
Nefrotický syndrom v dětském věku
Nephrotic syndrome in childhood
Nephrotic syndrome (NS) includes a group of diseases with increased permeability of glomerular filtration barrier, which causes heavy proteinuria, hypoalbuminemia and edema. Idiopathic NS is the most common type in children. Most of the children are steroid-sensitive, however, about 20% fail to respond to steroid treatment and are at significant risk for progressive renal failure. Extensive research has revealed the structure of permeability membrane and podocyte function. More than 40 genes associated with steroid-resistant NS have been discovered. While corticosteroids and other immunosupressives are used in patients with idiopatic NS, immunosupressive therapy should be stopped in patients with proven genetic disease.
KEY WORDS:
nephrotic syndrome – idiopathic, genetic
:
S. Skálová 1; T. Seeman 2; Š. Štolbová 2; J. Zieg 2
:
Dětská klinika FN a LF UK, Hradec Králové
1; Pediatrická klinika FN Motol a 2. LF UK, Praha
2
:
Čes-slov Pediat 2017; 72 (2): 88-98.
:
Nefrotický syndrom zahrnuje skupinu onemocnění charakterizovanou zvýšenou propustností glomerulární filtrační bariéry. Klinicky se onemocnění projevuje proteinurií v nefrotickém rozmezí, hypoalbuminémií a vznikem otoků. Idiopatický NS je nejčastějším typem v dětském věku. Léčba kortikoidy navodí u většiny dětí plnou remisi onemocnění, nicméně okolo 20 % nemocných na tuto terapii nezareaguje. Tito pacienti jsou ve zvýšeném riziku progresivního chronického selhání ledvin. V posledních letech nám výzkum umožnil lépe porozumět struktuře permeabilní membrány a funkci podocytu. Bylo objeveno více než 40 genů podmiňujících vznik kortikosteroid-rezistentního NS. Zatímco v případě idiopatického NS používáme v léčbě kortikoidy a další imunosupresiva, nález kauzální mutace u kortikosteroid-rezistentního NS je indikací k vysazení imunosupresivní léčby.
KĽÚČOVÉ SLOVÁ:
nefrotický syndrom – idiopatický, geneticky podmíněný
ÚVOD
Nefrotický syndrom (NS) představuje v dětském věku spektrum onemocnění s různými příčinami, projevy, histopatologickými nálezy a prognózou. Jedná se o klinický stav, který je definován proteinurií >960 mg/m2/24 hodin a hypoalbuminémií <25 g/l. Tyto dva laboratorní příznaky jsou pro diagnózu NS klíčové, ve většině případů bývají navíc doprovázeny otoky a hyperlipidémií. K definici nefrotické proteinurie se v klinické praxi v poslední době dává přednost stanovení poměru bílkoviny ke kreatininu z jednorázového vzorku moče, kde nefrotické rozmezí je definováno hodnotou >200 mg/mmol.
Incidence a prevalence
Incidence NS se udává 2–7 případů na 100 000 dětské populace ročně, závisí však na věku a je také ovlivněna etnickým původem a geografickými podmínkami. Prevalence představuje 16 případů na 100 000 dětí. Postižení chlapců je častější než dívek, poměr se pohybuje mezi 1,6–2 : 1, tento rozdíl však v adolescenci klesá a u dospělých pacientů je postižení mužů a žen 1 : 1 [1, 2]. Incidence NS je dlouhodobě stabilní, nicméně se ukazuje narůstající výskyt fokálně segmentální glomerulosklerózy (FSGS) u pacientů s NS. Udává se, že 70–80 % dětí s NS onemocní ve věku 2–6 let, výskyt NS na podkladě minimálních změn je v tomto věku až v 80 % případů.
Etiologie a klasifikace
NS můžeme podle etiologie dělit na primární a sekundární [3]. Podle věku manifestace rozlišujeme v prvním roce života NS na kongenitální a infantilní (tab. 1).

Primární NS, který se v dětském věku vyskytuje nejčastěji, dělíme do dvou podskupin:
- idiopatický NS, jehož histologickým podkladem je v naprosté většině případů nemoc minimálních změn glomerulů (minimal change disease = MCD), méně často FSGS.
- NS na podkladě primární glomerulonefritidy – membranózní nefropatie, membranoproliferativní, mezangiálně proliferativní glomerulonefritidy, IgM nefropatie, vzácně IgA nefropatie.
Sekundární NS mohou způsobit některé infekce, léky, či systémová onemocnění.
Kongenitální NS se manifestuje v prvních 3 měsících života. Jeho příčina je v 70–85 % genetická, ale vzácně mohou být příčinou i některé vrozené infekce (lues, toxoplazmóza, cytomegalovirus, virus rubeoly, HIV).
Infantilní NS se manifestuje mezi 4.–12. měsícem života a jeho etiologie je rovněž zhruba ve 40–45 % případů geneticky podmíněná.
Geneticky podmíněným formám NS je v tomto přehledu věnována samostatná kapitola.
IDIOPATICKÝ NEFROTICKÝ SYNDROM
Idiopatický nefrotický syndrom představuje nejčastější formu NS v dětském věku. Jak již bylo zmíněno, jeho histopatologickým podkladem je v naprosté většině případů MCD a podstatně méně často FSGS, vzácně difuzní mezangiální proliferace. Zatímco ve světelné mikroskopii má MCD typicky normální nález, elektronovou mikroskopií lze prokázat splynutí výběžků podocytů zvaných pedicely. Pro FSGS je charakteristický obraz segmentální obliterace glomerulárních kapilár extracelulární matrix vedoucí ke skleróze. Postižení je fokální, popsané změny nejsou přítomny ve všech glomerulech. Elektronová mikroskopie odhalí rovněž difuzní splynutí pedicel. Z hlediska klasifikace idiopatického NS má z praktického i prognostického hlediska největší význam dělení podle odpovědi na léčbu kortikosteroidy na kortikosteroid-senzitivní (SSNS) a kortikosteroid-rezistentní (SRNS).
Patogeneze
Základní a klíčovou patogenetickou abnormalitou je masivní proteinurie, která vzniká při zvýšené propustnosti glomerulární filtrační bariéry. Ta je za normálních okolností nepropustná pro látky s molekulovou hmotností větší než 69 kDa, tedy i pro bílkoviny typu albuminu a větší. Ke změně propustnosti dochází při ztrátě jejího negativního náboje či její struktury. Glomerulární filtrační bariéru tvoří tři vrstvy: speciální fenestrované endoteliální buňky, glomerulární bazální membrána (GBM) z vláken kolagenu a proteoglykanů a glomerulární epiteliální buňky podocyty, které jsou svými pedicely přichyceny a fixovány ke GBM. Výběžky sousedících podocytů jsou vzájemně propojeny a tato spojení tvoří tzv. štěrbinovou membránu („slit diaphragm“), která je důležitá pro funkčnost této třetí vrstvy filtrační bariéry [4]. V současné době je známa celá řada proteinů a jejich kódujících genů, jejichž mutace se podílí na etiopatogenezi geneticky podmíněného NS. V patogenezi idiopatického NS je zvažováno několik teorií týkajících se imunitního systému a jeho vlivu na podocyty [5]. K těmto hypotézám patří: 1. dysfunkce T-lymfocytů, která má za následek uvolnění cytokinů zvyšujících permeabilitu filtrační bariéry, 2. alterace imunitního systému, jež vede k produkci solubilního cirkulujícího faktoru, který zvyšuje propustnost filtrační bariéry (příkladem může být u pacientů s FSGS solubilní receptor pro plasminogen urokinázového typu – suPAR), 3. imunitní postižení podocytu se zvýšenou aktivací a expresí některých antigenů při poruše autoregulace (např. antigen CD80). Žádná z těchto hypotéz však zatím nebyla jednoznačně potvrzena a přesný mechanismus onemocnění tak není dosud znám [6, 7].
Patogenezi otoků při NS vysvětlují dvě teorie. První, tzv. „underfill“, předpokládá jako hlavní mechanismus hypoalbuminémii, která vede k poklesu onkotického tlaku plazmy, úniku tekutiny do intersticia a vzniku hypovolémie. Kompenzační reakcí organismu je aktivace systému renin-angiotenzin-aldosteron se sekundární retencí sodíku a vody. Druhá teorie, tzv. „overfill“, vysvětluje vznik otoků primární aktivací epiteliálního sodíkového kanálu v distálním tubulu, která vzniká již v úvodu při zvyšování proteinurie a jejíž důsledkem je primární retence sodíku a tím i vody v organismu [8, 9].
Hyperlipidémie, především hypercholesterolémie a hypertriacylglycerolémie, jsou důsledkem komplexní poruchy syntézy a degradace lipoproteinů, ke které vedou jejich zvýšené ztráty močí kompenzované zvýšenou produkcí v játrech. Předpokládá se zvýšená aktivita jaterní β-hydroxy-β-metylglutaryl – koenzym A reduktázy a acyl-cholesterol-koenzym A-acyl transferázy a snížení aktivity cholesterol 7α-hydroxylázy a lipoproteinové lipázy [10].
Klinické příznaky
V klinickém obraze jsou nejvýznamnějším nálezem otoky, které jsou přítomny v různém rozsahu u 95 % dětí s NS. Otoky jsou lokalizované zejména v obličeji periorbitálně, na dolních končetinách a v oblasti genitálu. V případě rozsáhlých otoků je běžným nálezem i ascites, pleurální a perikardiální efuze. K dalším příznakům patří oligurie a hypertenze, která bývá přítomná až u 30 % dětí s iniciální atakou NS. U dětí s hypovolémií můžeme při fyzikálním vyšetření zjistit naopak nízký krevní tlak, tachykardii a prodloužený kapilární návrat.
Nespecifické příznaky zahrnují letargii či naopak podrážděnost, mohou být přítomny i bolesti břicha a průjem, které jsou důsledkem edému střevní stěny. Některé děti mohou mít známky akutně probíhající infekce (pneumonie, sepse) vzhledem k narušené imunitní odpovědi při ztrátách imunoglobulinů do moči a porušené funkci T-lymfocytů. Manifestaci NS velmi často předchází infekce horních cest dýchacích.
Laboratorní nálezy
Vyšetření dítěte s první atakou NS je zaměřeno na potvrzení diagnózy NS, diferenciální diagnostiku a zhodnocení renálních funkcí. Základní je vyšetření moče chemicky, zhodnocení močového sedimentu, kvantitativní vyšetření proteinurie a vyšetření koncentrace sodíku a draslíku ve vzorku moče (tab. 2).
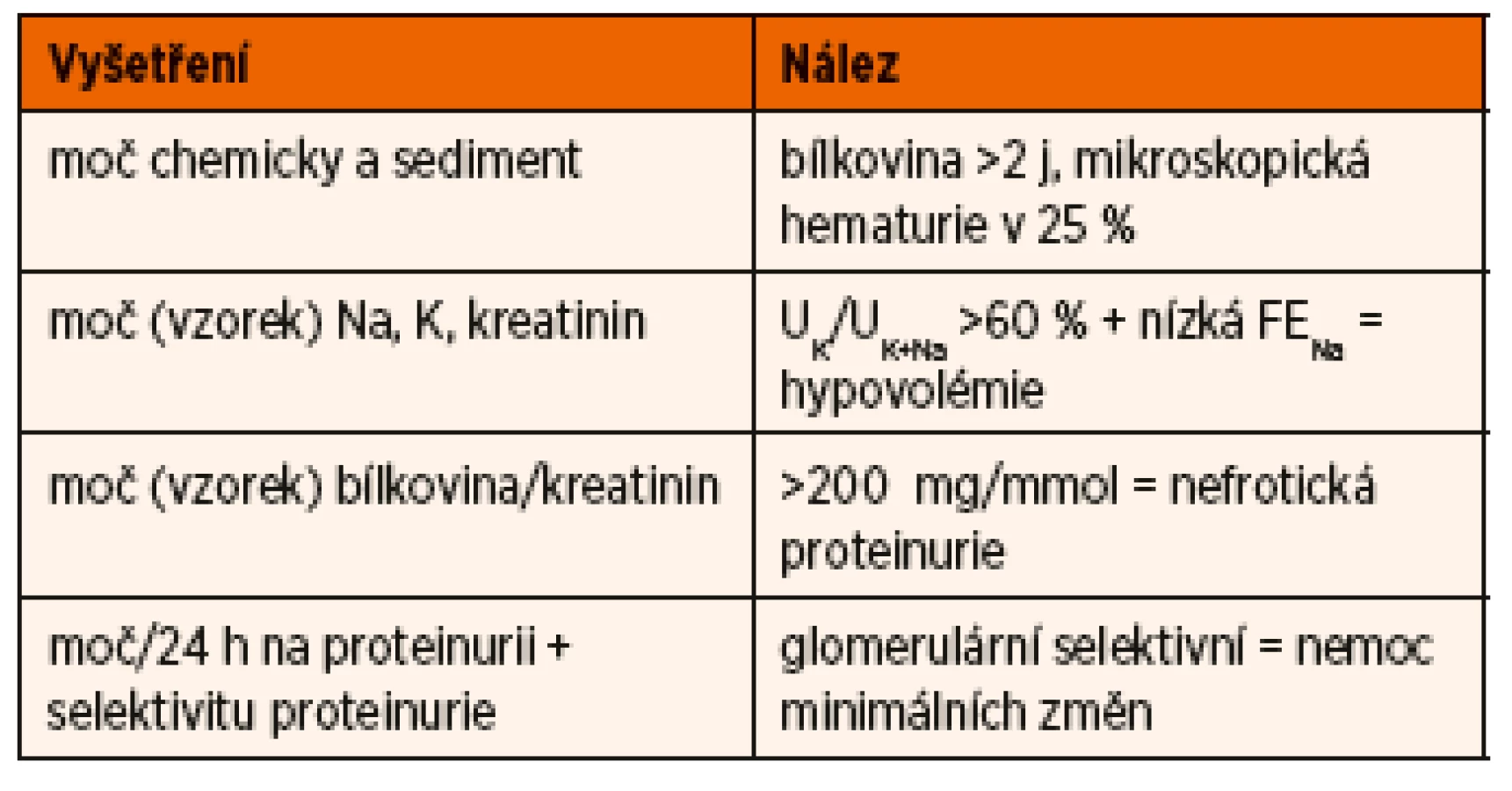
Kvantitativní zhodnocení proteinurie umožňuje stanovení poměru bílkoviny ke kreatininu z jednorázového vzorku moče, kde hodnota >200 mg/mmol odpovídá nefrotickému rozmezí, přesnější je ale sběr moče za 24 hodin s odpadem bílkoviny >960 mg/m2/24 hodin (aproximativně 1 g/m2/24 hodin). K potvrzení diagnózy NS je nutné vyšetření hladiny sérového albuminu, které prokáže hypoalbuminémii pod 25 g /l. Makroskopická hematurie není pro NS na podkladě MCD typická, ale mikroskopická hematurie může být přítomna až u 25 % nemocných [11]. Hodnoty kreatininu a urey v séru jsou závislé na náplni intravaskulárního prostoru, při redukci jeho objemu klesá efektivní filtrační tlak na úrovni glomerulu a výsledkem je pokles glomerulární filtrace a vzestup hladiny kreatininu i urey. Při pozdní diagnóze může stav přejít až do prerenálního selhání ledvin. Hyperlipidémie je běžná, hodnoty cholesterolu i triacylglycerolů často významně přesahují horní hranice normy.
Z imunologických vyšetření je v rámci diferenciální diagnostiky NS důležité stanovení C3 a C4 složky komplementu, antinukleárního faktoru (ANF), vyšetření k vyloučení hepatitidy B a hepatitidy C (HBsAg, anti-HCV protilátky), HIV. Laboratorní vyšetření u dítěte s iniciální atakou NS a typické či běžné nálezy jsou uvedeny v tabulkách 3 a 4.


Ultrazvukové vyšetření slouží k posouzení přítomnosti a rozsahu výpotků (ascites, hydrothorax, ev. perikardiální výpotek), dále posouzení parenchymu ledvin, důležitou roli hraje ultrazvuk také při podezření na trombotickou komplikaci.
Renální biopsie je u dětí s NS indikována na základě věku, klinických či laboratorních nálezů tehdy, pokud je malá pravděpodobnost MCD jako podkladu NS. K těmto indikacím patří:
- věk <1 rok a >12 let,
- makroskopická hematurie,
- mikroskopická hematurie a hypertenze,
- zvýšení hladiny kreatininu/urey, které není způsobeno hypovolémií,
- kortikosteroid-rezistentní NS, pokud není přítomna kauzální mutace pro geneticky podmíněný NS,
- kortikosteroid-dependentní NS vždy před zahájením léčby kalcineurinovými inhibitory a mykofenolát mofetilem [12].
Léčba iniciální ataky nefrotického syndromu
Kortikosteroidy jsou používány v léčbě NS od druhé poloviny 50. let 20. století, kdy bylo poprvé publikováno jejich úspěšné použití [13]. Původní používané schéma International Study of Kidney Diseases in Children (ISKDC) ze 60. let doporučovalo podávání prednisonu v dávce 60 mg/m2/den pod dobu 4 týdnů a dále 40 mg/m2/obden další 4 týdny. V r. 1993 bylo publikováno doporučení Arbeitsgemeinschaft fűr Pädiatrische Nephrologie (APN), které prokázalo snížení frekvence relapsů při prodloužení terapie kortikosteroidy z původních celkem 8 týdnů na 12 týdnů [14]. Snížení počtu relapsů bez zvýšení nežádoucích účinků léčby potvrdila i meta-analýza pěti randomizovaných studií, do kterých byly zařazeny děti s první atakou NS [15]. V současné době je tedy doporučována léčba prednisonem 60 mg/m2/den po dobu 6 týdnů s následným podáváním 40 mg/m2/obden dalších 6 týdnů. Maximální denní dávka prednisonu by neměla překročit 80 mg. Při této léčbě dosáhne remise přes 90 % pacientů s idiopatickým NS, 75 % dětí s MCD dosáhne zpravidla remise do 2 týdnů od zahájení léčby. Studie z posledních let, které se zabývaly léčbou iniciální ataky NS, neprokázaly vyšší efekt dalšího prodlužování kortikoterapie na ovlivnění počtu relapsů [16]. Jak již bylo uvedeno, je odpověď na léčbu jedním z důležitých kritérií klasifikace NS. Klasifikace a terminologie NS související s léčbou je uvedena v tabulce 5.

Léčba relapsu nefrotického syndromu
K relapsu NS dochází zhruba u 60 % kortikosteroid-senzitivních pacientů. Definice relapsu i rozdělení pacientů podle frekvence relapsů je uvedena v tabulce 5. K léčbě relapsu se používá prednison 60 mg/m2/den podávaný do dosažení remise a následně 3 dny, poté v dávce 40 mg/m2/obden alespoň další 4 týdny.
Léčba nefrotického syndromu s častými relapsy a kortikosteroid-dependentního nefrotického syndromu
U těchto dětí máme několik možností, jak dosáhnout dlouhodobé remise:
- Po dosažení remise pokračovat 6–12 měsíců v léčbě prednisonem v obdenním podávání s postupně se snižující dávkou (zpravidla v rozmezí 0,1–0,5 mg/kg), která je schopná udržet remisi. Cílem je nejen udržení remise, ale i minimalizace nežádoucích účinků kortikosteroidů, ke kterým patří závažná obezita, hypertenze, porucha růstu, katarakta, osteoporóza, ale i kosmeticky velmi nepříjemné a trvalé strie.
- Přechod na denní podávání prednisonu z obdenního režimu po dobu 5–7 dní v průběhu akutního respiračního onemocnění, případně nasazení malé dávky prednisonu (0,5 mg/kg) po dobu 5 dní u pacientů bez kortikoidní léčby.
- Léky umožňující snížit či vysadit kortikosteroidy, zejména při známkách toxicity a nežádoucích účincích léčby kortikosteroidy. Do skupiny těchto léků patří levamizol, alkylační látky, kalcineurinové inhibitory a mykofenolát mofetil. U pacientů s komplikovaným a opakovaně relabujícím NS lze použít monoklonální protilátku proti antigenu CD20 – rituximab či nově při rezistenci na rituximab ofatumumab. Před zahájením léčby imunosupresivy je doporučeno provedení renální biopsie. V současné době nejsou k dispozici výsledky randomizovaných studií srovnávajících účinek jednotlivých kortikosteroidy šetřících léků, a proto jejich výběr závisí na preferenci pracoviště dětské nefrologie a rodiny nemocného.
Levamizol je anthelmintikum s prokázanými imunomodulačními vlastnostmi, který ve srovnání s placebem či prednisonem vede ke snížení frekvence relapsů a je vhodný zejména u mírnějších forem NS s častými relapsy [17]. Podává se v dávce 2,5 mg/kg/obden a umožňuje snížení či vysazení prednisonu. Léčba by měla trvat 12 měsíců a je nutné během ní pravidelně kontrolovat krevní obraz pro riziko neutropenie a jaterní testy pro riziko hepatotoxicity. Nevýhodou je současná nedostupnost tohoto léku v celé řadě zemí včetně České republiky.
Alkylačními látkami užívanými k léčbě NS jsou cyklofosfamid a chlorambucil. V našich podmínkách se používá téměř výhradně cyklofosfamid, jehož účinek ve srovnání s chlorambucilem je vyšší a má méně nežádoucích účinků. Jeho efekt v navození dlouhodobé remise je vyšší u dětí s častými relapsy, kde 70 % pacientů je v dlouhodobé remisi po 2 letech od zahájení léčby ve srovnání s 25 % pacientů s kortikosteroid-dependentním NS [18].
Cyklofosfamid se podává v dávce 2–3 mg/kg/den jednou denně ráno po dobu 8–12 týdnů, neměla by být překročena kumulativní dávka 200 mg/kg. Současně s cyklofosfamidem se doporučuje alternativní podávání malé dávky prednisonu. K nežádoucím účinkům patří nauzea, zvýšené vypadávání vlasů, útlum kostní dřeně a hemoragická cystitida. Pozdním následkem léčby může být poškození gonád, riziko je vysoké zejména u prepubertálních chlapců a při překročení výše uvedené kumulativní dávky. Opakovaná kúra cyklofosfamidu je z výše uvedených důvodů kontraindikovaná. Při léčbě cyklofosfamidem je nutno kontrolovat krevní obraz a jaterní testy pro riziko hepatopatie a útlumu kostní dřeně.
Kalcineurinové inhibitory blokují aktivaci T-lymfocytů, modulují tak imunitní odpověď, navíc se ukazuje, že mají i vazoaktivní účinek a předpokládá se i přímý efekt na podocyty.
Cyklosporin A (CsA) je používán v léčbě NS od 90. let minulého století, dokáže navodit dlouhodobou remisi s možností vysazení kortikosteroidů u většiny pacientů s kortikosteroid-dependentním NS. Léčba se zahajuje po dosažení remise v dávce 5 mg/kg/den rozděleně ve dvou stejných dávkách. Výhodou je možnost kontrol plazmatické hladiny léku, kdy za optimální se v prvních měsících léčby považuje hodnota 80–150 ng/ml. Léčba by měla trvat minimálně 12 měsíců, většinou je však delší, neboť velká část pacientů má tendenci po jejím ukončení relabovat [19]. K nežádoucím účinkům CsA patří zejména nefrotoxicita, dále hypertenze, gingivální hyperplazie a hypertrichóza. Takrolimus (TAC) je dobrou alternativou CsA zejména u dívek, protože nevede k nežádoucím kosmetickým účinkům, je proto lépe tolerován. Je rovněž nefrotoxický a jeho podávání může vést ke vzniku diabetes mellitus, který se ale zatím rozvinul pouze u pacientů léčených TAC po transplantaci ledviny.
Mykofenolát mofetil (MMF) má antiproliferační efekt jak na T, tak B-lymfocyty. Používá se v dávce 1200 mg/m2/den (max. 2 g/den) rozděleně ve dvou dávkách. K nežádoucím účinkům, které jsou závislé na dávce, patří útlum kostní dřeně a gastrointestinální obtíže. Jeho výhodou ve srovnání s CsA je, že není nefrotoxický. Jeho efekt na dlouhodobou remisi je při vyšším dávkování srovnatelný s CsA [20]. Podobně jako u CsA je však u MMF po jeho vysazení riziko relapsu NS. MMF je teratogenní, proto je nutné u fertilních dívek podávat současně kontraceptiva.
Rituximab je chimérická monoklonální protilátka, která se váže na antigen CD20 exprimovaný pre-B a B-lymfocyty, to vede k jejich depleci. Působí také přímo na podocyty stabilizací cytoskeletu. Přestože zatím neexistuje dostatek randomizovaných studií, zdá se, že rituximab umožňuje u pacientů s kortikosteroid-dependentním či CsA-dependentním NS vysazení veškeré léčby, i když se ukazuje, že tento efekt je pouze dočasný. K udržení dlouhodobé remise je proto v některých případech nutné opakované podání tohoto preparátu [21]. Léčba rituximabem je určena pacientům se závažným průběhem onemocnění, které se nedaří dostat pod kontrolu výše zmíněnými imunosupresivy. Terapie může být vzácně spojena s nežádoucími účinky, jako jsou závažné infekce, plicní fibróza, fulminantní myokarditida, těžká kolitida či závažná alergická reakce.
Léčba kortikosteroid-rezistentního nefrotického syndromu
Léčba SRNS zůstává pro pediatry stále velkou výzvou a patří do rukou zkušených dětských nefrologů ve větších centrech. Definice rezistence na kortikosteroidy není zcela jednotná, ale předpokládá podávání prednisonu v dávce 60 mg/m2/den po dobu 4–8 týdnů bez dosažení plné remise základního onemocnění. V případě rezistence na kortikosteroidy je indikováno provedení renální biopsie a molekulárně genetické vyšetření k vyloučení geneticky podmíněných forem NS. Biopsie ledviny odliší případně jiné vzácné diagnózy (např. membranózní nefropatii nebo C3 glomerulonefritidu). Diagnostika genetických forem NS je naprosto zásadní, protože v těchto případech není indikováno podávání imunosupresivní léčby. Navzdory tomu, že některá kazuistická sdělení prokázala efekt cyklosporinu na snížení proteinurie u dětí s NS na podkladě mutací v genech NPHS2, WT1 [22, 23], výsledky rozsáhlé německé pediatrické studie, zabývající se účinkem CsA u dětí s kortikosteroid-rezistentním NS, podávání imunusupresivní léčby u pacientů s geneticky podmíněným NS nepodporují [24]. Efekt CsA lze vysvětlit spíše ovlivněním hemodynamiky, případně stabilizací cytoskeletu podocytu. Tito pacienti navíc přes léčbu CsA dospěli do terminální fáze chronického onemocnění ledvin. Pozoruhodný je i popsaný efekt imunosupresivní léčby u dvou pacientů s mutací v PLCE1 genu [25]. Dlouhodobé podávání kalcineurinových inhibitorů je ale všeobecně vzhledem k nefrotoxicitě a riziku dalších nežádoucích účinků u geneticky podmíněného NS nevhodné. Terapie geneticky podmíněného SRNS je tedy symptomatická a léky první volby jsou preparáty ovlivňující osu renin-angiotenzin-aldosteron, což jsou ACE-inhibitory, případně blokátory angiotenzinového receptoru, které mají antihypertenzní a antiproteinurický účinek. Při podávání těchto preparátů je nutno brát v potaz jejich teratogenitu. Další léčba je závislá na klinickém stavu nemocného a jeho laboratorních nálezech.
V případě idiopatického SRNS je naopak imunosupresivní terapie plně indikovaná. Studie Ehricha et al. [26] prokázala dobrý efekt kombinované léčby kortikoidy a CsA. Účinek byl významně vyšší při kombinaci parenterálního a perorálního podávání kortikoidů ve srovnání s terapií samotným prednisonem. Doporučuje se podání methylprednisolonu (3–8 pulzů obden v dávce 300–1000 mg/m2/den) a nasazení CsA, iniciální dávka je 150 mg/m2/den. Od počátku podáváme také perorální prednison v dávce 40 mg/m2/obden. Dávku prednisonu postupně snižujeme po dobu 6 měsíců, CsA ponecháváme v léčbě po dobu několika let. Odpověď na léčbu byla pozorována asi u 80 % pacientů, nicméně je obvykle postupná a lze ji očekávat i několik měsíců od zahájení. Namísto CsA můžeme podávat i TAC, který prokázal srovnatelný efekt v porovnání s CsA u kortikosteroid-rezistentního NS, i když nemáme zatím k dispozici tolik dat o jeho pozitivním účinku jako v případě CsA.
MMF v kombinaci s dexamethazonem prokázal v rámci FSGS Clinical Trial srovnatelný efekt s CsA v dosažení parciální nebo plné remise u dětských i mladých dospělých pacientů s kortikosteroid-rezistentním NS a nálezem FSGS v renální biopsii. MMF tedy můžeme považovat za alternativu CsA [27]. V indikovaných případech lze léčebně použít kombinaci MMF s kalcineurinovým inhibitorem a prednisonem [28]. Odpověď na léčbu rituximabem je u pacientů se SRNS významně nižší v porovnání se SSNS. Randomizovaná kontrolovaná studie italských nefrologů, do které bylo zařazeno 31 dětí léčených doposud prednisonem a CsA bez efektu na zlepšení průběhu nemoci, neprokázala účinek rituximabu na snížení proteinurie [29]. Recentní japonská práce naopak popsala velmi dobrý efekt léčby kombinací rituximabu s pulzním methylprednisolonem [30]. K ozřejmění role rituximabu v léčbě SRNS bude třeba provést další kontrolované studie.
Ofatumumab, plně humanizovaná protilátka, která se váže na antigen CD20, byla úspěšně podána u několika pacientů se SRNS a může být v budoucnu alternativou hlavně u pacientů s rezistencí nebo alergií na rituximab [31].
Také eliminační metody – plazmaferéza a imunoadsorpce – mohou mít příznivý léčebný efekt u pacientů se SRNS [32].
Symptomatická léčba nefrotického syndromu
Děti s otoky vyžadují pečlivé denní monitorování diurézy a příjmu tekutin, při významných otocích a oligurii se doporučuje restrikce tekutin a dieta s omezením soli. Diuretika používáme uvážlivě, své postavení mají v léčbě pacientů s významnou retencí tekutin a oligurií, u kterých byla vyloučena hypovolémie [33]. Nejčastěji používaným je furosemid podávaný perorálně či intravenózně v dávkách 1–2 mg/kg/dávku s maximem 10 mg/kg/den, lepšího efektu lze při refrakterních otocích dosáhnout jeho aplikací v kontinuální intravenózní infuzi. Z dalších diuretik lze při nedostatečném efektu furosemidu použít do kombinace hydrochlorothiazid v dávce 1–3 mg/kg/den či spironolakton v dávce 2–3 mg/kg/den. Aplikace infuze 20% albuminu při kompenzovaném stavu s normálním krevním tlakem a dostatečnou diurézou není v současnosti indikována, je vyhrazena pouze pro pacienty s intravaskulární hypovolémií. Pro tyto pacienty je typická nízká frakční exkrece natria (FENa) a současně hodnota indexu UK /(UNa + UK) nad 60 %.
Komplikace nefrotického syndromu
K nejčastějším komplikacím NS patří v dětském věku infekce a tromboembolické příhody. Infekce byly v éře před zavedením antibiotik a kortikosteroidů do léčby NS nejčastější příčinou úmrtí a umíralo na ně až 67 % dětí s NS. K nejčastějším infekčním komplikacím patří peritonitida, pneumonie a sepse. Z infekčních agens se nejvíce uplatňuje Streptococcus pneumoniae, dále v menší míře Streptococcus pyogenes, Hemophilus influenzae a Escherichia coli.
Tromboembolické komplikace (TE) jsou v dětském věku podstatně méně časté než u dospělých. Jejich incidence je udávána mezi 1,8–5,3 %, daleko vyšší je však u kongenitálních forem NS, kde dosahuje 10–13 %. Etiologie hyperkoagulačního stavu u NS je multifaktoriální, uplatňuje se snížená hladina antitrombinu III (ztráty močí, zvýšený katabolismus a konsumpce), zvýšená hodnota fibrinogenu (zvýšená proteosyntéza), snížená aktivita proteinu C a S, zvýšení počtu destiček a jejich agregace. K rizikovým faktorům pro vznik TE patří zejména hypovolémie, zvýšený hematokrit, infekce, delší doba imobilizace, léčba diuretiky a kortikoidy [34]. Vzhledem k absenci randomizovaných kontrolovaných studií prokazujících efekt profylaxe TE neexistují jednoznačná doporučení pro prevenci TE u dětských ani dospělých pacientů s NS. U vysoce rizikových dětí s NS z hlediska TE se doporučuje konzultace a spolupráce s dětským hematologem.
Geneticky podmíněný nefrotický syndrom
Genetická příčina NS se u kongenitálních a infantilních forem NS předpokládala již v dávné minulosti, avšak první skutečný důkaz o NS jako monogenní chorobě byl podán až v roce 1998, kdy finští vědci odhalili mutace v genu kódujícím glomerulární protein nefrin jako kauzální příčinu kongenitálního NS finského typu [35]. V roce 2000 byly francouzskou skupinou Antignac a spol. objeveny mutace v dalším glomerulárním proteinu zvaném podocin, který je nejčastější příčinou autozomálně recesivního SRNS u nefinské dětské populace [36]. V novém století bylo s rozvojem metod molekulární genetiky identifikováno do letošního roku již více než 40 genů, jejichž mutace způsobují NS. Tyto geny kódují téměř bez výjimky strukturní a regulační proteiny podocytů a štěrbinové membrány (slit diaphragma), která je lokalizována mezi výběžky podocytů zvané pedicely. Tím je narušena funkce glomerulární filtrační membrány, která je nezbytná pro zamezení nadměrného průniku proteinů do moči. Geneticky podmíněný NS je tedy způsoben téměř vždy poruchou struktury nebo funkce podocytárních proteinů a je tedy často označován jako „podocytopatie“.
Výskyt a klinická manifestace
Jak již bylo uvedeno, u dětí s kongenitálním NS tvoří geneticky podmíněné formy 70 % příčin NS, u dětí s infantilním SRNS 45 % a v celém dětském věku 20–30 % příčin SRNS (klesající výskyt v závislosti na stoupajícím věku dětských pacientů).
Největší zastoupení mají mutace v recesivních genech NPHS1 (40 % případů kongenitálního NS a 10 % případů infantilního NS) a NPHS2 (5–12 % případů) a v dominantním genu WT1 (2–12 % případů) [37]. V současnosti je identifikováno celkem 43 genů, jejichž mutace způsobují SRNS u dětí (tab. 6). Avšak 2/3 případů kongenitálního a infantilního NS jsou způsobeny mutacemi pouze 4 genů: NPHS1, NPHS2, PLCE1 a LAMB2 [38].

Klinickým projevem téměř všech geneticky podmíněných forem NS je SRNS [39]. Existují však raritně i familiární formy SSNS, (autozomálně recesivní i autozomálně dominantní způsoby dědičnosti). U nich byl doposud objeven pouze jeden gen v jedné turecké rodině (gen EMP2, epithelial membrane protein 2), jehož mutace způsobovala autozomálně recesivní familiární SSNS [40].
NPHS1 gen kódující nefrin
V roce 1998 bylo finskými vědci odhaleno, že mutace v genu kódující nový glomerulární protein zvaný nefrin, způsobují kongenitální NS (dříve označovaný jako finského typu). Tento protein komunikuje s dalšími proteiny štěrbinové membrány a zajišťuje funkčnost filtrační bariéry, zejména co se týká nepropustnosti pro sérové bílkoviny. Byly popsány dvě nejčastější mutace předčasně ukončující syntézu nefrinu (Fin-minor a zkracující truncating mutaci Fin-major), které tvoří asi 90 % všech mutací u finských dětí, přičemž nebyla nalezena korelace mezi typem mutace a fenotypem [41]. Tento typ NS se dědí autozomálně recesivně, jeho gen je lokalizován na 19. chromozomu, incidence se udává 1 : 10 000. Vyskytuje se u více než 90 % finských dětí s kongenitálním NS, zatímco u dětí nefinského původu pouze v 10–40 %.
Klinicky se manifestuje téměř vždy již v novorozeneckém věku nebo do 3 měsíců od narození. Často bývají již prenatální známky NS u plodu, zejména zvětšená placenta. Průběh je velmi těžký s extrémní proteinurií, hypoproteinémií s hypogamaglobulinémií. Děti jsou léčeny symptomaticky infuzemi albuminu a dietou obohacenou o bílkoviny. Vzhledem k významnému riziku závažných infekcí musí být včas a adekvátně léčeny. Pokud pacienti prospívají a jejich vývoj probíhá normálně, vyžadují pouze výše zmíněnou symptomatickou terapii. Nicméně u pacientů s opožděným růstem, ev. v případě dětí s častými infekcemi by měla být provedena časná nefrektomie (většinou oboustranná) s následným zahájením dialýzy, případně provedenou transplantací ledviny podle stavu dítěte [42]. Bohužel část pacientů s Fin-major mutací vyvine rekurenci v transplantované ledvině, která je způsobena vznikem antinefrinových protilátek.
NPHS2 gen kódující podocin
Gen NPHS2 kóduje glomerulární protein podocin, který se nachází na chromozomu 1 a je podobně jako nefrin součástí štěrbinové membrány. Mutace v tomto genu byly poprvé popsány v roce 2000 u skupiny dětí s časným začátkem autozomálně recesivní formy SRNS [36]. Přestože gen NPHS2 obsahuje pouze 8 exonů, bylo již popsáno 126 různých mutací v tomto genu, z toho zhruba polovina jich je asociovaných se SRNS. Tyto mutace ovlivňují funkci, strukturu nebo expresi podocinu, čímž způsobují poruchu prostorového uspořádání a stability podocytu [43]. Nejčastější skupinou mutací genu NPHS2 jsou missense mutace, tvoří 42 % z celkového počtu nalezených mutací. Ve 29 % je tvoří malé inzerce a delece, třetí nejčastější skupinou jsou nonsense mutace [37].
Mutace v podocinu způsobují SRNS, který se manifestuje nejčastěji před šestým rokem věku a velice rychle progreduje do chronického selhání ledvin. Na rozdíl od mutací v genu NPHS1 je prevalence rekurence SRNS ve štěpu téměř nulová [44]. Mutace v genu NPHS2 jsou nejčastější (10 %) u pacientů se SRNS s manifestací onemocnění mezi 1.–18. rokem života. U pacientů s manifestací onemocnění do 3 měsíců života jsou tyto mutace druhé nejčastější (11 %) po mutacích v genu NPHS1 (40 %) [37]. Doba klinické manifestace závisí především na typu mutace v genu NPHS2. Nejčastější mutací vyskytující se v severní Evropě a USA je R138Q. Homozygoti pro tuto mutaci mívají závažnější projevy onemocnění [43]. Bylo popsáno také několik polymorfismů včetně R229Q, který s určitými dalšími patogenními mutacemi na druhé alele genu NPHS2 způsobuje zvýšené riziko vzniku SRNS, jenž se vyskytuje častěji u dospělých pacientů a průběh onemocnění bývá méně závažný [37]. Bioptický nález u pacientů s mutacemi v NPHS2 může vykazovat jak FSGS, tak MCD, takže podle histologického obrazu nelze předpovědět, jestli bude gen NPHS2 mutovaný.
WT1 (Wilms tumor1) gen
Gen WT1 je tumor-supresorový gen umístěný na chromozomu 11, který kóduje transkripční faktor nezbytný pro vytváření genitouretrálního traktu a glomerulární filtrační membrány ve fetální ledvině [43]. Po ukončení nefrogeneze je exprese genu WT1 omezená pouze do oblasti podocytů. Gen WT1 také řídí gen SRY, který kóduje specifický faktor stojící na počátku kaskády, jež determinuje rozvoj mužského pohlaví [45]. Mutace v genu WT1, kterých je více než 95 % lokalizováno v oblasti exonu 8 a 9, způsobují širokou škálu fenotypů – Denysův-Drashův syndrom (DDS), Frasierův syndrom (FS), izolovaný Wilmsův tumor, nefrotický syndrom typ 4 (NPHS4), WAGR syndrom (Wilmsův tumor, aniridie, genitouretrální abnormality a mentální retardace) a extrémně vzácný Meachamův syndrom (genitouretrální abnormality, kongenitální srdeční vady). DDS je charakterizován časným začátkem SRNS s rychlou progresí do chronického renálního selhání, mužským pseudohermafroditismem, vysokým rizikem vzniku Wilmsova tumoru (WT) a nálezem difuzní mezangiální sklerózy (DMS) v renální biopsii. Byly popsány též inkompletní formy DDS [46]. FS je vzácné onemocnění definované jako mužský pseudohermafroditismus, SRNS a vysoké riziko vzniku gonadoblastomu. Riziko vzniku WT je nižší v porovnání s pacienty s DDS. Klinický začátek onemocnění je pozdější než u pacientů s DDS (mezi 2–6 lety života), progrese do chronického renálního selhání je pomalejší a typickým nálezem v renální biopsii je FSGS. NPHS4 je charakterizován časným začátkem onemocnění (4–12 měsíců věku), izolovanou DMS, SRNS a rychlou progresí do chronického renálního selhání [43]. Mutace v genu WT1 se dědí autozomálně dominantně, většina mutací vzniká de novo. Vzhledem k riziku vzniku nádorových onemocnění (WT, gonadoblastom) a genitouretrálních malformací se doporučuje u všech pacientů s prokázanou mutací v genu WT1 doplnit vyšetření karyotypu a onkologické sledování pacienta. Dnes převládá názor, že u pacientů s mutací WT1 genu, u nichž se dosud WT nemanifestoval, ale u nichž hrozí časný vznik WT, by měla být v době progrese onemocnění do konečného stadia chronického onemocnění ledvin indikována oboustranná nefrektomie a zahájena dialyzační léčba s následnou transplantací. Při manifestaci WT je nefrektomie samozřejmě indikována co nejdříve bez ohledu na poruchu renálních funkcí.
NPHS3 (PLCE1) gen
Tento gen leží na 10. chromozomu a kóduje enzym fosfolipázu C-epsilon 1 (PLCE1), která se podílí na nitrobuněčné signalizaci generováním druhých poslů. Mutace, které způsobí předčasné ukončení syntézy proteinu, se projevují histologickým obrazem difuzní mezangiální sklerózy (DMS). Bylo zjištěno, že skoro třetina případů DMS u dětí s NS je způsobena mutací v PLCE1 genu a je tedy třikrát častější genetickou příčinou DMS u dětí než mutace v genu WT1 [47]. Byla zjištěna korelace mezi genotypem a fenotypem u pacientů – věk při manifestaci NS byl dřívější u dětí se slice site mutacemi než u dětí s missense nebo C-terminálními truncating mutacemi [37]. Missense mutace se projevují spíše obrazem FSGS. Zajímavou skutečností je, že u dvou pacientů s mutacemi v PLCE1 genu došlo k remisi NS při léčbě kortikoidy a CsA [25].
LAMB2 gen
Mutace tohoto genu způsobují autozomálně recesivní Piersonův syndrom, případně mohou být asociovány s mírnějšími variantami tohoto syndromu či s izolovaným NS. Piersonův syndrom je charakterizován nejčastěji kongenitálním nebo infantilním NS, těžkou mentální retardací a typickým postižením oka s mikrokorií. Gen leží na 3. chromozómu a kóduje protein laminin β-2. NS je velmi těžký, srovnatelný s NS finského typu. Postižení většinou umírají v novorozeneckém nebo kojeneckém věku, avšak jsou popsány i případy s mírnějším průběhem s dobou přežití výjimečně až do dospělého věku. Byla zjištěna genotypicko-fenotypická korelace, kdy se pacienti s N-terminálními zkracujícími mutacemi manifestovali do 2 měsíců věku a pacienti s C-teminálními zkracujícími mutacemi až po 2 měsících věku [37].
SMARCAL1 gen
Mutace v tomto genu způsobují velmi závažné multisystémové onemocnění – Schimkeho imunooseální dysplazii (SIOD). Toto onemocnění se projevuje T-buněčným imunodeficitem, těžkou skeletální dysplazií s extrémně malým vzrůstem, typickou faciální dysmorfií a SRNS, který vede časně k terminální fázi chronického onemocnění ledvin. Mortalita pacientů se SIOD již v dětském věku je velmi vysoká, nejčastější příčinou úmrtí jsou buď závažné infekce při T-buněčném imunodeficitu v kombinaci s hypogamaglobulinémií, nebo cerebrovaskulární příhody při stenózách intrakraniálních arterií.
LMX1B gen
Autozomálně dominantní nail-patella syndrom je způsoben heterozygotními mutacemi v genu LMX1B. Tento syndrom je klinicky, jak z názvu vyplývá, typicky charakterizován dysplazií nehtů a hypoplazií až úplným chyběním pately. U 10–40 % pacientů je přítomna i nefropatie, která se může projevit proteinurií a/nebo hematurií, postižení ledvin je nejdůležitějším faktorem určujícím prognózu, neboť pacienti mohou dospět až do terminální fáze chronického onemocnění ledvin v mladém dospělém věku.
U dospívajících a mladých dospělých jsou nejčastějšími mutovanými geny, které jsou nalézány u 2–5 % pacientů se SRNS, ACTN4, TRPC6 a INF2 geny [37].
ACTN4 gen
Gen ACTN4 kóduje protein, který je součástí aktinového cytoskeletu a váže se s f-aktinem. Aktinová cytoskeletální síť je nutná pro správnou prostorovou konfiguraci buněk a jejich pohyb. Toto přesné prostorové uspořádání podocytů a jejich výběžků pravděpodobně umožňuje správnou funkci štěrbinové membrány. Mutace v tomto genu způsobují NS s histologickým obrazem FSGS, který se manifestuje u dospívajících neb u mladých dospělých většinou do 25 let. Onemocnění je autozomálně dominantně dědičné a označuje se jako familiární FSGS typ 1.
TRPC6 gen
Tento gen zodpovědný u malé části dospívajících a mladých dospělých za rozvoj SRNS s histologickým obrazem FSGS kóduje kalciový kanál (TRPC6, transient receptor potential cation channel 6). Mutace způsobují vyšší aktivaci tohoto kanálu a zvýšený vstup kalcia do podocytů. Přesné vysvětlení, jak může mutace v TRPC6 genu způsobit glomerulosklerózu, není zatím známo. Onemocnění je dědičné autozomálně dominantně a je označováno jako familiární FSGS typ 2.
INF2 gen
Heterozygotní mutace v genu inverted formin 2 (INF2) způsobují autozomálně dominantní periferní neuropatii – Charcot-Marie-Tooth syndrom (CMT). U malé části pacientů s tímto syndromem se může objevit proteinurie (většinou asymptomatická, vzácně i jako NS, který je rezistentní na kortikoidy) a část pacientů může dospět v dospělosti až do terminální fáze chronického onemocnění ledvin (tzv. CMT-asociovaná glomerulopatie, histologicky většinou FSGS). Mutace v tomto genu byla nalezena i u 2–5 % dospělých pacientů se SRNS, kteří zatím nejevili klinické projevy CMT. Tento gen kóduje protein inverted formin 2, který funguje jako regulační molekula při polymerizaci aktinu v podocytech.
Klinický obraz
Klinickým projevem téměř všech geneticky podmíněných forem NS je SRNS. Existují však raritně i familiární formy SSNS, autozomálně recesivní i autozomálně dominantní způsoby dědičnosti. U nich byl doposud objeven pouze jeden gen v jedné turecké rodině (gen EMP2, epithelial membrane protein 2), jehož mutace způsobovala autozomálně recesivní familiární SSNS [40].
Diagnostika
U každého dítěte se SRNS musí být dnes vždy indikováno molekulárně genetické vyšetření. A to i přesto, že mezinárodní doporučení KDIGO z roku 2012 toto ještě neuvádějí. Současný názor většiny odborníků je však takový, že u každého dítěte se SRNS by měla být provedena DNA analýza genů pro SRNS. Důvodů pro tato doporučení je několik [48]: DNA analýza jednoznačně a nezpochybnitelně zjistí skutečnou příčinu SRNS (nejedná se pak tedy již o idiopatický NS), může pacienty ušetřit biopsie ledviny a umožní prenatální i postnatální diagnostiku u dalších členů rodiny. Navíc je molekulárně genetické vyšetření důležité pro predikci extrarenálních postižení (např. Wilmsova tumoru, gonadoblastomu, imunodeficitu). Biopsie ledviny je obecně indikována u všech pacientů se SRNS, avšak histologický obraz neodliší idiopatický od geneticky podmíněného NS, a proto pokud známe jasnou příčinu genetického NS, diagnostickou renální biopsii provádět nemusíme. Obě nejčastější formy geneticky podmíněného NS se totiž mohou histologicky projevovat jako MCD, FSGS nebo DMS.
Pro pacienty z České republiky by měla být provedena DNA analýza nejméně 2 nejčastějších genů způsobující geneticky podmíněný SRNS, a to genu NPHS2 a exonů 8 a 9 genu WT1. Toto vyšetření odhalí více než dvě třetiny případů a je časově i finančně efektivní. Mutace v genu NPHS1 jsou v našem regionu vzácné. Pokud se vyšetří i gen NPHS1, odhalí se v evropské populaci dětí cca 90 % případů geneticky podmíněných NS. Ostatních 40 dosud známých genů jsou podle zkušeností z velkých světových kohort pacientů specifické pro velmi malé množství pacientů a jedná se o většinou familiárně unikátní geny.
PROGNÓZA DĚTÍ S NEFROTICKÝM SYNDROMEM
Dlouhodobé následky v dětství manifestovaného kortikosteroid-senzitivního NS byly obecně považovány za nezávažné. Zatímco studie z 80. let ukázala, že pouze 5,5 % pacientů s kortikosteroid-senzitivním NS relabovalo do dospělosti [49], další recentní studie ukazují, že přetrvávající relapsy v dospělosti má daleko více nemocných. Např. japonská retrospektivní práce uvádí, že polovina pacientů, kteří se manifestovali v dětství NS s častými relapsy, má časté relapsy i v dospělosti [50]. Stejně tak francouzská studie potvrdila, že 40 % pacientů s manifestací kortikosteroid-senzitivního NS v dětství trpělo relapsy NS i v dospělosti. Vyšší riziko bylo spojeno s nízkým věkem při manifestaci onemocnění, nutností užívání druhého imunosupresiva a těžším průběhem choroby [51]. Dobře jsou známé i následky užívání imunosupresivní medikace, hlavně kortikosteroidů. Nízká finální výška, obezita, osteoporóza a hypertenze byly popsány u pacientů s kortikosteroid-senzitivním NS v observačních studiích [50, 51]. Děti se SRNS mají nejzávažnější prognózu, 34–64 % z nich dospěje do 10 let od manifestace onemocnění do terminální fáze chronického onemocnění ledvin. Příznivým prognostickým faktorem u dětí s idiopatickým NS je pozitivní odpověď na léčbu [52].
Prognóza všech pacientů s geneticky podmíněným NS je výrazně horší než u pacientů s idiopatickým NS a téměř všichni, a relativně časně, progredují do terminální fáze chronického onemocnění ledvin většinou ještě v dětském věku. Navíc u syndromologických typů geneticky podmíněného SRNS je prognóza často negativně ovlivněna i extrarenálními projevy těchto syndromů, např. Wilmsovým tumorem či gonadoblastomem (WT1), očním postižením nebo mentální retardací (LAMB2), nebo imunodeficitem a cévními abnormalitami (SMARCAL1).
Naopak pozitivním faktem je, že geneticky podmíněné formy, na rozdíl od idiopatických forem s obrazem FSGS, nerekurují v transplantované ledvině, což významně zlepšuje prognózu přežívání funkce transplantované ledviny. Jedinou výjimkou jsou pacienti s Fin-major mutacemi v genu pro nefrin (v ČR zatím nebyl žádný takový pacient diagnostikován), kterým zcela chybí tento protein a kteří mohou po transplantaci vyvinout antinefrinové autoprotilátky a s nimi související proteinurii i po transplantaci ledviny [42].
Potvrzení geneticky podmíněného NS by mělo vést v rodinách s postiženým dítětem k prenatální diagnostice v případě dalšího těhotenství.
Z praktického hlediska je zásadní spolupráce s rodinou pacienta, u starších dětí i s nimi samotnými. Důležité je poučení o původu onemocnění, možnostech diagnostiky a léčby, včetně jejích možných vedlejších účinků a různých komplikací. Rodině by měla být včas podána informace o očekávané prognóze onemocnění a možnosti jeho dědičného přenosu při další graviditě.
ZÁVĚR
Nefrotický syndrom patří v dětském věku k onemocněním, se kterým se může setkat každý pediatr. Nejčastěji se jedná o idiopatický nefrotický syndrom na podkladě nemoci minimálních změn, jehož etiopatogenezi se zatím nepodařilo plně objasnit. Na druhé straně poznatky o geneticky podmíněných formách nefrotického syndromu přinesly za poslední dvě desetiletí celou řadu nových informací nejen o vlastním onemocnění, ale také o struktuře a funkci glomerulární filtrační bariéry. Léčba iniciální ataky nefrotického syndromu se v posledních několika dekádách významněji nezměnila a základem nadále zůstávají kortikosteroidy. Odpověď na tuto léčbu je považována za nejvýznamnější prognostický faktor. U všech dětí s kortikosteroid-rezistentním nefrotickým syndromem by mělo být provedeno genetické vyšetření. Dostupnost tohoto vyšetření je pro pacienty v ČR nejen přínosem v diagnostice, ale především v léčebném přístupu.
Podporováno grantem AZV MZ ČR reg. č. 15-31586A.
Doc. MUDr. Sylva Skálová, Ph.D.
Dětská klinika FN
a LF UK v Hradci Králové
Sokolská 581
500 05 Hradec Králové
e-mail: sylva.skalova@fnhk.cz
Sources
1. Dossier C, Lapidus N, Bayer F, et al. Epidemiology of idiopathic nephrotic syndrome in children: endemic or epidemic? Pediatr Nephrol 2016; 31 (12): 2299–2308.
2. Floege J, Amann K. Primary glomerulonephritides. Lancet 2016; 387 : 2036–2048.
3. Eddy AA, Symons JM. Nephrotic syndrome in childhood. Lancet 2003; 362 : 629–639.
4. Tesař V, Zima T, Kalousová M. Pathobiochemistry of nephrotic syndrome. Adv Clin Chem 2003; 37 : 173–218.
5. Cara-Fuentes G, Clapp WL, Johnson RJ, et al. Pathogenesis of proteinuria in idiopathic minimal change disease: molecular mechanisms. Pediatr Nephrol 2016; 31 (12): 2179–2289.
6. Davin JC. The glomerular permeability factors in idiopathic nephrotic syndrome. Pediatr Nephrol 2016; 31 (2): 207–215.
7. Ling C, Liu X, Shen Y, et al. Urinary CD80 levels as a diagnostic biomarker of minimal change disease. Pediatr Nephrol 2015; 30 (2): 309–316.
8. Bockenhauer D. Over - or underfill: not all nephrotic states are created equal. Pediatr Nephrol 2013; 28 (8): 1153–1156.
9. Kapur G, Valentini RP, Imam AA, Mattoo TK. Treatment of severe edema in children with nephrotic syndrome with diuretics alone–a prospective study. Clin J Am Soc Nephrol 2009; 4 (5): 907–913.
10. Vaziri ND. Disorders of lipid metabolism in nephrotic syndrome: mechanisms and consequences. Kidney Int 2016, 90 (1): 41–52.
11. Geier P. Nefrotický syndrm. Pediatrie pro praxi 2001; 2 (3): 120–123.
12. Skalova S, Podhola M, Geier P, Tichy T. Renal biopsy in children with steroid-dependent nephrotic syndrome. Bratisl lek Listy 2009; 110 (10): 647–649.
13. Arneil GC. Treatment of nephrosis with prednison. Lancet 1956; 270 (6920): 409–411.
14. Ehrich HH, Brodehl J. Long versus standard prednisone therapy for initial treatment of idiopathic nephrotic syndrome in children. Arbeitsgemeinschaft für Pädiatrische Nephrologie. Eur J Pediatr 1993; 152 (4): 357–361.
15. Hodson EM, Knight JF, Willis NS, Craig JC. Corticosteroid therapy in nephrotic syndrome: a meta-analysis of randomised controlled trials. Arch Dis Child 2000; 83 (1): 45–51.
16. Hoyer PF. New lessons from randomized trials in steroid-sensitive nephrotic syndrome: clear evidence against long steroid therapy. Kidney Int 2015; 87 (1): 17–19.
17. Sumegi V, Haszon I, Ivanyi B, et al. Long-term effects of levamisole treatment in childhood nephrotic syndrome. Pediatr Nephrol 2004; 19 : 1354–1360.
18. Latta K, von Schnakenburg C, Ehrich J. A meta-analysis of cytotoxic treatment for frequently relapsing nephrotic syndrome in children. Pediatr Nephrol 2001; 16 (3): 271–282.
19. Larkins N, Kim S, Craig J, Hodson E. Steroid-sensitive nephrotic syndrome: an evidence-based update of immunosuppressive treatment in children. Arch Dis Child. 2016 Apr; 101 (4): 404–408.
20. Gellermann J, Weber L, Pape L, et al. Mycophenolate Mofetil versus Cyclosporin A in children with frequently relapsing nephrotic syndrome. J Am Soc Nephrol 2013; 24 (10): 1689–1697.
21. Ravani P, Bonanni A, Rossi R, et al. Anti-CD20 antibodies for idio-pathic nephrotic syndrome in children. Clin J Am Soc Nephrol 2016; 11 (4): 710–720.
22. Malina M, Cinek O, Janda J, Seeman T. Partial remission with cyclosporine A in a patient with nephrotic syndrome due to NPHS2 mutation. Pediatr Nephrol 2009; 24 (10): 2051–2053.
23. Gellermann J, Stefanidis CJ, Mitsioni A, Querfeld U. Successful treatment of steroid-resistant nephrotic syndrome associated with WT1 mutations. Pediatr Nephrol 2010; 25 (7): 1285–1289.
24. Büscher AK, Beck BB, Melk A, et al. Rapid response to cyclosporin A and favorable renal outcome in nongenetic versus genetic steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol 2016; 11 (2): 245–253.
25. Hinkes B, Wiggins RC, Gbadegesin R, et al. Positional cloning uncovers mutations in PLCE1 responsible for a nephrotic syndrome variant that may be reversible. Nat Genet 2006; 38 (12): 1397–1405.
26. Ehrich JH, Geerlings C, Zivicnjak M, et al. Steroid-resistant idiopathic childhood nephrosis: overdiagnosed and undertreated. Nephrol Dial Transplant 2007; 22 (8): 2183–2193.
27. Gipson DS, Trachtman H, Kaskel FJ, et al. Clinical trial of focal segmental glomerulosclerosis in children and young adults. Clinical trial of focal segmental glomerulosclerosis in children and young adults. Kidney Int 2011; 80 (8): 868–878.
28. Gellermann J, Ehrich JH, Querfeld U. Sequential maintenance therapy with cyclosporin A and mycophenolate mofetil for sustained remission of childhood steroid-resistant nephrotic syndrome. Nephrol Dial Transplant 2012; 27 (5): 1970–1978.
29. Magnasco A, Ravani P, Edefonti A, et al. Rituximab in children with resistant idiopathic nephrotic syndrome. J Am Soc Nephrol 2012; 23 (6): 1117–1124.
30. Kamei K, Ishikura K. Rituximab treatment for refractory steroid-resistant nephrotic syndrome. Pediatr Nephrol 2016; 31 (2): 337–338.
31. Basu B. Ofatamumab for rituximab-resistent nephrotic syndrome.N Engl J Med 2014; 370 (13): 1268–1270.
32. Franke D, Zimmering M, Wolfish N, et al. Treatment of FSGS with plasma exchange and immunadsorption. Pediatr Nephrol 2000; 14 (10–11): 965–969.
33. Kapur G. Treatment of severe edema in children with nephrotic syndrome with diuretics alone — A prospective study. CJASN 2009; 4 (5): 907–913.
34. Kerlin BA, Haworth K, Smoyer WE. Venous thromboembolism in pediatric nephrotic syndrome. Pediatr Nephrol 2014; 29 (6): 989–997.
35. Kestilä M, Lenkkeri U, Männikkö M, et al. Positionally cloned gene for a novel glomerular protein--nephrin--is mutated in congenital nephrotic syndrome. Mol Cell 1998; 1 (4): 575–582.
36. Boute N, Gribouval O, Roselli S, et al. NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat Genet 2000 Apr; 24 (4): 349–543.
37. Sadowski CE, Lovric S, Ashraf S, et al. A single-gene cause in 29.5 % of cases of steroid-resistant nephrotic syndrome. J Am Soc Nephrol 2015; 26 (6): 1279–1289.
38. Hinkes BG, Mucha B, Vlangos CN, et al. Nephrotic syndrome in the first year of life: two thirds of cases are caused by mutations in 4 genes (NPHS1, NPHS2, WT1, and LAMB2). Pediatrics 2007; 119 (4): e907–919.
39. Ruf RG, Lichtenberger A, Karle SM, et al. Patients with mutations in NPHS2 (podocin) do not respond to standard steroid treatment of nephrotic syndrome. J Am Soc Nephrol 2004; 15 (3): 722–732.
40. Gee HY, Ashraf S, Wan X, et al. Mutations in EMP2 cause childhood-onset nephrotic syndrome. Am J Hum Genet 2014; 94 (6): 884–890.
41. Patrakka J, Kestilä M, Wartiovaara J, et al. Congenital nephrotic syndrome (NPHS1): features resulting from different mutations in Finnish patients. Kidney Int 2000; 58 (3): 972–980.
42. Holmberg C, Jalanko H. Congenital nephrotic syndrome and recurrence of proteinuria after renal transplantation. Pediatr Nephrol 2014; 29 (12): 2309–2317.
43. Joshi S, Andersen R, Jespersen B, Rittig S. Genetics of steroid-resistant nephrotic syndrome: a review of mutation spectrum and suggested approach for genetic testing. Acta Paediatr 2013; 102 (9): 844–856.
44. Bouchireb K, Boyer O, Gribouval O, et al. NPHS2 mutations in steroid-resistant nephrotic syndrome: A mutation update and the associated phenotypic spectrum. Hum Mutat 2014; 35 (2): 178–186.
45. Ahn YH, Park EJ, Kang HG, et al. Genotype-phenotype analysis of pediatric patients with WT1 glomerulopathy. Pediatr Nephrol 2017; 32 (1): 81–89.
46. Chernin G, et al. Genotype/phenotype correlation in nephrotic syndrome caused by WT1 mutations. Clin J Am Soc Nephrol 2010; 5 (9): 1655–1662.
47. Gbadegesin R, Hinkes BG, Hoskins BE, et al. Mutations in PLCE1 are a major cause of isolated diffuse mesangial sclerosis (IDMS). Nephrol Dial Transplant 2008; 23 (4): 1291–1297.
48. Lovric S, Ashraf S, Tan W, Hildebrandt F. Genetic testing in steroid-resistant nephrotic syndrome: when and how? Nephrol Dial Transplant 2016; 31 (11): 1802–1813.
49. Trompeter RS, Lloyd BW, Hicks J, et al. Long-term outcome for children with minimal-change nephrotic syndrome. Lancet 1985; 1 (8425): 368–370.
50. Ishikura K, Yoshikawa N, Nakazato H, et al. Morbidity in children with frequently relapsing nephrosis: 10-year follow-up of a randomized controlled trial. Pediatr Nephrol 2015; 30 (3): 459–468.
51. Fakhouri F, Bocquet N, Taupin P, et al. Steroid-sensitive nephrotic syndrome: from childhood to adulthood. Am J Kidney Dis 2003; 41 (3): 550–557.
52. Abeyagunawardena AS, Sebire NJ, Risdon RA, et al. Predictors of long-term outcome of children with idiopathic focal segmental glomerulosclerosis. Pediatr Nephrol 2007; 22 (2): 215–221.
Labels
Neonatology Paediatrics General practitioner for children and adolescentsArticle was published in
Czech-Slovak Pediatrics

2017 Issue 2
- What Effect Can Be Expected from Limosilactobacillus reuteri in Mucositis and Peri-Implantitis?
- The Importance of Limosilactobacillus reuteri in Administration to Diabetics with Gingivitis
Most read in this issue
- Thrombotic microangiopathies – hemolytic-uremic syndromes and thrombotic thrombocytopenic purpura
- Nephrotic syndrome in childhood
- Guidelines for the diagnosis and treatment of urinary tract infections in children and adolescents. Recommendations of the Working Group Pediatric Nephrology of the Czech Pediatric Society
- Neurofibromatosis for 5-year-old child – a diagnosis based on rinolalia