
Výskyt vrozených srdečních vad – dopad prenatální diagnostiky
The incidence of congenital heart defects – the impact of prenatal diagnosis
Objective:
To study of the occurrence of congenital heart defects (CHDs) and the effectiveness of their prenatal detection. To study the frequency of extracardiac anomalies. To determine the possibility of perinatal care in pathological newborns.
Methods:
The retrospective cohort study between 1999–2016. The observed region was the Moravian-Silesian region. The investigation of all significant heart defects, ultrasound examination of the fetal heart (fetal echocardiography) in the second trimester of pregnancy and postnatal standardized examination by a pediatric cardiologist, presence of pediatric cardiologist at all autopsies with a precise description of the defect, birth of a pathological newborn at a specialized center, genetic testing.
Results:
During the monitored 18years period, a total of 784 (3.7 cases per 1,000 fetuses) of significant CHDs were observed in the total population of 209,300 fetuses. There were 52% (406/784) CHDs detected prenatally and 48% (378/784) of cases were not prenatally recognized. The effectiveness of CHDs screening has improved progressively and the success rate of prenatal detection was 74% in the last three years. In the group of prenatally diagnosed significant CHDs, 53 % (215/406) of families decided for termination of gravidity. In most cases, the CHDs was diagnosed as an isolated anomaly, in 78 % (609/784) of fetuses. In 16% (125/784) of cases, concomitant genetic defect was observed, 6 % (50/784) of cases suffered from a different extra cardiac morphological pathology, without a genetic anomaly. Among the patients with genetic impairment, 70% (88/125) of cases were associated with a prenatal diagnostics, in case of extracardiac malformations, this number reached 86 % (43/50).
Conclusions:
CHDs were mostly manifested as an isolated anomaly. Concomitant genetic and other extracardiac pathology were diagnosed in one third of cases and these cases were significantly associated with a prenatal diagnosis. Prenatal care allows a more detailed examination of the pregnancy with a proper counseling for the affected family and the examination of pathological pregnancy and planning of delivery and postnatal interventions for the affected newborn. In our region, more than half of the families with a prenatally diagnosed CHD in the fetus decide to terminate the pregnancy and the incidence of significant CHDs is reduced in the newborn population.
Key words:
congenital heart defect, screening, fetal echocardiography, genetic abnormality, extracardiac malformation
Authors:
J. Pavlíček 1; E. Klásková 2; S. Kaprálová 2; T. Zaoral 3; B. Trávníček 3; E. Šilhánová 4; R. Kaniová 4; S. Polanská 1; A. Mužná 1; T. Gruszka 1
Authors‘ workplace:
Oddělení dětské a prenatální kardiologie, Klinika dětského lékařství, FN Ostrava
1; Dětská klinika FN a LF UP, Olomouc
2; Oddělení pediatrické a resuscitační péče, Klinika dětského lékařství, FN Ostrava
3; Oddělení lékařské genetiky, FN Ostrava
4
Published in:
Čes-slov Pediat 2018; 73 (5): 304-312.
Category:
Original Papers
Overview
Cíl práce:
Studium výskytu významných srdečních vad (VSV) a efektivity jejich prenatální diagnostiky, popis četnosti přidružených anomálií a stanovení možností perinatální péče u patologických novorozenců.
Metodika:
Retrospektivní studie 1999–2016. Sledování výskytu významných VSV v populaci Moravskoslezského kraje. V prenatální i postnatální péči došetření všech významných kardiálních patologií. Prenatálně ultrazvukové vyšetření srdce plodu – fetální echokardiografie provedené většinou ve II. trimestru gravidity, postnatálně standardizované komplexní vyšetření dětským kardiologem. Při umělém ukončení gravidity u autopsie vždy přítomnost dětského kardiologa s přesným popisem vady. Porod patologického novorozence na specializovaném pracovišti. Genetické došetření.
Výsledky:
Ve sledovaném 18letém období se v populaci 209 300 plodů vyskytlo celkem 784 (3,7 na 1000 plo-dů) významných VSV. V průměru bylo prenatálně detekováno 52 % (406/784) VSV, prenatálně nepoznáno 48 % (378/784). Efektivita prenatální detekce stoupala, v posledních třech letech bylo před porodem zachyceno 74 % významných VSV. U prenatálně detekovaných VSV se 53 % (215/406) rodin rozhodlo pro ukončení gravidity. Srdeční vada se vyskytla převážně jako izolované postižení v 78 % (609/784), ve spojení s genetickým postižením v 16 % (125/784) a s extrakardiální malformací bez genetické patologie v 6 % (50/784). Ze 125 chromosomálních anomálií bylo 70 % (88/125) spjato s prenatální diagnózou a 30 % (37/125) bylo zjištěno až postnatálně. U extrakardiálních patologií bylo ve spojení s VSV prenatálně 86 % (43/50) a postnatálně 14 % (7/50) případů.
Závěr:
Srdeční vada se většinou vyskytuje jako izolované postižení. Genetické a jiné extrakardiální anomálie se spojují přibližně s jednou třetinou kardiálních diagnóz a jsou významně svázány s prenatální diagnostikou. Prenatální péče umožňuje došetření patologické gravidity, správné poradenství postižené rodině a plánování porodu novorozence se srdeční vadou. Polovina rodin graviditu s fetálně poznanou VSV ukončí a je tak redukován výskyt VSV v populaci novorozenců.
Klíčová slova:
vrozená srdeční vada, screening, fetální echokardiografie, genetické abnormity, extrakardiální patologie
ÚVOD
Vrozené srdeční vady (VSV) jsou nejčastější kongenitální defekty v lidské populaci [1, 2] a jejich podíl na všech vrozených malformacích je až 40% [3]. Jejich výskyt je udáván v širokých rozmezích 4–50/1000 živě narozených, s obvyklými průměry 6/1000 až 18/1000 [4, 5]. Etiologie VSV je komplexní, roli hrají genetické i environmentální faktory [6]. Vztah genetických abnormalit a vznik VSV je částečně dobře znám, asi 20 % VSV je spojeno s přesnou genetickou příčinou [7], existuje řada studií a doporučení týkající se vztahu genetických patologií k VSV [8], jsou studovány nové geny, bodové mutace, možný vliv kryptických chromosomálních abnormalit [9, 10, 11]. Většina významných a kritických VSV je detekovatelná prenatálně a ultrazvukové vyšetření srdce plodu (fetální echokardiografie) umožňuje diagnostiku VSV před porodem [12]. S rostoucí kvalitou screeningu stoupá počet fetálně detekovaných VSV. Při prenatálně diagnostikované vadě část partnerů neakceptuje pokračování gravidity a těhotenství ukončí [13]. Cílem prenatální diagnostiky je celkové došetření patologické gravidity, správné a pečlivé poradenství a plánování porodu nemocného novorozence a jeho zajištění na adekvátním pracovišti.
Cílem práce je studium výskytu významných srdečních vad, efektivity jejich prenatální diagnostiky, popis četnosti přidružených anomálií a stanovení možností perinatální péče u patologických novorozenců.
SOUBOR DAT A METODIKA
Sledovanou oblastí byl Moravskoslezský kraj (1 200 000 obyvatel, průměrně 11 500 porodů ročně). Data byla hodnocena v retrospektivní 18leté studii v letech 1999–2016. Hodnocen byl výskyt VSV v populaci 209 300 plodů (živěrození, mrtvorození, ukončené gravidity pro VSV).
Autoři sledovali výskyt významných VSV. Významná srdeční vada byla ve studii definována jako patologie, která hemodynamicky nebo klinicky významně ovlivňuje zdraví novorozence nebo kojence a většinou je operována nebo katetrizačně řešena do roku věku dítěte. V přehledu byly ponechány srdeční tumory, které nejsou zahrnuty mezi VSV, ale představují pro dítě významná neurologická rizika pro rozvoj tuberózní sklerózy a dále izolovaná dextrokardie, kterou není bez přidružených VSV potřeba kardiochirurgicky řešit, ale může být svázaná s dalšími orgánovými patologiemi. Prenatálně je fetální echokardiografie jako primární screening provedena na pracovišti gynekologa, genetika nebo dětského kardiologa. Při podezření na patologii je ve sledovaném regionu v současnosti plod došetřen většinou na pracovišti FN Ostrava. Ultrazvukové vyšetření plodu – fetální echokardiografie byla na pracovišti autorů provedena na přístroji GE Vivid E9. Při ukončení gravidity byla autopsie vedena vždy za přítomnosti dětského kardiologa. Pokračující gravidita byla sledována a porod novorozence se srdeční vadou byl veden na specializovaném pracovišti.
Získaná data byla uložena a zpracována v programu Microsoft Excel. Stejný program byl použit i pro popisnou statistiku a základní grafy a tabulky.
VÝSLEDKY
Celkové hodnocení
Ve sledovaném 18letém období se v populaci 209 300 plodů vyskytlo celkem 784 (3,7 na 1000 plodů) prenatálně identifikovaných a postnatálně významných VSV. Prenatálně detekováno bylo 52 % (406/784) VSV, prenatálně nepoznáno 48 % (378/784). Podíl prenatální detekce na diagnostice jednotlivých typů vad ukazuje graf 1.
Vysvětlivky zkratek: VSD – defekt komorového septa, AVSD – defekt atrioventrikulárního septa. TOF – Fallotova tetralogie,
TGA – transpozice velkých arterií, COA – koarktace aorty, HLH – syndrom hypoplastického levého srdce, PS – stenóza
plicnice, DORV – dvojvýtoková pravá komora, AS – stenóza aorty, PAIVS – atrézie plicnice s intaktním komorovým
septem, PAVSD – atrézie plicnice s defektem komorového septa, CAT – společný arteriální trunkus, TA – trikuspidální
atrézie, SV – společná komora, EBST – Ebsteinova anomálie, IAA – interrupce aortálního oblouku, TAPVD – totální
anomální návrat plicních žil, TU – tumor/rabdomyom, CTGA – korigovaná transpozice velkých arterií, DXC – izolovaná
dextrokardie, MA – mitrální atrézie, ECT – srdeční ektopie
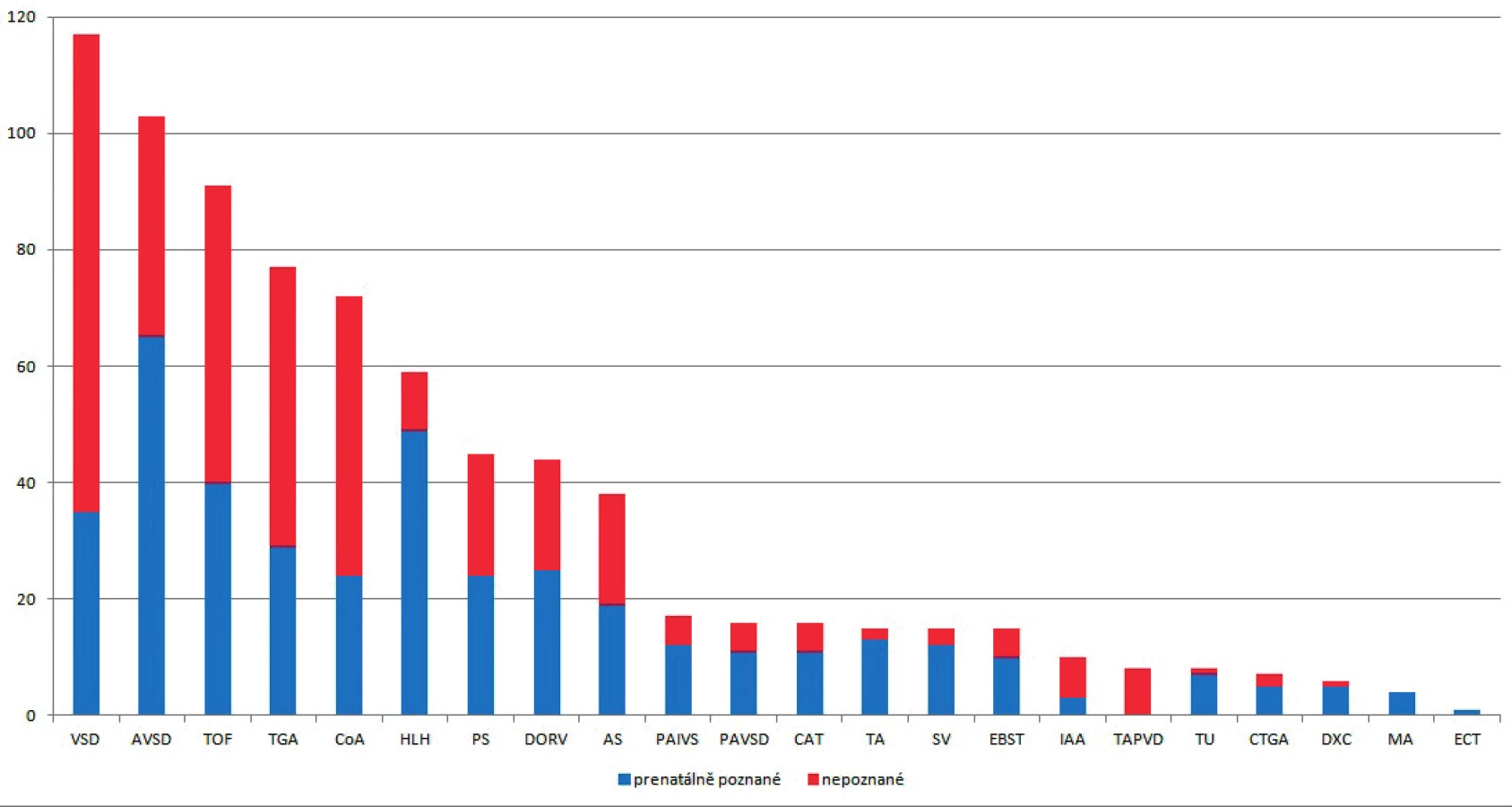
V průběhu studie se prenatální detekce VSV významně zlepšovala, z původních 20 % prenatálně detekovaných VSV v roce 1999 až na současných 74 % v roce 2016, u vad se změnami v morfologii komor (hypoplastické levé a pravé srdce, společná komora) byla v posledních 3 letech prenatální detekce 100 %. U prenatálně detekovaných VSV se 53 % (215/406) rodin rozhodlo pro ukončení gravidity. Srdeční vada se vyskytla převážně jako izolované postižení v 78 % (609/784), ve spojení s genetickým postižením v 16 % (125/784) a s extrakardiální malformací bez genetické patologie v 6 % (50/784). Ze 125 chromosomálních anomálií bylo 70 % (88/125) spjato s prenatální diagnózou a 30 % (37/125) bylo zjištěno až postnatálně. U extrakardiálních patologií bylo ve spojení s VSV prenatálně 86 % (43/50) a postnatálně 14 % (7/50) případů.
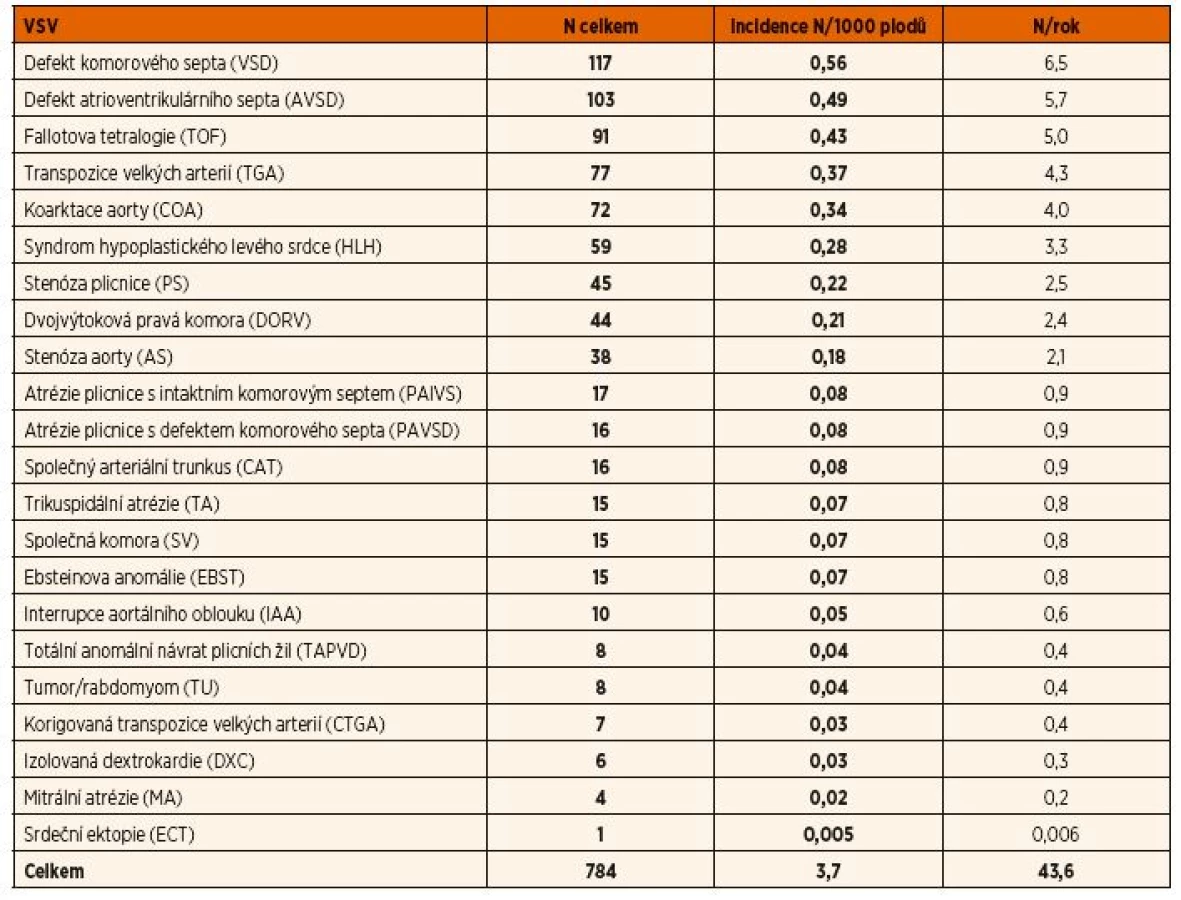
V tabulce 1 je uveden celkový počet VSV, jejich incidence na 1000 plodů a počet VSV ve sledovaném regionu za rok. V následující kapitole je rozbor jednotlivých skupin vad v sestupném pořadí podle celkového výskytu se zaměřením na podíl prenatální detekce, vazby na extrakaradiální anomálie, rozhodování rodičů o ukončení gravidity a perinatální přístup při pokračování těhotenství.
Jednotlivé vady
Defekt komorového septa
Defekt komorového septa je nejčastější VSV v regionu. Na rozdíl od níže uvedených vad je vzhledem k prenatální hemodynamice diagnostikován před narozením méně často, prenatální záchyt byl 30%. U prenatálně detekovaných defektů se 17 % partnerů rozhodlo pro ukončení gravidity, a to vždy ve vazbě na genetickou patologii, nejčastěji pro Downův syndrom. VSD většinou nepředstavuje kritickou srdeční vadu, perinatálně je dítě většinou bez rizik, je nutné diagnostikovat event. přidružené genetické abnormity. Velký a významný defekt komorového septa může způsobit časné srdeční selhávání a vývoj plicní hypertenze. Po vyšetření dětským kardiologem je vždy plánovaná intervence podle významnosti levopravého zkratu.
Defekt atrioventrikulárního septa
Defekt atrioventrikulárního septa je zásadní patologie, která se vyskytuje v několika formách (kompletní, inkompletní, přechodná) a představuje druhou nejčastější významnou vadu v regionu. Prenatální záchyt byl 63%. Podstatná je vazba na genetické syndromy v 49 %, s převahou Downova syndromu. I v této souvislosti se při prenatálně poznané vadě 70 % rodičů rozhodlo pro ukončení těhotenství. Perinatálně nejsou nutná zvláštní opatření, dítě je ale nutné sledovat dětským kardiologem od porodu, hrozí rychlý vývoj plicní hypertenze při velkém levopravém zkratu. Většinou u kompletních forem plánujeme operační korekci do 4 měsíců věku. Pokud nebyla provedena genetika prenatálně, vždy nabízíme genetické došetření.
Fallotova tetralogie
Fallotova tetralogie je v pořadí třetí nejčastější VSV a nejčastější cyanotickou vadou. I přes svou významnost a typickou morfologii byl prenatální záchyt pouhých 44 %. Při prenatálně detekované vadě se 45 % rodičů rozhodlo pro ukončení gravidity. Podstatnou informací je vazba na genetické abnormity v 28 %, s rovnocenným výskytem Downova a DiGeorgeova syndromu. Při zjištění této vady by mělo vždy následovat genetické poradenství. Při volbě porodu je klíčový závěr prenatálního diagnostika, podstatný je odhad významnosti stenózy plicnice a možné obstrukce výtokového traktu pravé komory. Vada se může chovat kriticky s cyanózou, ale může být i přítomna varianta s normálním prokrvením a levopravým zkratem na defektu komorového septa. Novorozence vždy rodíme pod dohledem dětského kardiologa. K chirurgické korekci dojde většinou v prvním roce života dítěte.
Transpozice velkých tepen
Transpozice velkých tepen je postnatálně kritická srdeční vada, bez korekce neslučitelná se životem. V podmínkách sledovaného regionu je čtvrtou nejčastější významnou VSV. Celková prenatální úspěšnost je za celé období jen 38%, v posledních třech letech se zlepšením k 75% záchytu před porodem. Po dobu sledování se s touto vadou nesdružovala žádná genetická patologie, přesto se při prenatálně poznané vadě 29 % partnerů rozhodlo pro ukončení těhotenství. Prenatální diagnostika je klíčová, plod je sledován do konce gravidity a porod vždy volíme v Dětském kardiocentru (DK) FN Motol v Praze. V případě porodu v regionu zajišťujeme novorozence prostaglandiny k udržení průchodnosti tepenné dučeje, pokud to stav vyžaduje, a dítě transportujeme do DK.
Koarktace aorty
Koarktace aorty s pátým místem v pořadí je potenciálně kritická srdeční vada, nemusí se projevit u novorozence a ke zhoršení stavu dochází až v domácích podmínkách. Prenatální diagnostika by byla klíčová, záchyt se ale pro obtížnou projekci aortálního oblouku a fetální hemodynamiku nedaří, je pouhých 33 %. Vazba na genetické abnormity je v 8 % (Turnerův a Edwardsův syndrom) a v těchto souvislostech se z prenatálně poznaných vad rozhodlo pro ukončení gravidity 20 % rodičů. Volba místa porodu není jednoduchá, při jasné koarktaci volíme DK, při fetálně nejasných případech je možné porodit dítě v perinatologickém centru, dále je nutné opakovaně vyšetřovat novorozence a hodnotit vývoj istmické části aorty při uzavírání tepenné dučeje. Při selhávání a vývoji kritické vady jsou indikovány prostaglandiny a transport do DK. Při nevýznamném zúžení nebo vývoji koarktace v dalším dětském věku je intervence plánována podle morfologie a zúžení descendentní aorty.
Syndrom hypoplastického levého srdce
Syndrom hypoplastického levého srdce, šestá nejčastější vada, je kritická VSV s nepříznivou anatomií hypoplazie a snížené funkce levé komory, stenózy nebo atrézie mitrálního nebo aortálního ústí a hypoplazií aortálního oblouku. Má vysokou prenatální detekci 83 %, v posledních třech letech 100 %. Prenatálně rodiče často neakceptují tuto vadu a ukončení je v 87 % případů, výrazně se tak redukuje výskyt této vady v populaci. Vazba na genetické abnormity je malá, v 7 % (Edwardsův, DiGeorgeův, Turnerův syndrom). Vada je duktus dependentní, postnatálně vždy vyžaduje prostaglandiny k udržení průchodnosti dučeje, nedoporučujeme oxygenoterapii. Porod volíme nejlépe v DK. Korekce je pak většinou třífázová směrem k jednokomorové cirkulaci.
Pulmonální stenóza
Pulmonální stenóza má menší prenatální detekci – 53 %, nevýznamné spojení s genetickou patologií ve 4 %. Perinatálně není nutný zvláštní přístup, kardiologické sledování se odvíjí podle postnatálního významu zúžení plicnice. V případě rychlé progrese, klinické a hemodynamické významnosti je možný katetrizační nebo operační zákrok.
Dvojvýtoková pravá komora
Dvojvýtoková pravá komora je charakterizována defektem komorového septa a odstupem obou velkých tepen z pravé komory. Má složitější morfologickou klasifikaci pro množství variant vztahu komorového defektu k velkým tepnám, morfologii výtokového traktu pravé komory a postavení velkých tepen. Časté jsou další přidružené VSV a extrakardiální anomálie. V našem souboru mělo s touto vadou genetické postižení 12 % VSV (Edwardsův, Jacobsenův a Pataův syndrom) a 11 % jinou extrakardiální malformaci s normálním karyotypem. Prenatální detekce 53 % a ukončení gravidity v 59 % se podobá Fallotově tetralogii a stejně důležité je i u této vady pečlivé genetické došetření a diagnostika extrakardiálních anomálií. Vzhledem k velkému množství anatomických variant a různým možnostem klinického průběhu je nutné frekventní sledování dětským kardiologem, perinatálně je většinou čas na posouzení a sledování. Podle plicního průtoku, objemové zátěže a klinického chování pak plánujeme korekci vady.
Aortální stenóza
Aortální stenóza se vyskytuje ve valvární, subvalvární a supravalvární formě. U významné formy může již fetálně dojít k selhávání levé komory a k vývoji její dysfunkce nebo hypoplazie. K fetální detekci je nutné použít vyjma běžného 2D obrazu i dopplerovské techniky. I proto je tato prenatální identifikace jen v 50 %, 33 % rodičů se při prenatálně detekované vadě rozhodlo pro ukončení, většinou při nepříznivém nálezu na funkci a morfologii levé komory. Z pediatrického pohledu je nutno myslet na event. přidružené genetické abnormity (Williamsův--Beurenův a Noonanové syndrom), v uvedeném souboru v 5 % z celkového počtu aortálních stenóz. Před porodem je klíčový odhad chování vady, echokardiografické vyšetření postnatálně provádíme denně pro možný velmi rychlý vzestup gradientu v aortě a možné kardiální selhávání dítěte. Terapie je pak katetrizační nebo operační.
Pulmonální atrézie s defektem komorového septa
Pulmonální atrézie s defektem komorového septa je komplikována různým zásobením plicního řečiště a může se projevovat nižším i zvýšeným plicním průtokem, na kterém závisí i postnatální přístup a další průběh onemocnění. Prenatální záchyt byl 69%, pro nepříznivou anatomii se 89 % rodičů rozhodlo pro ukončení gravidity. S genetickou patologií bylo spojeno 20 % této vady (DiGeorgeův a Pataův syndrom).
Pulmonální atrézie s intaktním komorovým septem
Pulmonální atrézie s intaktním komorovým septem se většinou projeví fetálně hypoplazií pravé komory, a proto má dobrý prenatální záchyt v průměru 70 %, v posledních letech 100 %. Žádná vada se nevázala s genetickou patologií, přesto při prenatálně poznané vadě se 100 % rodičů rozhodlo pro ukončení gravidity. Prenatální diagnostika je podstatná, vada je závislá na průchodné dučeji, při jejím uzavírání dochází k rychlému rozvoji hypoxémie.
Trikuspidální atrézie
Trikuspidální atrézie má většinou hypoplastickou pravou komoru, a proto je fetální diagnostika vysoká, vada byla detekována v regionu v průměru v 88 %, v posledních letech ve 100 % a 92 % rodičů se při prenatální diagnóze rozhodlo pro ukončení gravidity. Celkově se tato VSV spojovala s genetickou patologií ve 12 % (Edwardsův sy). Vada má většinou nízký plicní průtok, projevuje se cyanózou. Řešením jsou postupné korekce na jednokomorovou cirkulaci.
Arteriální trunkus
Arteriální trunkus je konotrunkální vada charakterizována jediným arteriálním kmenem nasedajícím nad komorový defekt a různými typy krevního zásobení plic. Fetálně byla diagnostikována v 69 % a 90 % rodin se rozhodlo pro ukončení. Vyšší je podíl genetických abnormit, v 31 % (DiGeorgeův a Pataův sy). Plicní průtok je většinou zvýšený, tomu odpovídají i projevy srdečního selhání při velkém levopravém zkratu, brzy se ale vyvíjí plicní hypertenze a prohlubuje se cyanóza. Postnatálně se korekce většinou provádí do 2. měsíce života.
Společná komora
Společná komora, dnes přesněji dvojvtoková, je komplexní vada s hlavní charakteristikou spojení síní s jedinou funkční komorou. Má mnoho variant: dominantní jedna z komor s rudimentem druhé komory, vzácněji nediferencovaná komora. Přítomna může být jedna nebo obě atrioventrikulární chlopně a různé postavení a malformace velkých tepen. Pro velké množství anatomických variant je klinicky stav závislý na velikosti plicního a systémového průtoku, vada je ale symptomatická téměř vždy brzy po narození. Pro jasnou patologii srdeční čtyřdutiny s chyběním komorového septa je fetální záchyt vysoký – v 80 % a na vadu je možno se perinatálně připravit, 45 % rodičů se rozhodlo prenatálně pro ukončení. Vada se v 6 % spojovala s genetickou patologií (Edwardsův sy), podstatný byl ale výskyt extrakardiálních malformací ve 23 % případů.
Ebsteinova anomálie
Ebsteinova anomálie je vada trojcípé chlopně s posunutím septálního a zadního cípu do vtokové části pravé komory a elongací předního cípu. Dochází k atrializaci pravé komory. Fetálním i postnatálním problémem je velká variabilita vady, u těžkých forem může dojít k intrauterinnímu úmrtí. Prenatálně byla vada detekována v 67 % a 60 % rodin se rozhodlo pro ukončení gravidity u těžkých forem. U této vady se obtížně odhaduje prognóza a komplikací jsou supraventrikulární tachykardie pro přídatné atrioventrikulární spojky. Nebyla zaznamenána žádná genetická patologie. Klinické projevy po porodu zahrnují široké spektrum, od kritického chování až po asymptomatické pacienty.
Interrupce aortálního oblouku
Interrupce aortálního oblouku je přes svou komplikovanost méně častá vada s 30 % prenatálního záchytu a 100% rozhodnutím rodin pro ukončení. Genetické postižení bylo v 30 % (DiGeorgeův sy). Tato vada je duktus dependentní, klinicky se projevuje jako koarktace aorty s defektem komorového septa. Při uzavírání dučeje dochází k rychlému rozvoji cyanózy a srdečnímu selhávání. Terapie je operační, nutná je rekonstrukce aortálního oblouku.
Totální anomální návrat plicních žil
Totální anomální návrat plicních žil nebyl dosud v regionu fetálně diagnostikován. Při došetření dětí nebyla shledána genetická patologie. Část novorozenců byla symptomatických, část dětí byla došetřena až jako kojenci při klinické dekompenzaci v domácích podmínkách.
Srdeční tumory
Srdečními tumory byly perinatálně vždy rabdomyomy, pro vícečetný výskyt mají vysoký podíl prenatální detekce v 88 % a 43 % rodičů se rozhodlo pro ukončení gravidity. Tyto tumory téměř vždy po porodu regredují, negativním důsledkem je vývoj tuberózní sklerózy a poškození centrální nervové soustavy dítěte. Hemodynamicky jsou většinou dobře tolerovány, vyjma morfologické patologie se sledují i poruchy srdečního rytmu.
Korigovaná transpozice
Korigovaná transpozice s atrioventrikulární a ventrikuloarteriální diskordancí se klinicky projevuje při přidruženém defektu komorového septa, obstrukci výtoku levé komory, vadě trojcípé chlopně a poruchách srdečního rytmu. Fetální záchyt byl v 71 % a 50 % rodin se rozhodlo pro ukončení gravidity pro extrakardiální malformace bez genetických abnormit.
Izolovaná dextrokardie
Izolovaná dextrokardie není pravou VSV, jde jen o polohovou anomálii. Pro abnormální vztah polohy srdce k břišním orgánům byl možný vysoký prenatální záchyt, a to v 83 %. Žádná z rodin nežádala o ukončení gravidity a všechny narozené děti byly asymptomatické.
Mitrální atrézie
Mitrální atrézie se vyskytovala vzácně, všechny 4 případy byly detekovány prenatálně a 1 rodina se rozhodla pro přerušení těhotenství. Vada se projevuje obrazem plicního městnání již v novorozeneckém a kojeneckém věku.
Ektopie srdeční
Ektopie srdeční je extrémně vzácná vada, byla prenatálně zachycena v 1 případě a rodiče se rozhodli pro ukončení gravidity, plod neměl související další extrakardiální patologii.
DISKUSE
Srdeční vady jsou nejčastější morfologické defekty v populaci a pro jejich incidenci jsou uváděna velmi široká rozmezí. V letech 2003–2007 byl výskyt všech vad ve 22 zemích Evropy 23,9 na 1000 narozených dětí [14]. Studie Dětského kardiocentra FN Motol v Praze uvedla výskyt VSV 6,4 na 1000 živě narozených [5], analýza incidence VSV v České republice v letech 1994–2008 uvádí celkem 19,8 hlášených VSV na 1000 živě narozených [3]. Podle dat ÚZIS ČR byl v roce 2013 celkový výskyt hlášených VSV 22,8/1000 a v roce 2014 22,2/1000 živě narozených. Za období 2000–2014 tvořily VSV 42 % všech vrozených vývojových vad. Při započtení všech nevýznamných forem VSV včetně malých komorových defektů stoupá incidence až k 75/1000 živě narozených [15].
Vývoj srdeční vady je pravděpodobně způsoben kombinací genetických a negenetických faktorů [16] a přes rychlý rozvoj lékařské genetiky zůstává příčina vzniku větší části VSV nejasná a hovoříme o multifaktoriální hypotéze. Srdeční vady se vyskytuji většinou jako izolované patologie, menší část je svázaná s chromosomální abnormitou nebo s extrakardiální malformací při normálním karyotypu plodu nebo dítěte. Nejčastější VSV je defekt komorového a síňového septa, kritických VSV je 35 % z celkového množství. Většina významných VSV je v současnosti detekovatelná prenatálně, při fetálně diagnostikované vadě se část rodičů rozhoduje pro ukončení gravidity a je tak redukován výskyt VSV v populaci. Zvyšující se úspěšnost prenatální diagnostiky v České republice snížila výskyt VSV z 6,7/1000 na 5,3/1000 (kritických VSV 2,4/1000) živě narozených [17].
Uvedená studie se zaměřuje na kritické a významné VSV, které zásadně ovlivňují klinický stav novorozence nebo časného kojence a jsou operovány nebo katetrizačně řešeny většinou do jednoho roku věku dítěte. Výskyt významných 3,7 VSV na 1000 plodů odpovídal jiným studiím [18]. Nejčastější VSV byl defekt komorového septa, druhou nejčetnější VSV byl defekt atrioventrikulárního septa, nejčastější cyanotickou vadou byla Fallotova tetralogie, nejčetnější kritická vada u novorozenců byla transpozice velkých tepen [17] a nejméně četnou VSV ektopie srdeční. V uvedeném souboru bylo 78 % VSV izolovaných, 16 % mělo genetickou abnormitu a 6 % jinou extrakardiální morfologickou patologii při současně normálním karyotypu [19]. Nejčastější VSV spojenou s genetickou patologií byl ve 49 % defekt atrioventrikulárního septa [20, 21] s dominancí Downova syndromu diagnostikovaném u 39 % případů [22]. Při přítomnosti chromosomální aberace se většinou tato vada vyskytuje jako izolovaná srdeční patologie [23, 24]. Vyšší genetický podíl v etiologii VSV měl ještě společný arteriální trunkus, interrupce aortálního oblouku a Fallotova tetralogie. Syndromy Downův, Edwardsův, Pataův a DiGeorgův zastupovaly 80 % všech genetických abnormit. Trisomie dalších autosomů ve spojení s VSV jsou vzácné a většinou viabilní jenom ve formě mozaiky [25]. Turnerův syndrom, který je nejčastější chromosomální aberací u ženského pohlaví [26, 27], byl spojen s koarktací aorty a hypoplazií levého srdce. Žádná genetická patologie nebyla prokázána u Ebsteinovy anomálie, pulmonální atrézie s intaktním komorovým septem, transpozice velkých tepen, totálního anomálního plicního návratu, tumorů typu rabdomyomu, korigované transpozici velkých tepen, izolované dextrokardii a u vzácné ektopie srdeční. Vyšší podíl extrakardiálních patologií při normální genetické výbavě byl diagnostikován u společné komory, korigované transpozice velkých tepen a pulmonální atrézie s intaktním komorovým septem. Zvláštní skupinou byly fetální tumory srdce, které – přestože se většinou neřadí k srdečním vadám – byly pro možné významné klinické komplikace v hodnocení ponechány. Všechny tumory byly rabdomyomy, které jsou nejčastějším fetálním primárním srdečním tumorem [28]. Riziko spojení s tuberózní sklerózou centrální nervové soustavy a významným neurologickým postižením dítěte je až 50% [29], v případě mnohočetných nádorů je toto riziko vyšší [30]. Fetální komplikací může být vývoj hydropsu, obstrukce výtokových traktů, arytmie nebo srdeční selhání [31]. Komplexní prenatální poradenství je možné, včetně genetického došetření mutací genů pro tuberózní sklerózu a magnetické rezonance mozku plodu [32].
Vývoj ultrazvukových technik a kvalita prenatálního screeningu umožňuje většinu významných VSV detekovat prenatálně. Fetální echokardiografie (FECHO) je velmi přesná metoda ke zjištění kardiálních malformací u plodu a při vyšetření zkušeným lékařem má vynikající výsledky [33]. Pro detekci významných VSV má FECHO vysokou specificitu a senzitivitu, detekce nevýznamných vad je ale limitovaná [34]. V podmínkách České republiky je screening proveden gynekologem, dětským kardiologem nebo genetikem většinou ve II. trimestru gravidity s největší výtěžností mezi 18.–22. týdnem gravidity [35], u rizikových gravidit je možné rodině nabídnout časnou fetální diagnostiku [36, 37, 38]. V případě rizikových faktorů (matky, plodu, gravidity) je na pracovišti autorů první vyšetření provedeno v 16. týdnu, vždy s kontrolou ve 20. týdnu. Prvotrimestrální screening VSV je možný, má pak významný dopad na spektrum VSV a výsledek těhotenství a vede k vyššímu počtu ukončených gravidit [39]. Časná fetální diagnóza u významných vad má za následek přerušení gravidit až nad 70 % [40]. Definitivní posouzení významnosti vady v nízkých týdnech by mělo příslušet pouze specializovaným prenatálním centrům a nutné je komplexní genetické poradenství. V uvedeném souboru jsou ojedinělé případy posouzení VSV ve 12.–13. týd-nu, vždy po odeslání jiným ošetřujícím lékařem. Plánovaný screening VSV v I. trimestru v podmínkách ostravského regionu zatím autoři této práce neplánují.
Patologická gravidita je celkově došetřena, jsou posouzeny extrakardiální anomálie a doplněno genetické vyšetření. Rodiče jsou vždy plně informováni a rozhodují o pokračování nebo ukončení gravidity. Úspěšnost prenatální diagnostiky VSV se ve světě obecně zlepšuje [41], přesto se ale výsledky mezi jednotlivými zeměmi liší [42]. Česká republika patří k zemím s vysokou prenatální detekcí VSV v Evropě. Prenatální detekce VSV se zvýšila z 0,6 % v roce 1986 na 36,5 % v roce 2009 [43], v období 2002–2009 se pohybovala od 70 do 83 % [44], v posledních letech u syndromu hypoplastického levého srdce bylo zachyceno až 95 % případů [45]. Ve studii autorů a sledovaném regionu byla poprvé prenatálně detekována více než polovina významných VSV v roce 2005, poté se výtěžnost zlepšovala až na současných 74 %.
Část rodičů se při prenatálně detekované vadě rozhoduje graviditu ukončit. V různých registrech se počet ukončených gravidit při diagnóze VSV výrazně liší od 0 do 50 %. Česká republika patří k zemím s nejvyšší četností ukončených gravidit na světě. V našich podmínkách je toto možné z genetické indikace do 24. týdne. Důležité je správně vedené poradenství a celkové došetření gravidity. Partneři jsou náležitě poučeni o typu vady a sami rozhodují o pokračování nebo ukončení těhotenství. Výskyt vady v rodině není pouze otázkou zdravotní, ale také sociální a ekonomickou. Na rozhodnutí rodičů má vliv významnost VSV, věk a přidružené anomálie plodu [46]. Ve sledovaném období se v našem souboru 53 % rodin při prenatálně zjištěné VSV rozhodlo pro ukončení gravidity a tento trend se v podmínkách Moravskoslezského kraje dlouhodobě nemění. Z ukončených gravidit mělo 30 % chromosomální aberaci a 13 % jiné extrakardiální postižení. U autopsie plodu je na našem pracovišti vždy přítomen dětský kardiolog, který s patologem a genetikem uzavírá celkové nálezy.
Cílem prenatální diagnostiky je ale celkové došetření patologické gravidity, správné a pečlivé poradenství a plánování porodu nemocného novorozence a jeho zajištění na adekvátním pracovišti. V závěru gravidity je těhotná vedena v rizikové poradně, porod novorozence s VSV je možný vaginálně, nutný je pečlivý perinatální přístup se spoluprací gynekologa, neonatologa a dětského kardiologa. Znalost VSV před porodem může snížit morbiditu a mortalitu u některých typů kritických a významných srdečních vad [47]. Ve sledovaném regio-nu je v posledních letech stále fetálně nepoznáno 20 % významných VSV. Otázkou je zavedení screeningu pulzní oxymetrií na novorozeneckých odděleních, tato metoda může odhalit významné VSV ještě u asymptomatických novorozenců [48].
ZÁVĚR
Srdeční vada se většinou vyskytuje jako izolované postižení. Genetické a jiné extrakardiální anomálie se spojují přibližně s jednou třetinou kardiálních diagnóz a jsou významně svázány s prenatální diagnostikou. Prenatální péče umožňuje došetření patologické gravidity, správné poradenství postižené rodině a plánování porodu novorozence se srdeční vadou. Polovina rodin graviditu s fetálně poznanou VSV ukončí a je tak redukován výskyt VSV v populaci novorozenců.
Tato studie byla podporována grantem MZ VES 2017 (Reg. No. NV17-29111A).
Došlo: 12. 12. 2017
Přijato: 8. 2. 2018
MUDr. Jan Pavlíček, Ph.D.
Oddělení dětské a prenatální kardiologie
Klinika dětského lékařství
Fakultní nemocnice Ostrava
17. listopadu 1790
708 52 Ostrava Poruba
e-mail: jan.pavlicek@fno.cz
Sources
1. Bolisetty S, Daftary A, Ewald D, et al. Congenital heart defects in Central Australia. Med J Aust 2004; 180 (12): 614–617.
2. van der Linde D, Konings E, Slager M, et al. Birth prevalence of congenital heart disease worldwide. J Am Coll Cardiol 2011; 58 (21): 2241–2247.
3. Šípek A, Gregor V, Šípek A Jr, et al. Incidence of congenital heart defects in the Czech Republic – current data. Ceska Gynekol 2010; 75 (3): 221–242.
4. Baspinar O, Karaaslan S, Oran B, et al. Prevalence and distribution of children with congenital heart diseases in the central Anatolian region, Turkey. Turk J Pediatr 2006; 48 (3): 237–243.
5. Šamánek M, Slavík Z, Zbořilová B, et al. Prevalence, treatment, and outcome of heart disease in live-born children: A prospective analysis od 91,823 live born children. Pediat Cardiol 1989; 10 (4): 205–211.
6. Shi H, O‘Reilly VC, Moreau JL, et al. Gestational stress induces the unfolded protein response, resulting in heart defects. Development 2016; 143 (14): 2561–2572.
7. Blue GM, Kirk EP, Sholler GF, et al. Congenital heart disease: current knowledge about causes and inheritance. Med J Aust 2012; 197 (3): 155–159.
8. Pierpont ME, Basson CT, Benson DW, et al. Genetic basis for congenital heart defects: current knowledge. Circulation 2007; 115 (23): 3015–3038.
9. Richards AA, Santos LJ, Nichols HA, et al. Cryptic chromosomal abnormalities identified in children with congenital heart disease. Pediatr Res 2008; 64 (4): 358–363.
10. Fahed AC, Gelb BD, Seidman JG, et al. Genetics of congenital heart disease. Circ Res 2013; 112 (4): 707–720.
11. Wessels MW, Willems PJ. Genetic factors in non-syndromic congenital heart malformation. Clin Genet 2010; 78 : 103–123.
12. Nayak K, Naveen Chandra GS, Shetty R, et al. Evaluation of fetal echocardiography as a routine antenatal screening tool for detection of congenital heart disease. Cardiovasc Diagn Ther 2016; 6 (1): 44.
13. Bull C. Current and potential impact of fetal diagnosis on prevalence and spectrum of serious congenital heart disease at term in the UK. Lancet 1999; 354 (9186): 1242–1247.
14. Dolk H, Loane M, Garne E. The prevalence of congenital anomalies in Europe. Adv Exp Med Biol 2010; 686 : 349–364.
15. Hoffman JI, Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol 2002; 39 (12): 1890–1900.
16. Riehle-Colarusso TJ, Patel SS. Maternal nongenetic risk factors for congenital heart defects. In: Congenital Heart Disease. Karger Publishers, 2015 : 57–69.
17. Chaloupecký V, et al. Dětská kardiologie. Praha: Galén, 2006 : 195–203.
18. Dadvand P, Rankin J, Shirley MD, et al. Descriptive epidemiology of congenital heart disease in Northern England. Paediatr Perinat Epidemiol 2009; 23 (1): 58–65.
19. Tegnader E, Williams O, JohansenS J, et al. Prenatal detection of heart defects in a non-selectes population of 30 149 fetuses-detection rates and outcome. Ultrasound Obstet Gynecol 2006; 27 : 252–265.
20. Huggon IC, Cook AC, Smeeton NC, et al. Atrioventricular septal defects diagnosed in fetal life: associated cardiac and extra-cardiac abnormalities and outcome. J Am Coll Cardiol 2000; 36 (2): 593–601.
21. Rasiah SV, Ewer AK, Miller P, et al. Outcome following prenatal diagnosis of complete atrioventricular septal defect. Prenat Diagn 2008; 28 (2): 95–101.
22. Tumanyan MR, Filaretova OV, Chechneva V, et al. Repair of complete atrioventricular septal defect in infants with Down syndrome: outcomes and long-term results. Pediatr Cardiol 2015; 36 (1): 71–75.
23. Fesslova V, Villa L, Nava S, et al. Spectrum and outcome od atrioventricular septal defect in fetal life. Cardiol Young 2002; 12 (1): 18–26.
24. Hajdú J, Beke A, Pete B, et al. Prenatal diagnosis of the atrioventricular septal defect and it’s effect on the outcome of the pregnancies. Orv Hetil 2005; 146 (34): 1775–1780.
25. Staníková A, Grochová I, Borek I. Úskalí prenatální diagnostiky trizomie 20 a její mozaiky. Čes-slov Pediat 2012; 67 (6): 390–392.
26. Kaprálová S, Zapletalová J, Vrtěl R, et al. Získaná kardiovaskulární onemocnění u Turnerova syndromu. Čes-slov Pediat 2017; 72 (1): 54–62.
27. Klásková E, Tüdös Z, Wiedermann J, et al. Postižení kardiovaskulárního systému u Turnerova syndromu. Čes-slov Pediat 2012; 67 (2): 103–111.
28. Bonnamy L, Perrotin F, Megier P, et al. Fetal intracardiac tumors: prenatal diagnosis and management: Three case reports. Eur J Obstet Gyn R B 2001; 99 (1): 112–117.
29. Yuan SM. Fetal primary cardiac tumors during perinatal period. Pediatr Neonatol 2017; 58 (3): 205–210.
30. Niewiadomska-Jarosik K, Stanczyk J, Janiak K, et al. Prenatal diagnosis and follow-up of 23 cases of cardiac tumors. Prenat Diagn 2010; 30 (9): 882–887.
31. Jóźwiak S, Kotulska K, Kasprzyk-Obara J, et al. Clinical and genotype studies of cardiac tumors in 154 patients with tuberous sclerosis complex. Pediatrics 2006; 118 (4): e1146–e1151.
32. Sarac Sivrikoz T, Has R, Kalelioglu IH, et al. The association of prenatally diagnosed cardiac rhabdomyoma and tuberosclerosis complex: a dismal combination? Ultrasound Obstet Gynecol 2015; 46 (S1): 108.
33. Yu ZB, Han SP, Guo XR. Meta-analysis of the value of fetal echocardiography for the prenatal diagnosis of congenital heart disease. Chin J Evid Based Pediatr 2009; 4 : 330–339.
34. Chu C, Yan Y, Ren Y, et al. Prenatal diagnosis of congenital heart diseases by fetal echocardiography in second trimester: a Chinese multicenter study. Acta Obstet Gyn Scan 2017; 96 : 454–463.
35. Campbell S, Allan L, Benacerraf B, et al. Isolated major congenital heart disease. Ultrasound Obstet Gynecol 2001; 17 : 370–379.
36. ACOG Committee on Practice Bulletins. AGOG practice bulletin No 58. Ultrasonography in pregnancy. Obstet Gynecol 2004; 58 : 1449–1458.
37. Bellotti M, Fesslova V, De Gasperi C, et al. Reliability of the first–trimester cardiac scan by ultrasound–trained obstetricians with high–frequency transabdominal probes in fetuses with increased nuchal translucency. Ultrasound Obstet Gynecol 2010; 36 (3): 272–278.
38. Comas Gabriel C, Galindo A, Martinez JM, et al. Early prenatal diagnosis of major cardiac anomalies in a high risk population. Prenat Diagn 2002; 22 (7): 586–593.
39. Jicinska H, Vlasin P, Jicinsky M, et al. Does first-trimester screening modify the natural history of congenital heart disease? Circulation 2017; 135 (11): 1045–1055.
40. Garne E, Stoll C, Clementi M. Evaluation of prenatal diagnosis of congenital heart diseases by ultrasound: experience from 20 European registries. Ultrasound Obstet Gynecol 2001; 17 (5): 386–391.
41. Khoshnood B, De Vigan C, Vodovar V, et al. Trends in prenatal diagnosis, pregnancy termination, and perinatal mortality of newborns with congenital heart disease in France, 1983–2000: a population-based evaluation. Pediatrics 2005; 115 : 95–101.
42. Garne E, Stoll C, Clementi M. Evaluation of prenatal diagnosis of congenital heart diseases by ultrasound: experience from 20 European registries. Ultrasound Obstet Gynecol 2001; 17 : 386–391.
43. Tomek V, Marek J, Jíčínská, et al. Fetal cardiology in the Czech Republic: Current management of prenatally diagnosed congenital heart disease and arrhythmias. Physiol Res 2009; 58 (Suppl 2): 159–166.
44. Jičínská H. Prenatální kardiologie v České republice. Čes-slov Pediat 2010; 65 (11): 623–625.
45. Marek J, Tomek V, Škovránek J, et al. Prenatal ultrasound screening of congenital heart disease in an unselected national population: a 21-year experience. Heart 2011; 97 (2): 124–130.
46. Chenni N, Lacroze V, Pouet C, et al. Fetal heart disease and interruption of pregnancy: factors influencing the parental decision-making process. Prenat Diagn 2012; 32 (2): 168–172.
47. Simpson JM. Impact of fetal echocardiography. Ann Pediatr Cardiol 2009; 2 (1): 41–50.
48. Maťašová K, Bukovinská Z, Jánoš M, et al. Skríning kritických vrodených chýb srdca u novorodencov pulznou oxymetriou v regióne severného Slovenska. Čes-slov Pediat 2011; 66 (3): 146–152.
Labels
Neonatology Paediatrics General practitioner for children and adolescentsArticle was published in
Czech-Slovak Pediatrics

2018 Issue 5
- What Effect Can Be Expected from Limosilactobacillus reuteri in Mucositis and Peri-Implantitis?
- The Importance of Limosilactobacillus reuteri in Administration to Diabetics with Gingivitis
Most read in this issue
- Mentální anorexie s raným začátkem, diagnostika a terapie
- Výskyt vrozených srdečních vad – dopad prenatální diagnostiky
- Prenatální detekce srdečních vad a její důsledky
- Lokálne reakcie po uhryznutí hadom – klinické skúsenosti