium/kreatinín v moči bol nízky a vylúčil hyperkalciúriu. V krvi boli zvýšené skeletálne markery kostnej resorbcie (C-terminálny telopeptid kolagénu typu I, CTx 1340 pg/ml; N 115–750 pg/ml) aj kostnej formácie (Prokolagén typ 1 amino-terminálny propeptid, P1NP 630 µg/l; N 45,2–552,5 µg/l a osteokalcin 143 µg/l; N 12,2–110,6 µg/l). Treba však uviesť, že interpretácia koncentrácií kostných markerov u detí je vzhľadom na ich „nepretržitý“ rast pomerne zložitá. Kým pre markery kostnej formácie existujú publikované referenčné hodnoty pre detský vek, v prípade markerov kostnej resorbcie sme vychádzali „len“ z referenčných hodnôt nášho laboratória [2]. V ďalšom postupe sme cielenými paraklinickými vyšetreniami u pacienta vylúčili štruktúrovú chybu srdca, poruchu sluchu a poruchu pľúcnych funkcií, ktoré sprevádzajú určité typy OI. Dôležitým prínosom pre diferenciálnu diagnózu bol stomatologický nález opisujúci špecifické postihnutie trvalých zubov označované ako dentinogenesis imperfecta. Diagnózu definitívne potvrdilo molekulovo-genetické vyšetrenie. DNA analýza identifikovala mutáciu c.605G>A(p.Gly202Asp) v géne COL1A2 v heterozygotnom stave, čím sa potvrdila diagnóza osteogenesis imperfecta typ IV [15].
Pacient v úvode hospitalizácie dostával analgetiká, po zlepšení klinického stavu začal s intenzívnou rehabilitáciou. V liečbe sme ordinovali suplementáciu kalcia s vitamínom D a následne bisfosfonáty (pamidronát). Vzhľadom na dokázaný deficit rastového hormónu a signifikantné zaostávanie telesného rastu sme opätovne iniciovali liečbu rekombinantným rastovým hormónom, s ktorou matka súhlasila. Možno predpokladať, že kombinovaná osteoanabolická a antiresorbčná liečba bude účinne predchádzať ďalším patologickým zlomeninám a významne zlepší kvalitu života.
Prípad nášho pacienta je zaujímavý najmä tým, že ide o vzácne sa vyskytujúcu asociáciu osteogenesis imperfecta s izolovaným deficitom rastového hormónu.
DISKUSIA
Osteogenesis imperfecta (OI) je zriedkavá dedičná porucha spojivového tkaniva charakterizovaná zvýšenou krehkosťou kostí. Ochorenie prvýkrát opísal Malebranch v roku 1678 na prípade 20-ročného pacienta. Hoci OI je predmetom bádania ortopédov, rádiológov, genetikov a pediatrov už vyše 300 rokov, detailné informácie o skutočnej podstate ochorenia priniesli až ostatné roky.
Pôvodná klasifikácia, ktorú zaviedol Sillence v roku 1979, rozlišovala štyri typy OI – nedeformujúci typ (I), stredne závažný typ (IV), závažný typ (III) a perinatálne letálny typ (II). Po objave ďalších klinických, röntgenologických a najmä genetických charakteristík bola pôvodná klasifikácia revidovaná a rozšírená až na 9 samostatných typov [3], niektoré literárne zdroje uvádzajú dokonca už 11 typov OI [19]. Drvivú väčšinu prípadov osteogenesis imperfecta zapríčiňujú dominantné mutácie v jednom z dvoch génov kódujúcich alfa reťazce kolagénu typu I (COL1A1 a COL1A2), ktoré spôsobujú rôznu kvantitatívnu alebo kvalitatívnu poruchu syntézy kolagénu typu I. Mutácie génu COL1A1 majú závažnejší fenotypový prejav ako mutácie génu COL1A2 [19]. Autozómovo recesívne formy OI sa vyskytujú zriedkavejšie, majú najťažšie klinické prejavy a môžu končiť letálne. Ich genetickou podstatou sú mutácie špecifických proteínov, ktoré pôsobia ako esenciálne kofaktory pri tvorbe intracelulárneho kolagén modifikujúceho komplexu.
Kolagén typu I je hlavný štruktúrový proteín spojivového tkaniva, ktorý tvorí viac ako 90 % celej organickej kostnej hmoty a je kľúčovou stavebnou bielkovinou aj pre šľachy, väzy, kožu a skléry. Pri syntéze kolagénu I v prvom kroku vznikajú alfa-1 a alfa-2 polypeptidové reťazce, ktoré po spojení dvoch alfa-1 reťazcov s jedným alfa-2 polypeptidovým reťazcom vytvárajú trojzávitnicu – prokolagén typ I. Po odštiepení N-amino a C-karboxy terminálnej skupiny z prokolagénu typu I účinkom špecifických proteáz vzniká tropokolagén, ktorý tvorí stavebný základ kolagénových vlákien. Najdôležitejšou aminokyselinou alfa reťazcov je glycín (Gly), ktorý premosťuje reťazce a zodpovedá za správne umiestnenie alfa reťazcov do štruktúry trojzávitnice. Až takmer 80 % všetkých štruktúrových defektov kolagénu I zapríčiňuje zámena glycínu za inú aminokyselinu [19]. Svedčí o tom aj molekulová analýza DNA nášho pacienta, ktorá identifikovala zámenu glycínu za asparagín na pozícii 202 v géne COL1A2 a exaktne potvrdila klinické podozrenie na osteogenesis imperfecta.
Závažnosť a klinické prejavy osteogenesis imperfecta sa líšia podľa jednotlivých typov ochorenia. Najťažšiu formu predstavuje perinatálne letálna osteogenesis imperfecta (typ II), kedy zlomeniny vznikajú už počas intrauterinného vývoja plodu. IV. typ osteogenesis imperfecta, ktorý sme potvrdili u nášho pacienta, patrí k stredne ťažkým formám a môže byť sprevádzaný postihnutím zubov, tzv. dentinogenesis imperfecta (podtyp B). Charakteristickým prejavom sú časté, nízko-traumatické zlomeniny, ktoré môžu viesť k devastujúcim deformitám skeletu a následne aj k poruche rastu. Prvé fraktúry vznikajú už v rannom detstve, farba sklér je variabilná (biela, šedá, modrá). Medzi ďalšie prejavy ochorenia patrí možná porucha sluchu, ktorá sa manifestuje zvyčajne až v dospelosti. Obávanou komplikáciou OI typ IV je bazilárna impresia, ktorá sa prejavuje bolesťou hlavy s maximom v okcipitálnej oblasti sprevádzanou pestrou neurologickou symptomatológiou (kŕče, vertigo, slabosť končatín, spánkové apnoe, trigeminová neuralgia, atď.) [3, 19]. Biochemické ukazovatele kostného a minerálneho metabolizmu u pacientov s OI sú zvyčajne v referenčnom rozmedzí, aj keď mierna hyperkalciúria sa pozoruje až u tretiny pacientov, a to bez ohľadu na závažnosť prejavov základnej choroby. U ťažko postihnutých jedincov môžu byť elevované aj markery kostnej resorbcie (napr. C-terminálny telopeptid kolagénu typu I, CTx) [3, 8, 9]. K röntgenologickým nálezom OI patrí osteoporóza, kostné deformity, kyfoskolióza, či bikonkávne stavce (tzv. rybie stavce). Epifýzy a metafýzy dlhých kostí môžu mať „popkornovú“ štruktúru. Wormianské kosti v oblasti lebečných švov síce nie sú patognomické len pre OI, keďže sa vyskytujú aj pri iných ochoreniach skeletu, no ich nález v spojitosti s charakteristickými klinickými príznakmi diagnózu osteogenézy značne podporuje [16, 19]. Denzitometrické vyšetrenie preukáže poruchu mineralizácie a hustotu kostí a rôzny stupeň osteoporózy. Diagnózu OI definitívne potvrdí molekulová analýza DNA [3]. Z tohto hľadiska je pozoruhodný pomerne rozsiahly klinický súbor 25 českých pacientov s kolagénovým typom OI (typ I – IV), z ktorých sa u troch probandov identifikovala mutácia COL1A1 (22-ročný muž s OI typ IA, 23-ročná žena s OI typ III a 53-ročná žena s OI typ IVB). Pacientka s OI IV. typu (identický typ ochorenia ako má náš proband) mala modré skléry, otosklerózu, dentinogenesis imperfecta (druhú dentíciu stratila už vo veku 20 rokov). Prvé zlomeniny sa u nej manifestovali už vo veku 2 rokov, posledná fraktúra bola v období puberty. Podobne ako u nášho pacienta, röntgenologické vyšetrenie odhalilo bikonkávne hrudné a driekové stavce, a navyše aj šabľovito deformované tíbie. Je prekvapivé, že medikamentózna liečba bisfosfonátmi bola u nej započatá až vo veku 42 rokov [19].
Kazuistika nášho pacienta je zaujímavá zriedkavou asociáciou osteogenesis imperfecta s deficitom rastového hormónu. Nižšia telesná výška detí s OI sa pripisuje najmä deformitám dlhých kostí, kompresívnym fraktúram stavcov a kyfoskolióze. Interpretácia poruchy rastu u pacientov s osteogenesis imperfecta však nie je jednoduchá a nedá sa vysvetliť iba skeletálnymi deformitami. Marini a spol. zistili mierne odchýlky v osi rastového hormónu u detí s OI, no tieto endokrinné zmeny nekorelovali s typom OI a/alebo stupňom rastového deficitu [13]. Vo všeobecnosti deti s OI majú normálnu odpoveď rastového hormónu na provokačné testy. Na druhej strane, väčšina pacientov s OI má koncentrácie IGF-1 na dolnej hranici fyziologického rozpätia pre daný vek a plochší vzostup IGF-1 po injekcii rastového hormónu. Tieto abnormality v tvorbe IGF-1 sú však iné ako u ostatných detí s chronickými ochoreniami. Hypotetické vysvetlenie sa upriamuje na faktory ovplyvňujúce defektnú kostnú matrix, ktorá môže viesť k porušenej odpovedi na rastové signály [18]. U nášho pacienta sa priekazné zaostávanie v raste zistilo už v predškolskom veku a predchádzalo devastujúcim zlomeninám. Výsledky štandardných provokačných testov svedčili pre izolovaný deficit rastového hormónu. Asociácia deficitu rastového hormómu a osteogenesis imperfecta je vzácna. Môže to byť dané nízkou incidenciou osteogenesis imperfecta, keďže sa predpokladá, že frekvencia výskytu deficitu rastového hormónu u chorých s OI je rovnaká ako v bežnej detskej populácii [18].
V liečbe sme okrem suplementácie vápnika a vitamínu D indikovali bisfosfonáty a rekombinantný rastový hormón. Je prekvapivé, že hoci sa bisfosfonáty považujú za štandardnú medikamentóznu liečbu OI, nie sú pre toto ochorenie oficiálne registrované. S výnimkou neridronátu, ktorý je schválený v Taliansku, sa tieto farmaká u detí indikujú „off label“.
Bisfosfonáty sú analógy pyrofosfátov, ktoré dokážu znížiť osteoklastickú aktivitu bez toho, aby poškodili kostnú architektúru a mechanickú silu kostného tkaniva. Bisfosfonáty (pamidronát, alendronát, risedronát) sú indikované takmer pri všetkých formách OI (výnimkou je len typ VI). Ide najmä o pacientov s deformitami dlhých kostí, s kompresívnymi zlomeninami stavcov a chorých, ktorí majú viac ako 3 zlomeniny ročne [4]. Výsledky observačných štúdií u detí s OI preukázali, že bisfosfonáty môžu významne znížiť výskyt zlomenín (až takmer o 100 %), účinne zmierňujú bolesť a zlepšujú mobilitu postihnutých jedincov [6, 7, 17]. Zdá sa, že dokonca môžu zvyšovať minerálnu denzitu kostí [11]. Na druhej strane, klinické skúsenosti s liečbou rekombinantným rastovým hormónom u pacientov s OI sú oveľa skromnejšie. Z priekopníckych intervenčných štúdií spomenieme pilotnú prácu Mariniho a spol., ktorí dosiahli po ročnej liečbe rHuGH signifikantný nárast rastovej rýchlosti u viac ako polovici z 26 prepubertálnych pacientov s OI III. a IV. typu [14]. Priaznivý efekt osteoanabolickej liečby potvrdili aj talianskí autori, ktorí v kontrolnej klinickej štúdii u 30 prepubertálnych detí s OI porovnávali účinok monoterapie bisfosfonátmi (neridronát) s kombinovanou terapiou bisfosfonát (neridronát) plus rHuGH. Po 12-mesačnej liečbe sa preukázalo, že skupina pacientov s kombinovanou liečbou (bisfosfonát plus rHuGH) dosiahla lepšiu rastovú rýchlosť a nárast kostnej denzity ako deti liečené iba samotným bisfosfonátom [1].
ZÁVER
Osteogenesis imperfecta je zriedkavá dedičná porucha spojivového tkaniva charakterizovaná zvýšenou krehkosťou kostí. 90 % prípadov spôsobujú mutácie génov kódujúcich alfa-1 a alfa-2 reťazce kolagénu typu I. Spektrum fenotypových prejavov OI je mimoriadne široké. Kým ťažko postihnutí pacienti zomierajú už v perinatálnom období, alebo ich kvalitu života znižujú početné závažné netraumatické fraktúry, mierne formy OI sa môžu prejaviť pomerne nenápadne, „len“ predčasnou osteoporózou.
Klinické štúdie u detí s osteogenesis imperfecta preukázali priaznivý longitudinálny účinok bisfosfonátov v prevencii patologických zlomenín. Doterajšie skúsenosti naznačujú, že vyšší benefit na kostnú denzitu a telesný rast detí s OI možno dosiahnuť kombinovanou terapiou bisfosfonátmi s rastovým hormónom.
U nášho pacienta sme molekulovou DNA analýzou identifikovali kauzálnu mutáciu v géne COL1A2 svedčiacu pre osteogenesis imperfecta (typ IV) a súčasne sme dokázali izolovaný deficit rastového hormónu. Naša kazuistika demonštruje, že porucha rastu u detí s osteogenesis imperfecta nemusí byť iba „na vrub“ primárneho ochorenia skeletu, ale treba pátrať aj po iných príčinách rastovej retardácie.
Došlo: 22. 1. 2014
Přijato: 24. 2. 2014
Adresa pre korešpondenciu:
Prof. MUDr. Ľudmila Podracká, CSc.
Klinika detí a dorastu LF UPJŠ a DFN
Tr. SNP 1
040 11 Košice
Slovenská republika
e-mail: podracka@upjs.sk
Sources
1. Antoniazzi F, Monti E, Venturi G, et al. GH in combination with bisphosphonate treatment in osteogenesis imperfecta. Eur J Endocrinol 2010; 163 : 479.
2. Bayer M. Reference values of osteocalcin and procollagen type I N-propeptide plasma levels in a healthy Central European population aged 0–18 years. Osteoporos Int 2014; 25 : 729–736.
3. Beary J, Chines A. Osteogenesis imperfecta: Clinical features and diagnosis. www.uptodate.com; 07/2013.
4. Beary J, Chines A. Osteogenesis imperfecta: Management and prognosis. www.uptodate.com; 08/2013.
5. Bianchi LM. Official positions of the International Society for Clinical Densitometry (ISCD) on DXA evaluation in children and adolescents. Pediatr Nephrol 2010; 25 : 37–47.
6. Bishop N. Characterising and treating osteogenesis imperfecta. Early Hum Dev 2010; 86 : 743.
7. Castillo H, Samson-Fang L. Effects of bisphosphonates in children with osteogenesis imperfekta. Dev Med Child Neurol 2009; 51 (1): 17–29.
8. Chines A, Petersen DJ, Schranck FW, Whyte MP. Hypercalciuria in children severely affected with osteogenesis imperfecta. J Pediatr 1991; 119 : 51.
9. Chines A, Boniface A, McAlister W, Whyte M. Hyper-calciuria in osteogenesis imperfecta: a follow-up study to assess renal effects. Bone 1995; 16 : 333.
10. Cundy T. Recent advances in osteogenesis imperfecta. Calcif Tissue Int 2012; 90 : 439–449.
11. Fleisch H. Bisphosphonates in Bone Disease: From the Laboratory to the Patient. 4th ed. London: Academic Press, 2000 : 1–212.
12. Kovács L. Osteogenesis imperfecta. Pediatr Prax 2012; 13 (3): 141.
13. Marini JC, Bordenick S, Heavner G, Rose S, Chrou-sos GP. Evaluation of growth hormone axis and responsiveness to growth stimulation of short children with osteogenesis imperfecta. Am J Med Genet 1993 Jan 15; 45 (2): 261–264.
14. Marini JC, Hopkins E, Glorieux FH, et al. Positive linear growth and bone responses to growth hormone treatment in children with types III and IV osteogenesis imperfecta: high predictive value of the carboxyterminal propeptide of type I procollagen. J Bone Miner Res 2003; 18 : 237.
15. Marini JC, et al. Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum Mutat 2007; 28 (3): 209–221.
16. Renaud A. Radiographic features of osteogenesis imperfecta. Insights Imaging 2013; 4 : 417–429.
17. Salehpour S, Tavakkoli S. Cyclic pamidronate therapy in children with osteogenesis imperfecta. J Pediatr Endocrinol Metab 2010; 23 : 73.
18. Samson-Fang L. Growth in Osteogenesis Imperfecta. Handbook of Growth and Growth Monitoring in Health and Disease. Springer Science + Business Media, 2012 : 1829–1840.
19. Šormová L, et al. Analýza mutací genu COL1A1 českých pacientu s diagnózou osteogenesis imperfecta, typ I-IV. EJBI 2012; 8 (5): 30–37.
Labels
Neonatology Paediatrics General practitioner for children and adolescentsArticle was published in
Czech-Slovak Pediatrics

2014 Issue 2
- What Effect Can Be Expected from Limosilactobacillus reuteri in Mucositis and Peri-Implantitis?
- The Importance of Limosilactobacillus reuteri in Administration to Diabetics with Gingivitis
Most read in this issue
- Complications of surgical treatment of hydrocephalus
- The results of expanded newborn screening in the Czech Republic
- Osteogenesis imperfecta associated with isolated growth hormone deficiency
- Anthropometric, hormonal and metabolic changes in prepubertal children born as SGA (small for gestational age), with decreased growth, treated with the growth hormone, after the first year of therapy