
Intrafamiliárna fenotypová variabilita klasického Marfanovho syndrómu
Intrafamiliar phenotype variability of classic Marfan syndrome
Marfan syndrome is a rare systemic disorder of the connective tissue affecting skeletal, cardiovascular system and also eyes. The estimated prevalence within different ethnic groups is 1 : 3–5,000.
We provide intra-familiar phenotype variability, new diagnostic criteria and autosomal dominant inheritance information within casuistic of a 16-year old male subject with fully developed signs of Marfan syndrome and within gentle phenotype of his father. We managed to provide evidence of identical, up to now undefined, "splicing" mutation c.7820-1G>A within FBN1 gene in its heterozygous state. The actual usage of genetic diagnostic tests in order to confirm exact diagnosis significantly contributes to improvement of comprehensive health care management provided to subjects with this potentially fatal disorder.
Key words:
Marfan syndrome, phenotype variability, gene FBN1, revised diagnostic criteria
Authors:
S. Machalová 1; E. Čmelová 1; D. Ďurovčíková 1; M. Pechová 1; M. Hikkelová 2
Authors‘ workplace:
Klinika lekárskej genetiky SZU a UNB, Bratislava
prednostka doc. MUDr. D. Ďurovčíková, CSc.
1; MedGen, s. r. o., Bratislava
vedúci MUDr. RNDr. A. Genčík, CSc.
2
Published in:
Čes-slov Pediat 2015; 70 (5): 287-292.
Category:
Case Report
Overview
Marfanov syndróm je zriedkavé, systémové ochorenie spojivového tkaniva s postihnutím skeletu, kardiovaskulárneho systému a očí. Odhadovaná prevalencia sa v rôznych etnikách pohybuje od 1 : 3–5000.
Na kazuistike 16-ročného chlapca s plne manifestnými známkami Marfanovho syndrómu a miernom fenotype u otca prezentujeme intrafamiliárnu fenotypovú variabilitu, nové diagnostické kritériá a autozómovo dominantný typ dedičnosti. U oboch sme potvrdili identickú, v literatúre dovtedy nepopísanú ,,splicing“ mutáciu c.7820-1G>A v géne FBN1 v heterozygotnom stave. Použitie genetických diagnostických testov pre definitívne potvrdenie diagnózy prispieva k zlepšeniu manažmentu komplexnej zdravotnej starostlivosti o pacientov s týmto potencionálne letálnym ochorením.
Kľúčové slová:
Marfanov syndróm, fenotypová variabilita, FBN1 gén, revidované diagnostické kritériá
Úvod
Marfanov syndróm je systémové ochorenie spojivového tkaniva s vysoko variabilným fenotypovým prejavom. Spôsob dedičnosti je autozómovo dominantný [1]. Približne v 1/3 prípadov je ochorenie spôsobené mutáciou „de novo“ [2]. Marfanov syndróm bol prvý krát popísaný pred viac než 100 rokmi, v roku 1896 francúzskym profesorom, pediatrom A. B. Marfanom. Odhadovaná prevalencia sa v rôznych etnikách pohybuje od 1 : 3–5000 [3]. Etiopatogeneticky je klasický Marfanov syndróm podmienený mutáciami vo fibrilínovom géne FBN1. Samotný gén FBN1 sa podarilo identifikovať v roku 1991. K najčastejším typom kauzálných mutácií v géne FBN1 patria mutácie typu: „nonsence“, delécie, inzercie, „splicing“ mutácie a „missence“ mutácie. Doteraz bolo popísaných viac ako 1750 mutácií FBN1 génu.
Gén FBN1 je lokalizovaný na chromozóme č. 15 oblasť 15q21.1 [4]. Tvorený je 65 exónmi, kóduje syntézu štrukturálneho proteínu – fibrilínu 1 [5]. Fibrilín 1 je významný komponent extracelulárnej matrix. Zohráva dôležitú úlohu v udržiavaní homeostázy extracelulárnej matrix a významne participuje na regulácii aktivácie TGFβ (transforming growth factor β) signálnej dráhy prostredníctvom interakcie s TGFβ prekurzormi, ktoré udržiava v inaktívnej forme [3]. TGFβ je polypeptid secernovaný do extracelulárnej matrix, účastní sa procesu bunko-vej proliferácie, diferenciácie, apoptózy a formovaní samotnej matrix [5]. TGFβ je potencionálny stimulátor zápalu a fibrózy, podieľa sa na aktivácii proteináz v matrixe [6].
U pacientov s Marfanovým syndrómom fibrilín chýba, alebo je defektný, v dôsledku čoho dochádza k dysregulácii TGFβ signálnej dráhy [6, 7]. Táto dysregulácia v zmysle nadmernej aktivácie prekurzorov a následne aj samotného TGFβ spôsobí v cieľovom spojivovom tkanive poruchu bunkovej proliferácie, diferenciácie a apoptózy. Narušené je aj formovanie samotnej matrix. Klinicky sa uvedená dysregulácia v cieľovom spojivovom tkanive prejaví fenotypovým postihnutím hlavne skeletu, kardiovaskulárneho systému a očí (tab. 2). Genotypovo-fenotypová korelácia ochorenia nie je jednoznačná, podľa dostupných literárnych údajov bývajú najťažšie formy Marfanovho syndrómu spojené s mutáciami zahŕňajúcimi oblasť exónov 24 až 32 FBN1 génu [5, 4].
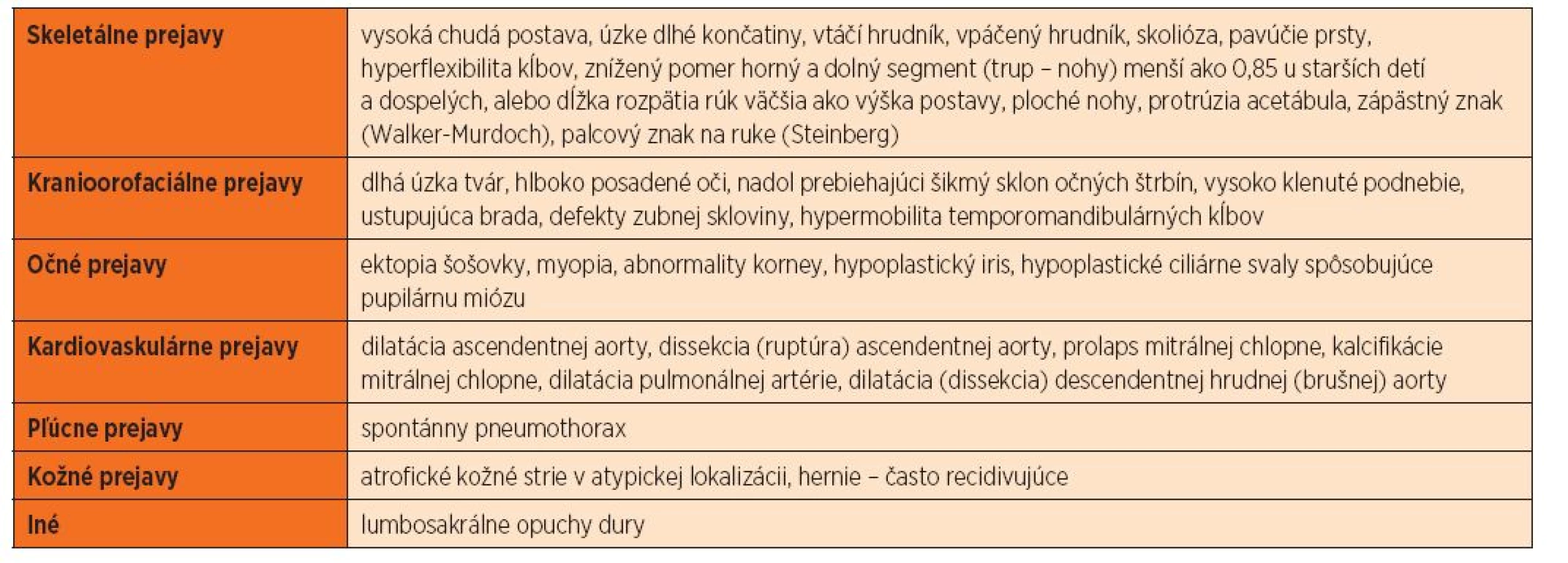
Nové revidované diagnostické kritériá (tab. 1a, 1b) spolu s využitím molekulárnych techník umožňujú potvrdiť diagnózu klasického Marfanovho syndrómu u viac než 95 % pacientov [8]. Na príklade z praxe poukazujeme na intrafamiliárnu fenotypovú variabilitu ochorenia u otca a syna s identickou kauzálnou „splicing“ mutáciou vo fibrilínovom géne FBN1.
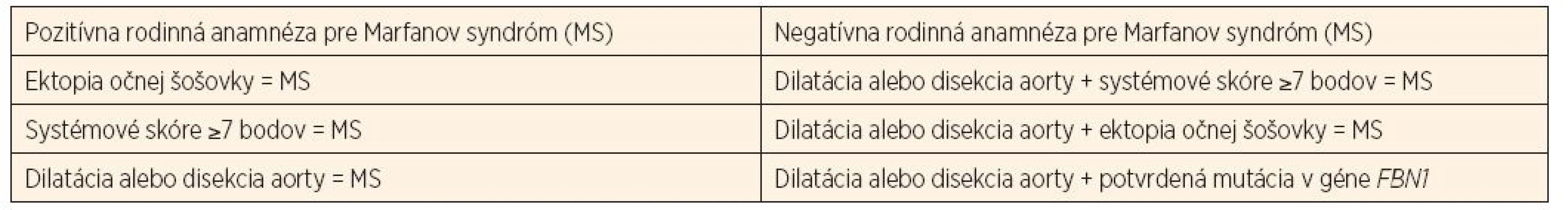
![Tab. 1b. Systémové skóre, Z skóre [10].](https://pl-master.mdcdn.cz/media/image/e01dd4e2007f748844d568015c645410.jpg?version=1537797278)
Kazuistika
Na genetické vyšetrenie odoslal 16-ročného probanda kardiológ pre podozrenie na Marfanov syndróm. Je narodený z druhej rizikovej gravidity pre krvácanie matky, pôrod v termíne, spontánny, záhlavím, pôrodná hmotnosť 3050 g, dĺžka 52 cm, bezprostredná popôrodná adaptácia v norme. Ako 2-týždňový bol operovaný na skrotálnu herniu (resekované aj črevo, pravostranná orchidektomia). V minulosti sledovaný kardiológom (bližšie údaje matka nevedela poskytnúť, lieky neužíval žiadne, kolapsové stavy nemával, neomodrával, fyzickú záťaž zvládal adekvátne), dlhodobo sledovaný u ortopéda, vzhľadom k výraznej deformite hrudníka plánované operačné riešenie. Pri predoperačnom vyšetrení, vo veku 16 rokov kardiológ diagnostikoval u probanda aortálnu insuficienciu II. stupňa, dilatáciu koreňa aorty 4,2 cm a dilatáciu ascendentnej aorty 4,0 cm. Očné vyšetrenie potvrdilo obojstranný astigmatizmus s myopiou.
Pri klinicko-genetickom vyšetrení probanda s vysokým vzrastom (výška 196 cm, nad 97. percentilom, rozpätie rúk 210 cm, pomer rozpätie rúk/výška = 1,07, pomer horný segment/dolný segment = 0,78) nachádzame viaceré fenotypové príznaky Marfanovho syndrómu: dlhú, úzku tvár, malú ustupujúcu bradu a nadol prebiehajúci šikmý sklon očných štrbín, vysoko klenuté podnebie, výrazne deformovaný, vpáčený hrudník, nápadne dlhé, úzke horné aj dolné končatiny, pavúčie prsty, pozitívny zápästný a palcový príznak, ploché nohy (obr. 1–3, tab. 3). Podľa nových revidovaných diagnostických kritérií mal proband systémové skóre 9, Z skóre 4,2 cm. Pre splnené diagnostické kritériá (klasický typ Marfanovho syndrómu) sme indikovali diagnostické testovanie kauzálneho FBN1 génu. DNA analýza FBN1 génu bola uskutočnená pomocou mutačného DGGE („denaturing gradient gel electrophoresis“) skríningu a následným priamym sekvenovaním. U probanda sa detekovala dovtedy v literatúre nepopísaná „splicing“ mutácia c.7820-1G>A v géne FBN1 v heterozygotnom stave. Genealogickým vyšetrením získaným od matky probanda, ktorá nevykazovala známky Marfanovho syndrómu, sme zistili sporadický výskyt ochorenia v rodine. Vrodené vývojové chyby srdca ani iné geneticky podmienené choroby v rodine neudala. Vzhľadom ku genealogickým údajom sme predpokladali, že u probanda vznikla čerstvá mutácia „de novo“. Overenie tejto hypotézy si vyžiadalo testovanie FBN1 génu s podrobným klinicko-genetickým vyšetrením oboch rodičov.
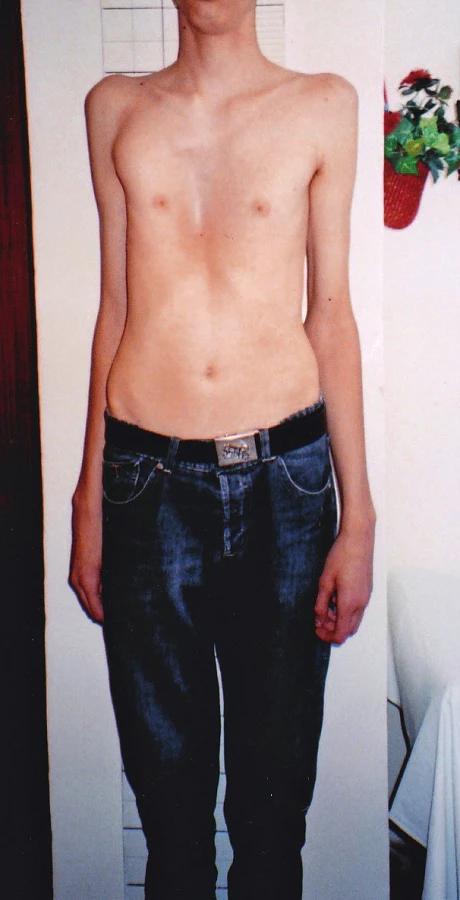

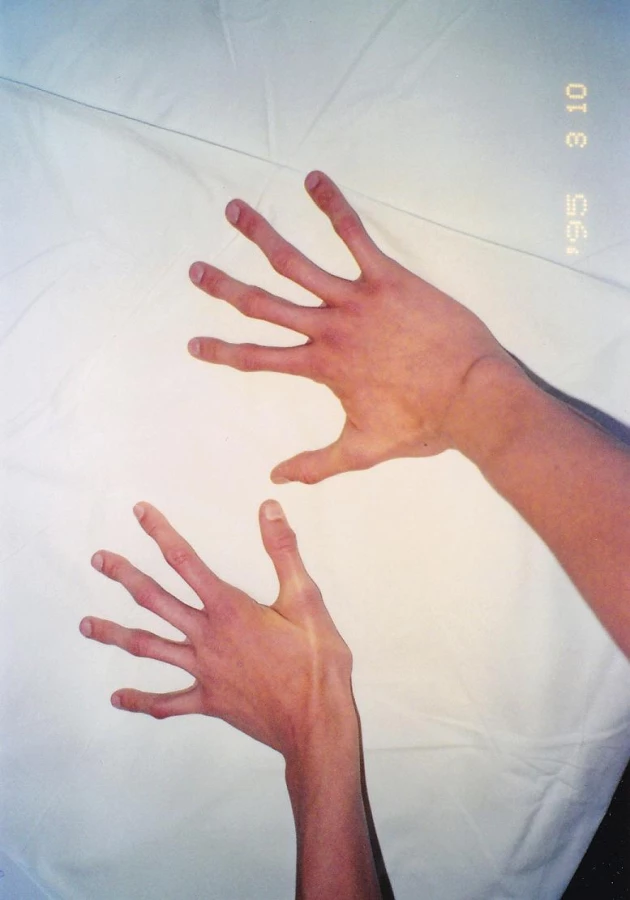
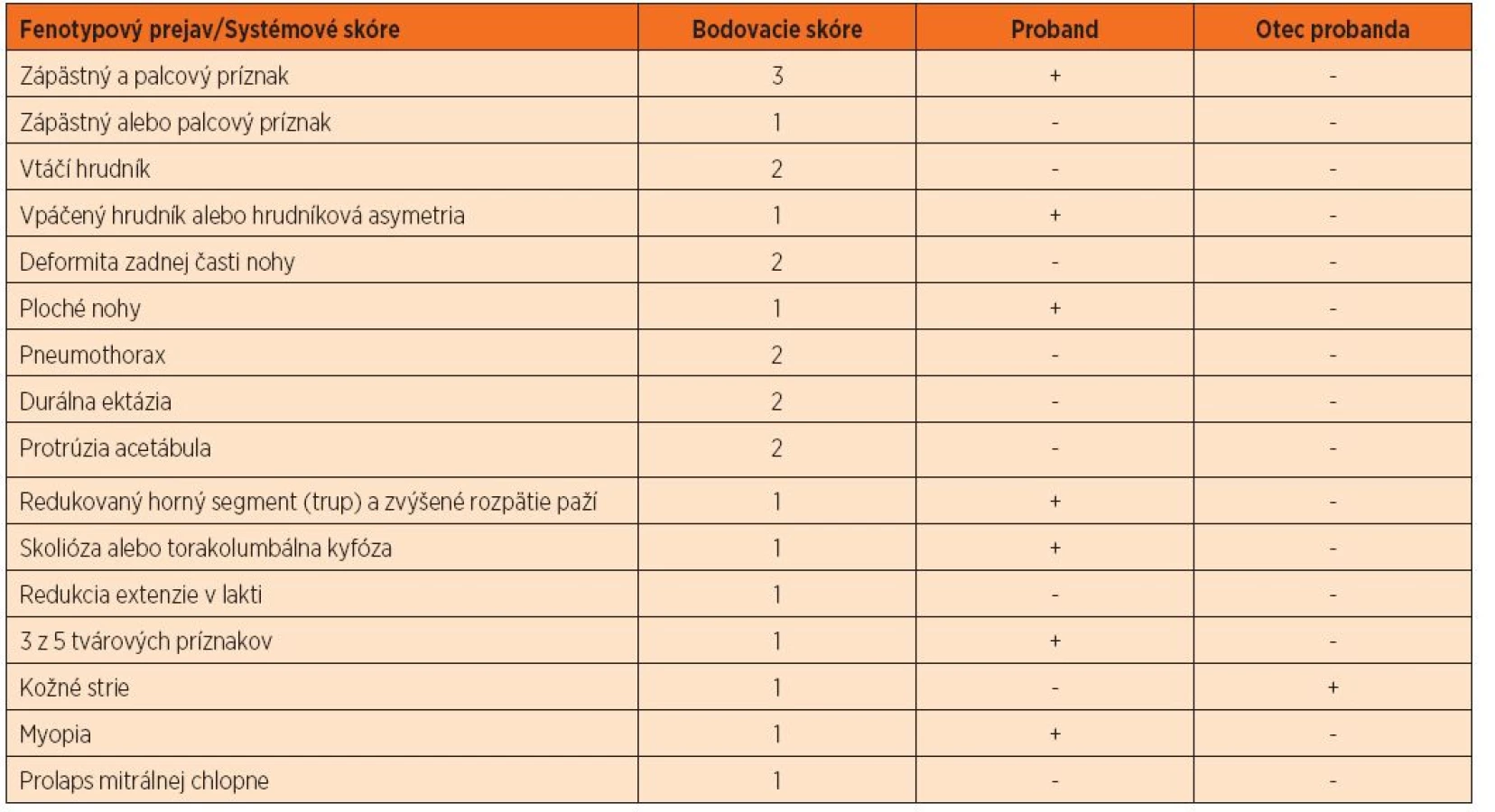
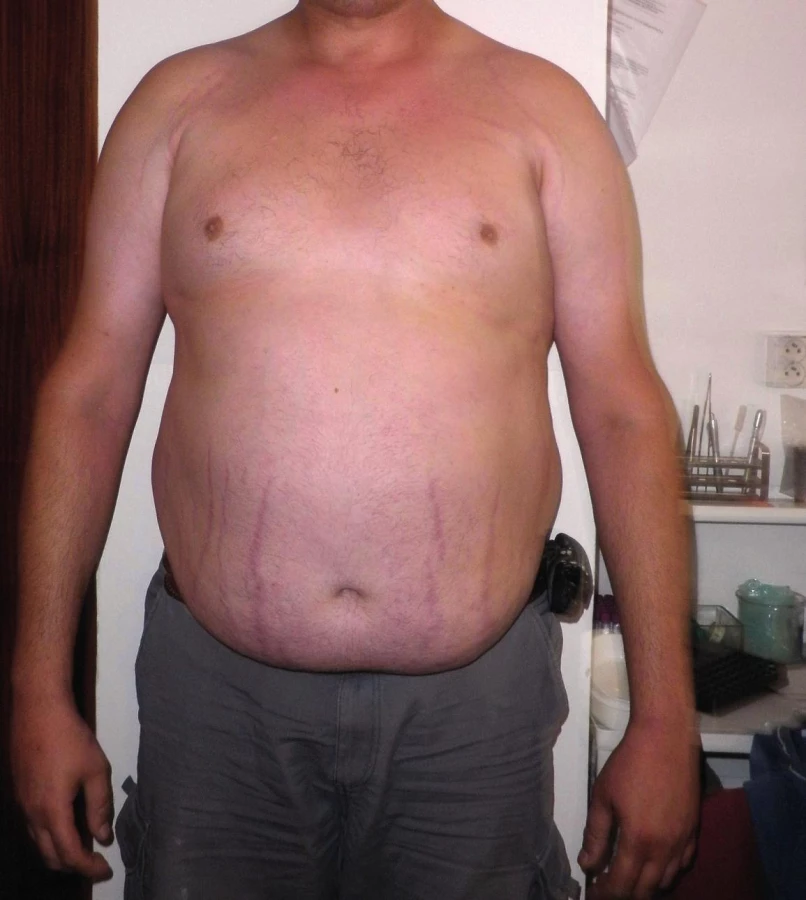
40-ročný otec probanda v anamnéze udával operáciu inguinálnej hernie v detstve, dlhodobé bolesti lumbosakrálnej časti chrbtice a tŕpnutie dolných končatín. Problémy so srdcom neudával, kardiológom nebol vyšetrený. Klinicko-genetickým vyšetrením nachádzame u otca parciálne známky Marfanovho syndrómu: mnohopočetné atrofické kožné strie (oblasť trupu), strie v atypickej lokalizácii (pravé rameno), vysoko klenuté, úzke podnebie a menšiu mierne ustupujúcu bradu (obr. 4–6, tab. 3). Systémové skóre mal menej ako 7 a Z skóre nebolo merané. Molekulárno-genetickým vyšetrením sa u otca potvrdila identická mutácia v géne FBN1 ako u jeho syna. U 39-ročnej matky probanda, ktorá nemala žiadne fenotypové prejavy Marfanovho syndrómu, táto mutácia nebola detekovaná. Na základe uvedených vyšetrení sme potvrdili paternálny pôvod zárodočnej mutácie v géne FBN1 a autozómovo dominantný typ dedičnosti.
Diskusia
Na príklade z praxe sme dokumentovali intrafamiliárnu fenotypovú variabilitu Marfanovho syndrómu. Fenotypová variabilita je typickým znakom ochorení s autozómovo dominantným typom dedičnosti [9]. Riziko odovzdania patologického génu potomkom postihnutého je 50 %. Práve fenotypová variabilita však môže spôsobovať určité diagnostické problémy. Z hľadiska diferenciálnej diagnostiky pri Marfanovom syndróme treba brať do úvahy podobné fenotypy asociované s mutáciami v iných génoch, takzvané Marfan-like syndrómy. Spomenúť možno hlavne Loeysov-Dietzov syndróm, v minulosti označovaný ako MS typ II. Etiologicky je podmienený mutáciami v génoch TGFBR1, TGFBR2, SMAD3, TGFB2. TAAD syndróm (thoracic and aortic aneurysms and aortic dissections syndrome) podmienený mutáciou v géne ACTA2, kongenitálna kontraktúrna arachnodaktýlia podmienená mutáciou v géne FBN2 a Ehlersov-Danlosov syndróm spôsobený mutáciami v génoch kódujúcich syntézu kolagénu.
Podobné fenotypové prejavy ako pri Marfanovom syndróme možno nájsť aj pri homocystinúrii a Shprintzenovom-Goldbergovej syndróme (podmienený mutáciou v géne SKI). S mutáciami v samotnom kauzálnom fibrilínovom géne FBN1 sú asociované aj iné typy fibrilinopatií, ktoré však majú odlišný fenotypový prejav, aj keď niektoré dyzmorfné známky môžu byť podobné ako pri Marfanovom syndróme. Ide hlavne o tieto typy fibrilinopatií: izolovaná ectopia lentis, Weillov-Marchesaniho syndróm, MASS syndróm a Stiff skin syndróm.
Marfanov syndróm je potenciálne letálne ochorenie. Z hľadiska pediatrickej praxe je dôležité včas zachytiť dyzmorfné známky u dieťaťa, venovať im dostatočnú pozornosť, všímať si možné fenotypové prejavy aj u rodičov a súrodencov postihnutého. Pri podozrení na genetické ochorenie odoslať dieťa na vyšetrenie ku klinickému genetikovi, využiť pri diagnostike ochorenia interdisciplinárnu spoluprácu medzi odborníkmi z jednotlivých medicínskych odborov. Použitie genetických diagnostických testov pre definitívne potvrdenie diagnózy prispieva k zlepšeniu manažmentu komplexnej zdravotnej starostlivosti o pacientov s týmto potencionálne letálnym ochorením.
Záver
Marfanov syndróm je zriedkavým, multisystémovým ochorením s vysokým stupňom fenotypovej variability. K diagnóze klasického Marfanovho syndrómu okrem samotného klinického nálezu s prihliadnutím na nové revidované diagnostické kritériá významne prispieva aj molekulárno-genetické vyšetrenie kauzálneho fibrilínového génu FBN1. Vzhľadom k uvedenému, ako aj k širokej problematike diferenciálnej diagnostiky Marfanovho syndrómu, stanovenie správnej diagnózy si vyžaduje interdisciplinárnu spoluprácu medzi odborníkmi z jednotlivých medicínskych odborov (genetik, kardiológ, oftalmológ, ortopéd, pediater).
Správna diagnostika a včasné predchádzanie vzniku komplikácií, hlavne zo strany kardiovaskulárneho a skeletálneho systému spolu so symptomatickou liečbou umožňujú zvýšiť kvalitu života pacientov s týmto potenciálne letálnym ochorením.
Došlo: 21. 5. 2013
Přijato: 29. 7. 2015
MUDr. Slávka Machalová
Klinika lekárskej genetiky SZU a UNB
Limbová 5
833 40 Bratislava
Slovenská republika
e-mail: slavka.machalova@gmail.com
Sources
1. Redlinger Jr. RE, Rushing GD, Moskowitz AD. Minimally invasive repair of pectus excavatum in patients with Marfan syndrome and marfanoid features. J Pediatr Surg 2010; 45 : 193–199.
2. Van Dijk FS, Hamel BC, Hilhorst-Hofstee Y. Compound-heterozygous Marfan syndrome. Eur J Med Genet 2009; 52 : 1–5.
3. Franken R, Wan den Hartog A, Singh M, Plus G. Marfan syndrome: Progress report. Progress in Pediatric Cardiology 2012; 34 : 9–14.
4. Lebreiro A, Martins E, Cruz C, Almeida J. Genotypic characterization of a Portuguese population of Marfan syndrome patients. Revista Portuguesa de Cardiologia (English Edition) 2011; 30 : 649–654.
5. Gen Gao L, Luo F, Hui Ru-Tai. Recent molecular biological progress in Marfan syndrome and Marfan-associated disorders. Ageing Res Rev 2010; 9 : 363–368.
6. Ahimastos A, Aggarual A, Savarirayan R, Dant AM. A role for plasma transforming growth factor - B and matrix metalloproteinases in aortic aneurysm surveillance in Marfan syndrome. Atherosclerosis 2010; 209 : 211–214.
7. Baker J De, Loeys B, Paepe A De. Marfan and Marfan-like syndromes. Artery Res 2009; 3 : 9–16.
8. Loeys BL, Dietz HC, Brauerman AC. The revised ghent nosology for the Marfan syndrome. J Med Genet 2010; 47 : 476–485.
9. Pritchard D, Korf B. Základy lékařské genetiky. 1. vyd. Praha: nakladatelství Galén, 2007 : 1–224. ISBN 978-80-7262-449-2.
10. http://www.marfan.org/marfan/2280/about-marfan-syndrome, 14. 03. 2013.
Labels
Neonatology Paediatrics General practitioner for children and adolescentsArticle was published in
Czech-Slovak Pediatrics

2015 Issue 5
- What Effect Can Be Expected from Limosilactobacillus reuteri in Mucositis and Peri-Implantitis?
- The Importance of Limosilactobacillus reuteri in Administration to Diabetics with Gingivitis
Most read in this issue
- Metabolické kostní onemocnění při nezralosti
- Vybrané špecifiká ultrasonografie pľúc v detskom veku
- Vzácný případ DiGeorgeova syndromu s anomáliemi končetin: přínos vyšetření metodou SNP microarrayí?
- Asociácia genetických polymorfizmov metyléntetrahydrofolát reduktázy s vrodenými chybami srdca v slovenskej populácii