
Turnerův syndrom a anomálie aortálního oblouku čtyřikrát jinak
Turner syndrome and anomalies of the aortic arch in four different ways
Turner syndrome (TS) is the most common chromosomal disorder in women, which is caused by absence or structural abnormality of X chromosome. The most common features of TS are short stature, gonadal dysgenesis and congenital heart and kidney diseases. Spectrum of congenital heart diseases is highly varied and most often the left side of the heart is affected. Bicuspid aortic valve and coarctation of the aorta are the most frequent of them. Authors present case reports of four patients with TS and the anomaly of the aortic arch but with clinically different manifestation and a brief overview of this topic.
Key words:
Turner syndrome, congenital heart diseases, coarctation of the aorta, elongated transverse aortic arch, aortic dis - section, hypertesion
Authors:
S. Kaprálová 1; J. Zapletalová 1; Z. Tudos 2; P. Hecht 3; J. Pavlíček 4; E. Klásková 1
Authors‘ workplace:
Dětská klinika Fakultní nemocnice Olomouc a Lékařské fakulty Univerzity Palackého, Olomouc
1; Radiologická klinika Fakultní nemocnice Olomouc a Lékařské fakulty Univerzity Palackého, Olomouc
2; Dětské kardiocentrum, FN Motol, Praha
3; Klinika dětského lékařství Fakultní nemocnice a Lékařské fakulty Ostravské univerzity, Ostrava
4
Published in:
Čes-slov Pediat 2018; 73 (5): 324-330.
Category:
Review
Overview
Turnerův syndrom (TS) je nejčastější chromosomální aberací u žen. Jeho příčinou je absence nebo strukturální abnormalita jednoho X chromosomu. Hlavními příznaky jsou malý vzrůst, gonadální dysgeneze a vrozené vývojové vady kardiovaskulárního a uropoetického systému.
Spektrum vrozených srdečních vad je velmi široké a nejčastěji postihuje levostranné srdeční oddíly. Mezi nejzastoupenější patří bikuspidální aortální chlopeň a koarktace aorty. Autoři předkládají kazuistiky čtyř pacientek s TS a anomálií aortálního oblouku, ale s klinicky rozdílnou manifestací a stručný přehled problematiky.
Klíčová slova:
Turnerův syndrom, vrozené srdeční vady, koarktace aorty, elongace transverzálního aortálního oblouku, aortální disekce, hypertenze
ÚVOD
Turnerův syndrom (TS) je svou četností 50/100 000 živě narozených dívek ročně jednou z nejčastěji se vyskytujících chromosomálních abnormalit [1, 2]. Příčinou TS je nejčastěji absence jednoho X chromosomu (45,X) nebo strukturální abnormalita. Numerická nebo strukturální odchylka může postihovat všechny buňky nebo jen jejich část ve formě chromosomální mozaiky.
TS je spojen se zvýšením předčasné kardiovaskulární morbidity i mortality na podkladě vrozených srdečních vad (VSV) a získaných kardiovaskulárních onemocnění. Kromě VSV a klasických fenotypických projevů (malá postava, nízká vlasová hranice, široký hrudník s oddálenými prsními bradavkami, kostní deformity, absence spontánní puberty – obr. 1) je pro TS typický vysoký výskyt vrozených vad uropoetického systému a také signifikantně vyšší četnost autoimunitních onemocnění (tyreoiditida, celiakie, diabetes mellitus 1. typu nebo idiopatické střevní záněty).
Fig. 1. The most common clinical features of Turner syndrome
(authors archive).

VSV má 22‒70 % nositelek TS, jejich klinická manifestace je ale velmi variabilní, od asymptomatických až po život ohrožující, a to již v novorozeneckém věku [3]. Nejčastěji zastoupenou vadou je bikuspidální aortální chlopeň (BAV), která je diagnostikována u 15‒30 % pacientek s TS v porovnání s její frekvencí v běžné populací (1‒2 %) [3].
Druhou nejčastější VSV se signifikantně vyšším zastoupením je koarktace aorty (CoA), vyskytující se u 17 % pacientek s TS v porovnání s obecným výskytem 0,04 % [3]. CoA ale není jedinou anomálií aortálního oblouku, až polovina pacientek s TS má elongaci transverzálního aortálního oblouku (ETA – elongated transverse arch of the aorta) se sifonovitým prohnutím oblouku v místě aortálního isthmu (tzv. kinking). Další anomálií aortálního oblouku může být jeho hypoplazie, která se často vyskytuje v kombinaci s CoA [3].
Ke kritickým srdečním vadám u TS řadíme syndrom hypoplastického levého srdce nebo kritickou aortální valvární stenózu [3]. Do spektra vrozených srdečních vad s klinicky příznivějším průběhem patří defekt septa síní a komor, parciální anomální návrat plicních žil, perzistující levostranná horní dutá žíla nebo pulmonální valvární stenóza [3].
KLINICKÉ PŘÍPADY
Popisujeme čtyři případy pacientek s TS a anomáliemi aortálního oblouku odlišující se klinickým průběhem. Cílem je poukázat na výraznou variabilitu klinické manifestace uvedených anomálií u nositelek TS.
1. Kritická koarktace aorty v novorozeneckém věku
Dívka z druhé fyziologické gravidity, porozena spontánně v termínu s fyziologickou časnou poporodní adaptací. Pro nález kožních duplikatur podél krku (tzv. pterygia colli) a lymfedému dolních končetin bylo po porodu vysloveno podezření na TS. V rámci pátrání po vrozených srdečních vadách byla provedena echokardiografie (ECHO) s nálezem CoA, tlakový gradient mezi horními a dolními končetinami činil 35 mmHg a s diagnózou korelovaly značně oslabené pulzace femorálních arterií, poslechově byl přítomen systolický šelest v intenzitě 2/6. Pacientka byla zajištěna infuzí prostaglandinu k udržení průchodnosti Botallovy dučeje a desátý den života byla odeslána na vyšší pracoviště.
Diagnóza těsné juxtaduktální CoA s hypoplastickým aortálním obloukem byla potvrzena, koarktační lišta byla lokalizována těsně před odstup levé a. subclavia. Funkce levé komory nebyla globálně postižena, ale kontraktilita zadní stěny levé komory byla snížená. Morfologie aortální chlopně byla zhodnocena jako dvojcípá. Pacientka podstoupila resekci CoA s rozšířenou anastomózou end-to-side a podvaz s resekcí levostranné a. subclavia. Pooperační průběh byl komplikován přechodnou iontovou dysbalancí. Genetickým vyšetřením byl ve věku jednoho měsíce potvrzen TS (45,X).
Pacientka je i nadále pravidelně sledována dětským kardiologem. Dosud (t. j. 13 let od operace) nebyly při echokardiografickém vyšetření prokázány známky rekoarktace. V rámci kardiologického sledování byla doplněna magnetické rezonance (MRI) srdce a velkých cév, která echokardiografické nálezy potvrdila včetně nálezu BAV. Provedena byla také 24hodinová monitorace krevního tlaku (ABPM), a to bez záchytu hypertenze.
V pravidelných intervalech je pacientka sledována i endokrinologem, byla léčena růstovým hormonem s dobrým efektem, žádné z asociovaných autoimunitních onemocnění s TS se u naší pacientky dosud neprojevilo. Ultrazvukové vyšetření vyloučilo i vrozenou vývojovou vadu urotraktu.
2. Asymptomatická koarktace aorty v dospělosti
Pacientka s TS (45,X). Diagnóza byla stanovena v šesti letech věku pro stigmatizaci (malý vzrůst, štítový hrudník). Byla v pravidelné péči endokrinologa včetně terapie růstovým hormonem, nebylo u ní prokázáno sdružené autoimunitní onemocnění ani vrozená vývojová vada ledvin.
Podrobné kardiologické vyšetření bylo provedeno až před plánovanou graviditou ve věku 31 let. Součástí vyšetření byla i MRI srdce a velkých cév, kde byla popsána hemodynamicky středně významná CoA (obr. 2), dále BAV a také dilatace ascendentní aorty s aortic size index (ASI) 2,4 cm/m2, který odpovídá téměř 99. percentilu. Klinický nález u pacientky byl nenápadný díky rozsáhlému kolaterálnímu oběhu, nebyl patrný žádný šelest, pulzace femorálních tepen byly dobře hmatné a tlakový rozdíl mezi pravou horní a dolní končetinou nepřekročil 20 mmHg. ABPM vyšetření nezachytilo hypertenzi.
Fig. 2. Magnetic resonance imaging in native coarctation of
the aorta (authors archive).
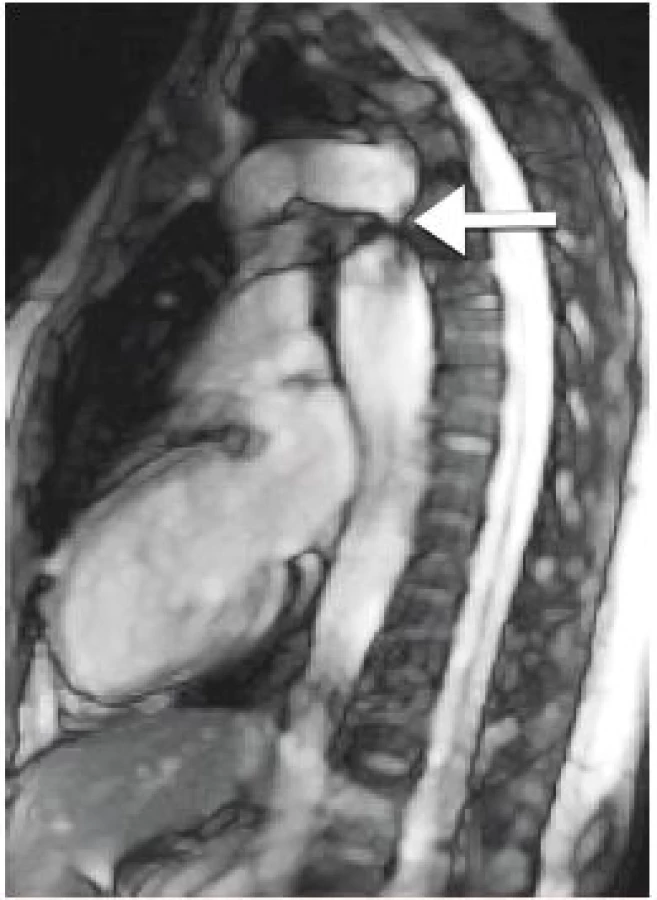
Pacientka byla odeslána ke konzultaci na vyšší pracoviště, kde bylo doplněno zobrazení hrudní aorty CT angiografií se shodnými nálezy jako při MRI. Byla zahájena léčba beta-blokátory vzhledem k dilataci ascendentní aorty, intervence na aortálním oblouku nebyla doporučena z důvodu CoA ani z důvodu dilatace ascendentní aorty. Případná gravidita byla vzhledem k vysokému riziku aortální disekce zhodnocena jako velmi riziková a nebyla doporučena.
3. Hypoplastický aortální isthmus s vývojem do méně významné koarktace aorty u kojence
Ve třetím případě se jedná o pacientku s diagnózou TS stanovenou již prenatálně ve formě mozaiky se zastoupením 45,X v 56 % amniocytů, fetální echokardiografie provedena nebyla. Jednalo se o první graviditu a předčasný porod (33+5 týden gestace) hypotrofického novorozence císařským řezem. Poporodní adaptace byla komplikována těžkou hypotonií a bradykardií pod 60/min s postupnou úpravou stavu. Postnatálně byla diagnóza TS ve formě mozaiky potvrzena cytogeneticky z lymfocytů periferní krve se zastoupením 45,X v 7,4 % buněk. Metodou fluorescenční in situ hybridizace (FISH) byla monozomie prokázána ve 13 % lymfocytů ze vzorku periferní krve a v buňkách bukální sliznice ve 35 %.
ECHO vyšetření v prvních dnech života prokázalo užší aortální isthmus s gradientem v descendentní aortě 27 mmHg ale s fyziologickým pulzatilním tokem v břišní aortě.
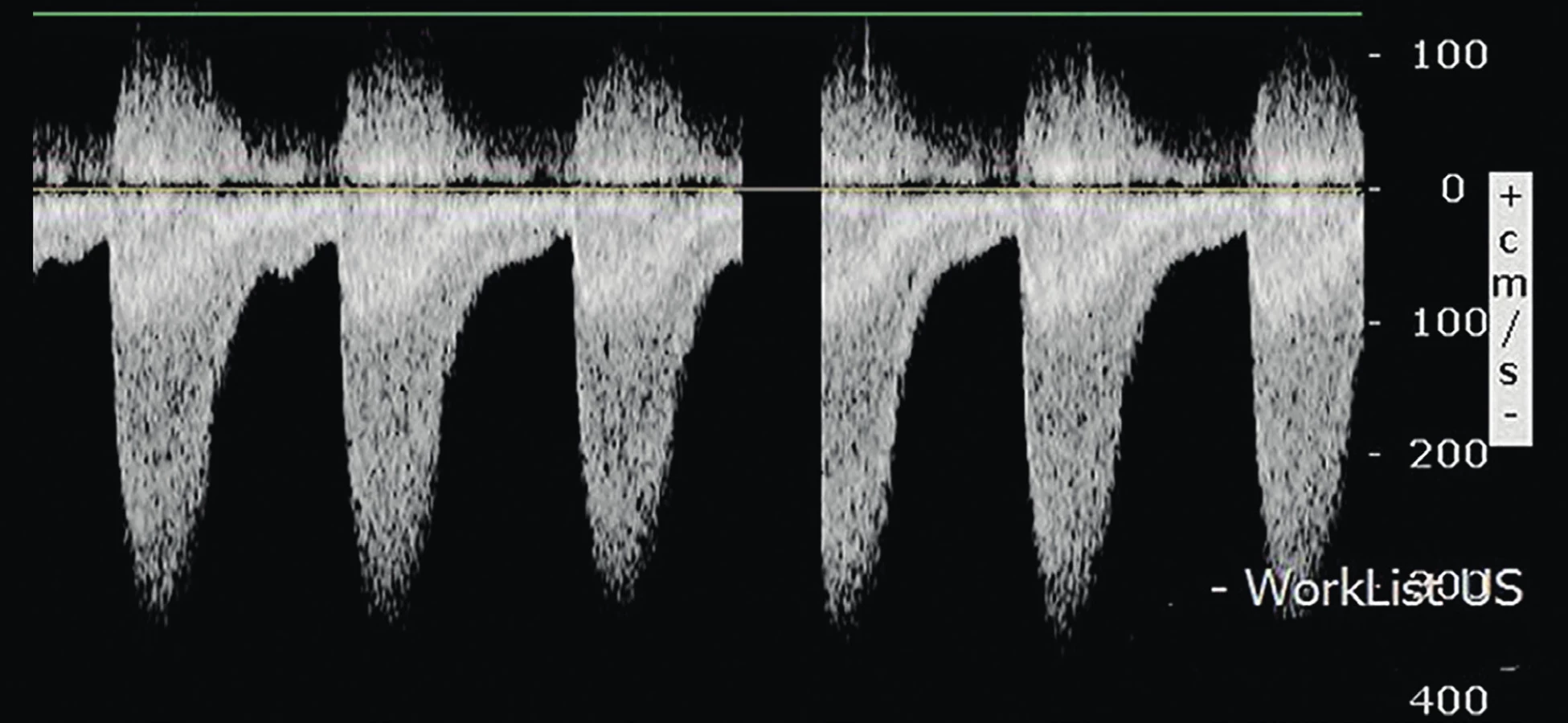
Pacientka byla pravidelně echokardiograficky sledována a devátý týden života byla v oblasti isthmu detekována typická koarktační lišta (1,9 mm) s turbulentním tokem při barevném dopplerovském zobrazení, s gradientem v descendentní aortě 45 mmHg a typickým pilovitým tokem s naznačeným "diastolickým tailem" (= tok protažený do diastoly) při pulzním dopplerovském měření (obr. 3 a 4). Zaznamenána byla i mírná deprese funkce levé komory (ejekční frakce levé komory 51 %). Klinicky s echokardiografickým nálezem korelovaly oslabené pulzace femorálních arterií, poslechově systolický šelest o intenzitě 2/6. Pacientka byla odeslána ke kontrole na vyšší pracoviště se závěrem méně hemodynamicky významné CoA a byl doporučen konzervativní postup. Pacientka je i nadále kardiologicky sledována, bez nutnosti intervence, vzhledem k postupnému růstu aortálního oblouku v oblasti isthmu.
Fig. 3. Turbulent flow at the site of coarctation in color-flow
mapping (authors archive).
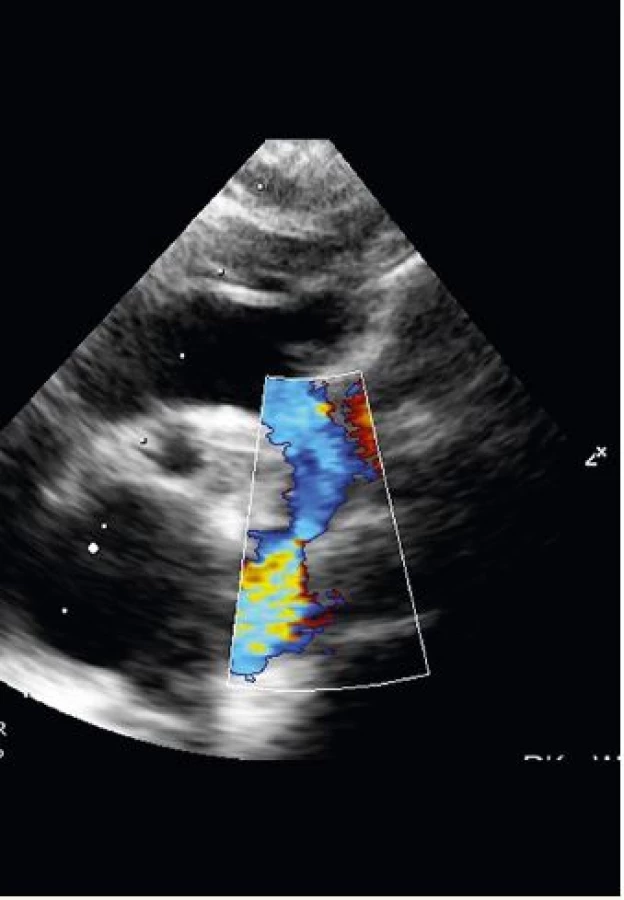
Fig. 4. Typical sawtooth pattern with continuous flow wave Doppler interrogation in coarctation of the aorta with „diastolic
tail“(authors archive).
Pacientka je vedena i v endokrinologické poradně, dosud bez zátěže sdruženým autoimunitním onemocněním. Vyloučeny byly i vrozené vývojové vady urotraktu.
4. Elongace transverzálního aortálního oblouku se sifonovitým prohnutím oblouku v místě aortálního isthmu
Ve čtvrtém případě uvádíme pacientku s anomálii aortálního oblouku, která se vyskytuje až u 50 % pa-cientek s TS. Jedná se o dívku z první rizikové gravidity, pro podezření na vrozenou vývojovou vadu centrálního nervového systému a srdce byla provedena ve 30. týdnu gestace amniocentéza s nálezem mozaiky trizomie chromosomu 8. Porod proběhl v termínu spontánně s nekomplikovanou poporodní adaptací. Postnatálním genetickým vyšetřením byla zjištěna mozaika trizomie chromosomu 8 s monozomií 45,X.
ECHO vyšetření u této dívky bylo značně limitováno nepříznivým akustickým oknem způsobeným závažnou skoliózou a deformitou hrudníku, proto byla časně, již v necelých deseti letech života, doplněna MRI srdce a velkých cév s nálezem elongace transverzálního aortálního oblouku a tzv. kinkingem (sifonovité prohnutí oblouku v místě aortálního isthmu), aortální chlopeň byla popsána jako dvojcípá (obr. 5). ABPM vyšetření u pacientky prokázalo diastolickou hypertenzi, proto byla zahájena léčba antihypertenzivy.
Fig. 5. MRI reconstruction image demonstrating elongated
transverse aortic arch (authors archive).

Pacientka má na rozdíl od předchozích nositelek TS řadu komorbidit, patří mezi ně závažná skolióza páteře, mentální retardace, alergie na bílkovinu kravského mléka. Současně má jedno z nejčastěji se vyskytujících autoimunitních onemocnění u TS, a to celiakii. Léčba růstovým hormonem nebyla u této pacientky z důvodu závažné progredující skoliózy indikována.
DISKUSE
Koarktací aorty je v užším slova smyslu nazývána lokální lišta vyklenující se ze zadní a laterální stěny aorty proti ústí tepenné dučeje. Podle vztahu k vyústění dučeje můžeme rozlišovat typ koarktace na preduktální, juxtaduktální a postduktální [4].
V běžné populaci je výskyt CoA více vázán k mužskému pohlaví (2‒5 : 1) a patří mezi jednu z nejčastěji se vyskytujících VSV s incidencí 36 na 100 000 živě narozených ročně [4]. Výskyt CoA je u pacientek s TS asi 400násobně vyšší oproti její frekvenci v běžné populaci [5]. Asociace mezi TS a CoA je obecně velmi dobře známá, recentně byly publikovány práce, které hodnotily vztah reciproční, a to prevalenci TS u dívek s CoA. V jedné ze studií byla prevalence stanovena na 5,3 % a v druhé studii na 12,6 % [6, 7]. Tyto výsledky vyvolaly diskusi o tom, zda by nemělo být provedeno genetické vyšetření u všech pacientek s CoA s cílem vyloučit TS jako její příčinu. Autoři výše zmíněných prací považují tento postup za opodstatněný vzhledem k tomu, že po TS pátráme i v případech malého vzrůstu, kde je prevalence TS jako příčiny růstové retardace pouze 2‒4 % oproti téměř 13 % v případě CoA [8, 9].
Příčina vzniku VSV včetně CoA u TS stále není plně objasněna, Clarkova teorie zvažuje jako příčinu rozšíření lymfatických cév omezující krevní průtok ve vyvíjejícím se srdci plodu [10]. Předpokládaný lokus pro tzv. lymfogenní gen, jehož haploinsuficience by mohla být zodpovědná za abnormální vývoj lymfatického systému, je v pseudoautozomální oblasti 1 (PAR1) na krátkém raménku X chromosomu – Xp11.4 [11]. Publikovány jsou ale také práce na podkladě epidemiologických studií a rozborů rodokmenů prokazující, že VSV se vyskytují i u těch pacientek s TS, u kterých nejsou patrné stigmatizace způsobené městnáním lymfy (pterygia colli, štítovitý široký hrudník, nízká vlasová hranice apod.). Autoři těchto prací se přiklání k teorii, že VSV nejsou vázány na abnormální vývoj lymfatického systému a jsou nezávislým fenotypickým projevem TS [12, 13].
Zmíněnou problematikou se zabývá také práce autorky Bondy z roku 2013, která analyzovala výskyt BAV a CoA u 185 pacientek s TS s karyotypy 45,X, 46,X,del(Xp), 46,X,del(Xq) a 46,X,i(Xq) [14]. Výskyt CoA a BAV byl nejvyšší u pacientek s monozomií 45,X, v případě CoA 12,5 % a u BAV 34 %. U pacientek s delecí krátkého raménka X chromosomu byla CoA zjištěna u 6,7 % a BAV v necelých 29 % případů. Ani jedna z VSV nebyla nalezena u pacientek s karyotypem 46,X,del(Xq). Autoři na základě těchto výsledků předpokládají, že haploinsuficience genů na krátkém raménku X chromosomu je za vznik VSV postihujících aortální chlopeň a oblouk aorty zodpovědná. Ani u jedné pacientky s BAV a delecí krátkého raménka X chromosomu ale nebyly popsány změny odpovídající lymfedémům, proto se lze přiklonit k teorii, že VSV a lymfedémy jsou na sobě dva nezávislé fenotypické projevy TS [14].
Klinická manifestace CoA je variabilní, závislá na hemodynamické významnosti vady. Kritická koarktace v novorozeneckém věku se projeví po uzávěru tepenné dučeje těžkým oběhovým selháním s hypoxémií a metabolickou acidózou. U části pacientů se projeví koarktace až v kojeneckém věku neprospíváním, tachypnoií a k srdečnímu selhání dochází postupně. U dětí a dospívajících s hemodynamicky středně a méně významnou koarktací patří mezi klinické projevy bolesti hlavy při sekundární hypertenzi, slabosti a klaudikace dolních končetin. V souboru našich klinických případů je právě tato variabilita dokumentována. I pacientky se stejným karyotypem, 45,X, se mohou v klinické manifestaci lišit. U pacientky z prvního případu se jednalo o CoA, která by po úplném uzávěru dučeje vedla ke zhroucení cirkulace. U druhé dívky byla diagnóza CoA náhodným nálezem při kardiologickém vyšetření, které by mělo být provedeno u všech dívek s TS, zejména pak před plánovanou graviditou. Anomálie aortálního oblouku u nositelek TS nejsou zákonitě svými klinickými projevy homogenní vrozenou srdeční vadou.
Diagnostický algoritmus začíná u fyzikálního vyšetření, při kterém jsou zásadními známkami CoA oslabené nebo zcela nehmatné pulzace femorálních tepen v případě, že je uzavřena tepenná dučej. Auskultační nález je u kritické CoA necharakteristický, u starších dětí je šelest slabší až střední intenzity slyšet nejlépe v mezilopatkovém prostoru nebo u levého horního okraje sterna. Součástí fyzikálního vyšetření je i měření krevního tlaku na horních a dolních končetinách, kde gradient více než 20 mmHg je považován za známku koarktace [4].
Základní zobrazovací modalitou je ECHO. V břišní aortě při dopplerovském vyšetření můžeme zaznamenat patologicky nízkou pulzatilitu. Zásadní je ale zobrazení celého aortálního oblouku včetně isthmu (ve 2D zobrazení bývá typicky patrná koarktační lišta). Pilovitý tok je charakteristickým znakem CoA při použití kontinuálního dopplerovského vyšetření [4]. Při ECHO vyšetření nesmí být opomenuto i zhodnocení funkce levé komory. Jde o neinvazivní a rychlé vyšetření, ale ne vždy je akustické okno optimální, zejména pak u pacientek s TS (pozůstatky fetálního lymfedému). V těchto případech je s výhodou využívána MRI srdce a velkých cév, která rovněž umožňuje zhodnocení morfologie aortální chlopně a stanovení rozměrů ascendentní aorty. Začleněním MRI srdce a velkých cév do diagnostiky u pacientek s TS se významně zvýšil záchyt BAV [15].
K dalším pomocným vyšetřením patří EKG záznam, kde mohou být patrné známky hypertrofie levé komory v případě významné koarktace v pozdějším věku. Na rentgenovém snímku pacienta s kritickou koarktací bude patrná bohatší plicní kresba a kardiomegalie. U dětí starších s méně významnou vadou může být patrná deformita tvaru trojky tvořená prestenotickou a poststenotickou dilatací aorty spolu se zářezem na levé kontuře stínu aorty v místě koarktace. A známým rentgenovým korelátem u starších pacientů s koarktací jsou usurace dolních okrajů žeber způsobené rozšířením interkostálních arterií v rámci kolaterálního oběhu [4].
Typ intervence se v případě CoA odvíjí od řady proměnných, jednou z nich je její významnost. Kritická koarktace aorty je řešena chirurgickou resekcí s end-to-end anastomózou urgentně, stejný operační postup je volen i u méně významných koarktací elektivně. V případě nepříznivých anatomických poměrů v oblasti aortálního oblouku může být proveden extra-anatomický bypass (cévní protéza mezi ascendentní a descendentní aortou) [4]. Diskutovanou a na některých pracovištích využívanou metodou je intervence katetrizační v podobě balónkové angioplastiky nebo implantace stentu. Obecným rizikem těchto metod je rekoarktace a vznik aneuryzmat. Tato rizika jsou u pacientek s TS zvýšená vzhledem k difuzní aortopatii [16]. Jedna z pracovních skupin publikovala výsledky po implantaci stentu u šesti pacientek s nativní CoA a u čtyř pacientek po předchozí balónkové angioplastice. Efekt intervence byl srovnatelný s chirurgickou metodou a během 30měsíčního sledování nedošlo ani u jedné z pacientek k rekoarktaci, pouze u dvou pacientek vzniklo aneuryzma [17].
CoA je příčinou zvýšené kardiovaskulární morbidity a mortality u pacientek s TS. Zároveň patří mezi rizikové faktory pro další kardiovaskulární rizika TS, jako je aortální disekce, která pacientky s TS ohrožuje až šestinásobně oproti běžné populaci, a to již v daleko mladším věku [18]. CoA byla popsána u 89 % pacientek s disekcí aorty [19]. Vyšším rizikem aortální disekce jsou ohroženy také pacientky s BAV, hypertenzí a dilatací ascendentní aorty [3]. Vztah rizikových faktorů pro často fatální aortální disekci je komplexní, sama CoA může vést k sekundárnímu zvýšení krevního tlaku a tak dál potencovat riziko. I jiné abnormality aortálního oblouku, jako je jeho elongace se sifonovitým prohnutím v místě isthmu, mohou vést k hypertenzi zřejmě na podkladě narušení funkce baroreceptorů nebo narušení kontinuity krevního toku [20]. I naše pacientka z posledního klinického případu s diagnózou ETA a kinkingu má dokumentovanou diastolickou hypertenzi při ABPM vyšetření.
Pacientky s TS mohou s vývojem možností reprodukční medicíny otěhotnět pomocí dárcovských oocytů. Riziko aortální disekce ale v graviditě významně narůstá, podle doporučení The Practice Committee of the American Society for Reproductive Medicine z roku 2012 je těhotenství v případě signifikantních abnormalitit (CoA, BAV, dilatace ascendentní aorty – ASI nad 2 cm/m2) kontraindikováno [21]. V případě doporučení Francouzské gynekologické společnosti je nález BAV považován pouze za relativní kontraindikaci ke graviditě [22]. Součástí komplexní péče o pacientky s TS je i na základě recentních doporučení vydaných v roce 2017 pravidelné kardiologické sledování, a to nejen v graviditě [23].
ZÁVĚR
VSV se u pacientek s TS podílejí 8 % na jejich celkové mortalitě [24]. CoA je jednou z nejčastějších. Naše čtyři případy pacientek s TS a anomálií aortálního oblouku dokumentují v literatuře popsanou variabilní klinickou manifestaci, ta sahá od kritické CoA v novorozeneckém věku manifestující se srdečním selháním s kardiogenním šokem, až po CoA projevující se v pozdějším dětství nebo v dospělosti hypertenzí či klaudikacemi končetin. U 5‒8 % pacientek s TS zůstává diagnóza CoA nebo jiné anomálie aortálního oblouku neodhalena z důvodu minimálních klinických projevů [3].
Echokardiografie zůstává prozatím první volbou v diagnostickém procesu, ale nezřídka je limitována neoptimálním akustickým oknem. MRI srdce a velkých cév má v těchto případech nezastupitelnou roli, zejména pokud jde o stanovení morfologie aortální chlopně a zhodnocení rozměrů ascendentní aorty.
Typ intervence v případě CoA je volen individuálně podle hemodynamické významnosti vady, přidružených VSV a zkušenostech pracoviště. V případě nativní CoA zůstává první volbou chirurgický výkon. Katetrizační intervence jsou prozatím využívány spíše v případě rekoarktace.
Tato práce byla podpořena grantem MZ VES 2017 (Reg. No. NV17-29111A).
Došlo: 13. 2. 2018
Přijato: 12. 6. 2018
MUDr. Sabina Kaprálová
Dětská klinika FN Olomouc
a Lékařské fakulty Univerzity Palackého
I. P. Pavlova 6
779 00 Olomouc
e-mail: sabina_s@email.cz
Sources
1. Elsheikh M, Conway GS, Wass JAHDDB. Turner’s syndrome in adults. Endocr Rev 2002; 23 : 120–140.
2. Stochholm K, Juul S, Juel K, et al. Prevalence, incidence, diagnostic delay, and mortality in Turner syndrome. J Clin Endocrinol Metab 2006; 91 : 3897–3902.
3. Mortensen KH, Andersen NH, Gravholt CH. Cardiovascular phenotype in Turner syndrome – integrating cardiology, genetics, and endocrinology. Endocr Rev 2012; 33 : 677–714.
4. Chaloupecky V, et al. Dětská kardiologie. 1. vyd. Praha: Galén, 2006 : 1–437.
5. Grech V. Diagnostic and surgical trends, and epidemiology of coarctation of the aorta in a population-based study. Int J Cardiol 1999; 68 : 197–202.
6. Wong SC, Burgess T, Cheung M, et al. The prevalence of Turner syndrome in girls presenting with coarctation of the aorta. J Pediatr 2014; 164 : 259–263.
7. Eckhauser A, South ST, Meyers L, et al. Turner syndrome in girls presenting with coarctation of the aorta. J Pediatr 2015; 167 : 1062–1066.
8. Cicquel C, Gaston V, Cabrol S, et al. Assesment of Turner’s syndrome by molecular analysis of the X chromosome in growth-retarded girls. J Clin Endocrinol Metab 1998; 83 : 1472–1476.
9. Grote FK, Oostdijk W, De Muinck Keizer-Schrama SM, et al. The diagnostic work up of growth failure in secondary health car, an evalution of consensus guidelines. BMC Pediatr 2008; 8 : 21.
10. Clark EB. Neck web and congenital heart defects: a pathogenic association in 45 X-0 Turner syndrome? Teratology 1984; 29 : 355–361.
11. Boucher CA, Sargnet CA, Ogata T, et al. Breakpoint analysis of Turner patients with partial Xp deletions: implications for the lymphoedema gene location. J Med Genet 2001; 38 : 591–598.
12. McBride K, Pignatelli R, Lewin M, et al. Inheritance analysis of congenital left ventricular outflow tract obstruction malformations: segrefation, multiplex relative risk, and heritability. Am J Med Genet Part A 2005; 134A: 180–186.
13. Loffredo CA, Chokkalingam A, Sill AM, et al. Prevalence of congenital cardiovascular malformations among relatives of infants with hypoplastic left heart, coractation of the aorta, and d-transposition of the great arteries. Am J Med Genet A 2004; 124A: 225–230.
14. Bondy C, Bakalov VK, Cheng C, et al. Bicuspid aortic valve and aortic coarctation are linked to deletion of the X chromosome short arm in Turner syndrome. J Med Genet 2013; 50 : 662–665.
15. Klásková E, Tüdös Z, Wiedermann J, et al. Postižení kardiovaskulárního systému u Turnerova syndromu. Čes-slov Pediat 2012; 67 : 103–111.
16. Lin AE, Lippe BM, Geffner ME, et al. Aortic dilatation, dissection, and rupture in patients with Turner syndrome. J Pediatr 1986; 109 : 820–826.
17. Zanjani KS, Thanopoulos BD, Peirone A, et al. Usefulness of stenting in aortic coarctation in patients with the Turner syndrome. Am J Cardiol 2010; 106 : 1327–1331.
18. Gravholt CH, Landin-Wilhelmsen K, Stoccholm K, et al. Clinical and epidemiological description of aortic dissection in Turner’s syndrome. Cardiol Young 2006; 16 : 430–436.
19. Sybert VP. Cardiovascular malformations and complications in Turner sydrome. Pediatrics 1998; 101 : 11–17.
20. De Groote K, Devos D, Van Herck K, et al. Abnormal aortic arch morphology in Turner syndrome patients is a risk factor for hypertension. Heart Vessels 2015; 30 : 618–625.
21. The practice Committee of American Society For Reproductive Medicine. Increased maternal cardiovascular mortality associated with pregnancy in women with Turner syndrome. Fertil Steril 2012; 97 : 282–284.
22. Cabanes L, Chalas C, Christin-Maitre S, et al. Turner syndrome and pregnancy: clinical practice. Recommendations for the management of patients with Turner syndrome before and during pregnancy. Eur J Obstet Gynecol Reprod Biol 2010; 152 : 18–24.
23. Gravholt CH, Andersen NH, Conway GS, et al. Clinical practice guidelines for the care of girls and women with Turner syndrome: proceedings from the 2016 Cincinnati International Turner Syndrome Meeting. Eur J Endocrinol 2017; 177: G1–G70.
24. Schoemaker NJ, Swerdlow AJ, Higgins CD, et al. Mortality in women with Turner syndrome in Great Britain: a national cohort study. J Clin Endocrinol Metab 2008; 93 : 4735–4742.
Labels
Neonatology Paediatrics General practitioner for children and adolescentsArticle was published in
Czech-Slovak Pediatrics

2018 Issue 5
- What Effect Can Be Expected from Limosilactobacillus reuteri in Mucositis and Peri-Implantitis?
- The Importance of Limosilactobacillus reuteri in Administration to Diabetics with Gingivitis
Most read in this issue
- Mentální anorexie s raným začátkem, diagnostika a terapie
- Výskyt vrozených srdečních vad – dopad prenatální diagnostiky
- Prenatální detekce srdečních vad a její důsledky
- Lokálne reakcie po uhryznutí hadom – klinické skúsenosti