
Těžký kombinovaný imunodeficit s atypickým fenotypem – kazuistika
Severe combined immunodeficiency with atypical phenotype – case report
Severe combined immunodeficiency (SCID) is clinically and genetically heterogeneous group of the most severe inborn errors of immunity. The disease typically presents as a failure of antimicrobial defences due to various defects of lymphocyte development and functions. Being a life-threatening condition, SCID requires a prompt diagnosis in order to implement an appropriate management aimed towards early hematopoietic stem cell transplantation. Besides the classic forms of SCID, atypical or leaky phenotypes exist, that may manifest later in life or/and with milder symptoms.
In this report, we describe a 5-months-old infant with autosomal recessive atypical RAG2 SCID, who is the only patient diagnosed with SCID in the Czech Republic in the years 2018 and 2019.
Keywords:
SCID – severe combined immunodeficiency – lymphopenia – SCID screening – TREC – KREC – hematopoietic stem cell transplantation
Authors:
M. Purkartová 1; R. Formánková 2; O. Hrušák 2,3; M. Nováková 2,3; H. Grombiříková 4; T. Freiberger 4; M. Svaton 2,3; A. Šedivá 5; M. Magner 1,5; M. Bloomfield 1,6
Authors‘ workplace:
Pediatrická klinika 1. LF UK a Thomayerovy nemocnice, Praha
1; Klinika dětské hematologie a onkologie 2. LF UK a FN Motol, Praha
2; CLIP – Childhood Leukaemia Investigation Prague
3; Centrum kardiovaskulární a transplantační chirurgie a LF MU, Brno
4; Klinika pediatrie a dědičných poruch metabolismu 1. LF UK a VFN, Praha
5; Ústav imunologie 2. LF UK a FN Motol, Praha
6
Published in:
Čes-slov Pediat 2020; 75 (6): 356-361.
Category:
Overview
SCID (Severe combined immunodeficiency) neboli těžký kombinovaný imunodeficit je klinicky i geneticky heterogenní skupina nejzávažnějších vrozených poruch imunity. Toto vzácné onemocnění vede především k selhávání obrany proti infekcím na základě abnormálního vývoje lymfocytů a jejich funkcí. Jako život ohrožující stav vyžaduje co nejčasnější diagnostiku k zajištění adekvátního managementu směřujícímu k časné transplantaci kmenových buněk krvetvorby. Kromě klasických forem SCID, manifestujících se v časném kojeneckém věku, existují také formy, jež se mohou projevit později a/nebo mohou mít mírnější klinické příznaky.
V tomto článku prezentujeme kazuistiku 5měsíčního kojence s autosomálně recesivním RAG2 SCID s atypickým fenotypem, který je jediným diagnostikovaným pacientem se SCID v České republice v letech 2018 a 2019.
Klíčová slova:
SCID – těžká kombinovaná imunodeficience – lymfopenie – SCID screening – TREC – KREC – transplantace kmenových buněk krvetvorby
ÚVOD
SCID (Severe combined immunodeficiency) je nejzávažnější vrozenou poruchou imunity postihující současně buněčnou a humorální složku imunitního systému. Příčinou jsou mutace v genech ovlivňujících vývoj a funkce T lymfocytů, někdy také B lymfocytů a NK buněk. V současnosti je známo přes 20 genů asociovaných se SCID. Onemocnění je až v 50 % X-vázané (mutace v IL2RG genu kódujícím společný γ řetězec podjednotky receptoru pro řadu cytokinů nezbytných k vývoji lymfocytů), v ostatních případech je autosomálně recesivní (například mutace v RAG1/2, AK2, ADA, JAK3) [1]. Incidence je celosvětově odhadována na 1 : 50 000–100 000. Klasickými klinickými projevy jsou neprospívání, chronické průjmy, dermatitidy a především závažné infekce od prvních týdnů života, které mají atypický průběh a hůře odpovídají na léčbu. Kromě běžných patogenů, z nichž obzvláště komplikovaně probíhají herpetické infekce (CMV, EBV, VZV, HSV), jsou pacienti zvýšeně náchylní k oportunním mikroorganismům, například Pneumocystis jirovecii, mykózám (Candida spp., Aspergillus spp.) či atenuovaným vakcinačním kmenům, například BCG [2].
Z neinfekčních komplikací se mohou u SCID vyskytnout příznaky maternofetálního engraftmentu (přítomnost mateřských lymfocytů v periferní krvi dítěte). Transplacentární přestup malého počtu mateřských lymfocytů je fyziologický, tyto buňky jsou však za normálních okolností eliminovány funkčním imunitním systémem dítěte. U SCID mohou mateřské lymfocyty v krvi kojence přežít a rozpoznávat HLA antigeny (human leukocyte antigen) dítěte jako „cizí“. Jejich přítomnost se pak projeví pod obrazem reakce štěpu proti hostiteli (graft versus host disease = GVHD) jako různě závažné dermatitidy, enteropatie, hepatopatie, lymfadenopatie či cytopenie [3]. Podobné „autoreaktivní“ fenomény mohou provázet i atypický (leaky) SCID. Při něm je na podkladě hypomorfních mutací v genech asociovaných se SCID možný částečný vývoj lymfocytů. Ty jsou však dysfunkční, mají omezený repertoár rozpoznávaných antigenů (jsou tzv. oligoklonální) a mohou reagovat vůči vlastním tkáním (definováno jako Omennův syndrom = generalizovaná dermatitida, SCID-like imunofenotyp, přítomnost oligoklonálních autoreaktivních lymfocytů, eozinofílie a elevace celkového IgE). Formy SCID se setrvalou T lymfopenií bez přítomnosti kauzální mutace jsou pak označovány jako idiopatická T lymfopenie (dříve tzv. variantní SCID) [2].
Diagnózu SCID stanovujeme na základě klinického projevu, osobní a rodinné anamnézy, imunologického a genetického vyšetření. V rodinné anamnéze pátráme cíleně po údajích o časné kojenecké úmrtnosti, vazbách obtíží na mužskou linii či možných příbuzenských partnerstvích. Z laboratorních vyšetření je klíčový diferenciální krevní obraz, v němž věnujeme pozornost lymfopenii a eventuálně eozinofílii. Dále stanovujeme rozpočet lymfocytárních subpopulací, podle nějž SCID definujeme jako T–, B+/–, NK+/– (přičemž počet autologních CD3+ T lymfocytů je u typického SCID <0,3x109/L, u atypického SCID 0,3–1,5x109/L), vyšetřujeme proliferační schopnost lymfocytů po stimulaci nespecifickými mitogeny a sérové hladiny imunoglobulinů [2,4], jejichž hodnotu interpretujeme s ohledem na věk dítěte.
Při klinickém podezření na SCID jsou v ČR dostupné také rychlé screeningové testy TREC (T-cell receptor excision circles) a KREC (kappa–deleting recombination excision circles). Jedná se o genetická vyšetření z malého množství krve definující schopnost vlastní produkce nejmladších vývojových forem T a B lymfocytů podle přítomnosti sestřihových zbytků DNA z procesu tzv. V(D)J rekombinace [5]. Podobnou informaci je možné získat cytometrickým vyčíslením nejnaivnějších forem T lymfocytů v periferní krvi, tzv. recent thymic emigrants (RTE). Přítomnost maternálních T lymfocytů v krvi dítěte je možné detekovat molekulárně genetickým vyšetřením chimérismu (např. dle HLA nebo tandemových repetic) či převahou vývojově vyzrálejších forem T lymfocytů (paměťových CD45RO+) [3]. Diagnózu SCID podporuje nález kauzální mutace. Do diferenciální diagnostiky SCID patří mimo jiné HIV infekce, kongenitální rubeola, DiGeorgeův syndrom, MHC II deficience či CD3 deficience. Diagnostická kritéria SCID uvádí obrázek 1.

Terapií SCID je co nejčasnější transplantace kmenových buněk krvetvorby, přičemž u některých forem je dostupná také enzymová a genová terapie (např. u deficitu adenosindeaminázy). Před transplantací je potřeba minimalizovat riziko infektu, pacient je proto izolován, je zastaveno očkování, zahájena adekvátní antiinfekční terapie/profylaxe (širokospektrá antibiotika, antimykotika, virostatika, ev. antituberkulotika) a substituce imunoglobulinů, v případě nutnosti podání krevních derivátů jsou používány pouze ozářené preparáty (riziko GVHD). Prognóza dětí s klasickým SCID je bez terapie infaustní, k úmrtí dochází zpravidla do dvou let věku [3, 4].
KAZUISTIKA
Prezentovaný chlapec je z 2. fyziologické gravidity, narozený v 35. týdnu těhotenství sekcí pro dehiscenci v jizvě dělohy po předchozím operativním porodu. Poporodní adaptace byla komplikována pouze novorozeneckým ikterem z nezralosti s nutností dvoudenní fototerapie, v 11. dni byl propuštěn domů. Rodinná anamnéza byla bez zátěže.
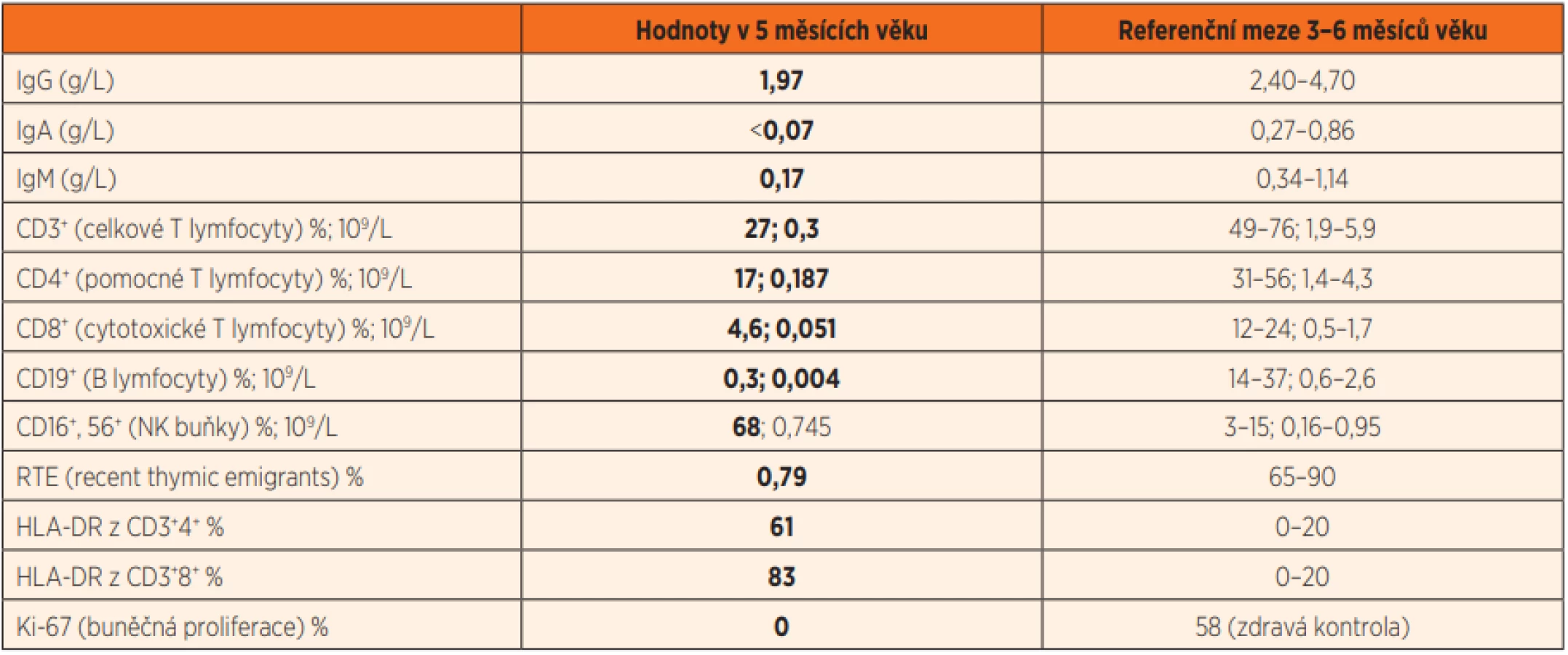
Ve 2 měsících věku byl jinak dobře prospívající kojenec krátce hospitalizován na Pediatrické klinice Thomayerovy nemocnice pro zahlenění, diskrétní konjuktivitidu a řídké stolice, vedlejším nálezem byla mírná dermatitida vzhledu ekzému. Laboratorně byla v diferenciálním krevním obraze patrna lymfopenie (8,5 %, abs. 0,58x109/L; norma 2,3–13,8x109/L), trombocytóza (717x109/L; norma 150–450x109/L) a eozinofilie (17,7 %, abs. 1,2x109/L; norma 0–0,7x109/L), CRP bylo 4,5 mg/L, výtěr z rekta s nálezem běžné flóry. Chlapcův klinický stav se po 3 dnech symptomatické terapie zlepšil natolik, že mohl být propuštěn, závěrem byla gastroenteritida nezjištěné etiologie. V dalším průběhu prospíval, dermatitida kompletně regredovala, neprodělal žádné další aparentní infekce, psychomotorický vývoj byl v normě, byl nekomplikovaně očkován dvěma dávkami hexavalentní vakcíny a jednou dávkou konjugované pneumokokové vakcíny. V 5. měsíci věku byl přijat na JIP Pediatrické kliniky pro den trvající kašel, dušnost, subfebrilie a poslechový nález odpovídající bronchiolitidě. Objektivnímu nálezu dominovala tachydyspnoe s difuzními chrůpky v inspiriu i exspiriu při normální saturaci krve kyslíkem (SpO2). Ve vstupní laboratoři byla opět patrná lymfopenie, zároveň trombocytóza, mírná eozinofílie a neutrofílie (tab. 1), CRP bylo 4,8 mg/L, na RTG plic byla snížená transparence obou plicních křídel (obr. 2a). PCR vyšetřením aspirátu z horních cest dýchacích byly prokázány rhinoviry.

Zahájili jsme symptomatickou léčbu, ale záhy si progredující dušnost a klesající SpO2 vyžádaly oxygenoterapii (potřeba systému vysokoprůtokové terapie), přechodně jsme pro obstrukční poslechový nález zařadili i inhalační bronchodilatancia a kortikosteroid. Na kontrolním RTG plic bylo při trvale nízké laboratorní zánětlivé aktivitě patrno zhoršení nálezu ve smyslu dalšího snížení transparence oboustranně, v pravém horním plicním laloku až s náznakem splývavé kresby (obr. 2b).

a) Snížená transparence levého plicního křídla a výraznější zastření pravého plicního křídla, zejména v pravém horním laloku (průkaz rhinovirů v nasofaryngu
metodou PCR)
b) Akcentace snížené transparence levého plicního
křídla a další výraznější zastření pravého plicního křídla s odstupem 10 dnů (průkaz Pneumocystis jirovecii
v nasofaryngu metodou PCR)
Fig. 2. Chest X-ray of 5-months-old RAG2 SCID patient.
a) Decreased transparency of the left lung and increased density of the right lung, particularly in right
upper lobe (rhinovirus PCR positivity in nasopharynx)
b) Accentuation of decreased transparency of the
left lung with progression of density of the right lung
after 10 days (Pneumocystis jirovecii PCR positivity in
nasopharynx)

Doplnili jsme echokardiografické vyšetření, které bylo s normálním nálezem a nasadili antibiotickou terapii klaritromycinem. Postupně docházelo k pozvolné úpravě stavu, nicméně protrahovaný průběh a opakované lymfopenie vyvolaly podezření na možnou imunopatologii. To se dále zvýšilo při následném záchytu genomu Pneumocystis jirovecii při opakovaném vyšetření sekretu z dýchacích cest (PCR). Prvotní imunologické vyšetření prokázalo výraznou T a B lymfopenii s normálním absolutním počtem NK buněk a hypogamaglobulinémii ve třídách IgA, IgM i IgG (tab. 2). Podrobnějším vyšetřením byla detekována nízká produkce TREC, nízký počet RTE a nulová schopnost proliferace lymfocytů po stimulaci phytohemaglutininem. Zároveň byla patrná vysoká aktivace přítomných cirkulujících T lymfocytů (dle povrchové exprese HLA DR znaku) (tab. 2) a jejich nápadná oligoklonalita (obr. 3). Maternofetální engraftment nebyl detekován.
Fig. 3. Graphic representation of individual T-lymphocyte clones
with complete T-cell receptor β (TRB) rearrangement of
a healthy newborn (top) compared to CD4+
T-lymphocytes
of RAG2 SCID patient (bottom). The rearrangement of TCR
gene segments via the process of V(D)J recombination creates a pool of unique, antigen-specific cell clones. In a healthy
person, a broad receptor repertoire diversity ensures the
ability to recognize a high number of antigens. A markedly
decreased T-lymphocyte repertoire diversity (oligoclonality)
is clearly shown in the patient with disturbed V(D)J recombination process due to RAG2 dysfunction. Each box represents
a unique clone (characterized by an amino acid sequence of
the CDR3 region of TRB-VDJ recombination) and its size represents its relative distribution amongst all identified clones.
The data for the repertoire analysis were generated using
next-generation sequencing [9].
![Grafické znázornění zastoupení jednotlivých klonů T lymfocytů s kompletní přestavbou T-lymfocytárního receptoru β
(TRB) u zdravého novorozence a CD4+
T lymfocytů RAG2
SCID pacienta. Přestavbou segmentů genu kódujícího
T-lymfocytární receptor procesem V(D)J rekombinace vznikají jedinečné, antigenně specifické buněčné klony. U zdravého člověka zajišťuje existence obrovského množství různých
klonů vysokou diverzitu imunitního repertoáru, neboli schopnost rozpoznat co největší počet antigenních struktur. U pacienta je patrná snížená diverzita repertoáru (oligoklonalita)
T lymfocytů v důsledku poruchy funkce RAG2 a neefektivního procesu V(D)J rekombinace. Každý obdélník reprezentuje
unikátní klon (charakterizovaný aminokyselinovou sekvencí
CDR3 oblasti kompletní TRB-VDJ přestavby) a jeho velikost
odpovídá relativnímu zastoupení mezi všemi identifikovanými klony. Analýza repertoáru byla provedena na datech
získaných metodou sekvenování nové generace (NGS) [9].<br>
Fig. 3. Graphic representation of individual T-lymphocyte clones
with complete T-cell receptor β (TRB) rearrangement of
a healthy newborn (top) compared to CD4+
T-lymphocytes
of RAG2 SCID patient (bottom). The rearrangement of TCR
gene segments via the process of V(D)J recombination creates a pool of unique, antigen-specific cell clones. In a healthy
person, a broad receptor repertoire diversity ensures the
ability to recognize a high number of antigens. A markedly
decreased T-lymphocyte repertoire diversity (oligoclonality)
is clearly shown in the patient with disturbed V(D)J recombination process due to RAG2 dysfunction. Each box represents
a unique clone (characterized by an amino acid sequence of
the CDR3 region of TRB-VDJ recombination) and its size represents its relative distribution amongst all identified clones.
The data for the repertoire analysis were generated using
next-generation sequencing [9].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image_pdf/bf966042a87c6ffde24a96fa5473f04f.jpeg)
Pacient byl ihned po vyslovení podezření na SCID přeložen na Kliniku dětské hematologie a onkologie FN Motol, kde byla při opakovaných vyšetřeních naměřena absolutní hodnota CD3+ T lymfocytů až 0,67x109/L a stanovena tak diagnóza atypického T–B–NK+ SCID (u typického SCID bez maternofetálního engraftmentu nepřesahuje počet T lymfocytů 0,3x109/L). Genetickým vyšetřením metodou Sangerova sekvenování bylo zjištěno, že pacient je složeným heterozygotem pro mutace v RAG2 genu (missense p.Arg73Cys; nonsense p.Glu404*), každý ze zdravých rodičů je přenašečem jedné z mutací, u nevlastního bratra maternální mutace nalezena nebyla (obr. 4).
Fig. 4. a) RAG2 genomic localization on short arm of chromosome
11 (arrow, credit: Genome Decoration Page/NCBI).
b) Familial segregation of the RAG2 mutations. The patient
(arrow) is a compound heterozygo3te for a missense
and a nonsense mutations in positions p.Arg73Cys and
p.Glu404*. His mother carries the p.Arg73Cys and father
the p.Glu404*. In the patient’s maternal half-brother
(II./2.) no variants in RAG2 were identified.

Na hematoonkologickém pracovišti byl chlapec od počátku hospitalizován v chráněném prostředí sterilního boxu transplantační jednotky. Byla zahájena terapie vysokodávkovaným trimetoprimem (20 mg/kg/den), cefepimem a micafunginem a pravidelná substituce i.v. imunoglobulinů. V nazofaryngeálním výtěru byly opakovaně prokázány silně pozitivní rhinoviry (PCR), nicméně klinický stav i RTG nález na plicích se postupně zlepšovaly. Chlapec byl po potvrzení diagnózy indikován k urgentní alogenní transplantaci kmenových buněk krvetvorby, která proběhla 2,5 měsíce od stanovení diagnózy, tj. ve věku 7,5 měsíců, po myeloablativním přípravném režimu štěpem od vhodné nepříbuzné dárkyně z řeckého registru. Přihojení granulocytů bylo dokumentováno 16. den po transplantaci.
Potransplantační průběh byl komplikován mukositidou, febrilní neutropenií, engraftment syndromem a následně mírnou kožní reakcí štěpu proti hostiteli s dobrou odpovědí na kortikoterapii. Chlapec byl propuštěn do domácí péče v dobrém klinickém stavu na standardní potransplantační perorální imunosupresivní, antimikrobiální a podpůrné terapii 38 dní po transplantaci.
DISKUSE
Většina forem SCID se projeví velmi časnou náchylností k infekcím a jejich závažným průběhem. Existují ale i mírnější fenotypy. Příkladem jsou hypomorfní mutace v recombination–activating genes 1 a 2 (RAG1/RAG2). Jedná se o enzymy, které se účastní přeskupování genových segmentů, tzv. V(D)J rekombinace, v procesu tvorby antigenních receptorů T a B lymfocytů [1]. RAG1/RAG2 mutace mohou mít široké spektrum projevů – od klasického SCID, přes Omennův syndrom, atypický SCID s mírnějšími projevy, až po obraz převážně protilátkového imunodeficitu typu CVID (Common Variable Immunodeficiency) [6].
V případě našeho pacienta se jednalo o atypický SCID na základě hypomorfního genotypu, který se odrazil ve vyšším zastoupení autologních T lymfocytů, než nacházíme u typického SCID (do 0,3x109/L). Věk první závažné infekce v 5. měsíci spadá do období, kdy se mohl významněji uplatnit také protilátkový deficit, neboť v tomto období tzv. fyziologické hypogamaglobulinémie odeznívá ochranný vliv transplacentárně přenesených maternálních protilátek. Zajímavé je, že přes vysokou aktivaci cirkulujících lymfocytů neměl pacient významnější projevy autoreaktivity proti vlastním tkáním, což je u atypického SCID častou komplikací.
Diagnóza SCID patří mezi pediatrické urgentní stavy a jako taková by měla být stanovena co nejdříve z důvodu nutnosti rychlého managementu a co nejčasnějšího provedení transplantace kmenových buněk krvetvorby. S rostoucím věkem, a tudíž i s přibývajícími infekčními i neinfekčními komplikacemi, úspěšnost transplantace klesá [4, 7, 8]. Celosvětově je pětileté přežití pacientů transplantovaných do 3,5 měsíce věku souhrnně 80–95% [7]. Na Klinice dětské hematologie a onkologie FN Motol bylo do března 2020 transplantováno pro SCID celkem 23 dětí, medián věku v době transplantace byl 6,7 měsíců (1,5–58 měsíců) a celkové přežití 82 %.
Výhodu včasné diagnózy, ale zároveň i nové otázky, přineslo nedávné zavedení celoplošného novorozeneckého screeningu SCID pomocí TREC/KREC esejí v některých zemích světa (např. USA, Norsko, Švýcarsko, Izrael a další). Nízké hodnoty TREC mohou odhalit i jiné syndromy se závažnou poruchou T lymfocytů – např. kompletní DiGeorgeův syndrom či ataxia teleangiectasia, mohou být nízké také u nedonošených dětí narozených před 37. týdnem gestace či u dětí matek léčených imunosupresivy [4]. Od zahájení screeningu je pozorován nárůst incidence SCID, což svědčí o jeho dosavadním poddiagnostikování. Dilema však vyvolávají screeningové záchyty dětí s T lymfopeniemi bez následně prokázané SCID mutace, u nichž nelze předvídat, jak závažný bude klinický fenotyp. V těchto případech musí být management péče o pacienta zcela individuální včetně indikace k transplantaci kmenových buněk krvetvorby. Znalost konkrétní mutace a její rodinné segregace také umožňuje poskytnout rodinám genetické poradenství vč. prenatální diagnostiky. V našem konkrétním případě autosomálně recesivního RAG2 SCID je riziko pro další potomky páru 25%.
Fakt, že molekulárně genetická diagnóza byla u našeho pacienta stanovena za necelých 72 hodin od vyslovení klinického podezření, je důkazem špičkové úrovně vyšetřovacích algoritmů a vynikající národní multioborové koordinace. Přes všechna komplexní vyšetření bylo prvotním klíčovým momentem diagnostiky správné vyhodnocení krevního obrazu, kde byla opakovaně zaznamenávána závažná lymfopenie.
ZÁVĚR
Cílem tohoto sdělení je připomenout vrozené poruchy imunity jako vzácnou, ale možnou příčinu závažněji či netypicky probíhajících infekcí u dětí a upozornit na dostupnost rychlého screeningového vyšetření TREC/KREC při podezření na závažné buněčné a kombinované defekty. V České republice se toto vyšetření provádí v laboratoři CLIP při Klinice dětské hematologie a onkologie FN Motol v Praze a v genetické laboratoři Centra kardiovaskulární a transplantační chirurgie v Brně. V případě podezření na vrozenou poruchu imunity je vhodné oslovit klinického imunologa, který se věnuje problematice vrozených poruch imunity a posoudí indikaci, rozsah a urgenci imunologického a genetického vyšetření; jmenný seznam podle pracovišť v ČR je dostupný na www.imunodeficience.cz.
Podpořeno granty AZV NV18-05-00162 a GAUK 954218.
Korespondující autor:
MUDr. Markéta Bloomfield
Pediatrická klinika 1. LF UK
a Thomayerovy nemocnice
Vídeňská 800
140 59 Praha 4
e-mail: marketa.bloomfield@ftn.cz
Sources
1. Kumrah R, et al. Genetics of severe combined immunodeficiency. Genes Dis 2020; 7 (1): 52–61.
2. Sponzilli I, Notarangelo LD. Severe combined immunodeficiency (SCID): from molecular basis to clinical management. Acta Biomed 2011; 82 (1): 5–13.
3. Wahlstrom J, et al. Transplacental maternal engraftment and posttransplantation graft-versus-host disease in children with severe combined immunodeficiency. J Allergy Clin Immunol 2017; 139 (2): 628–633.
4. Formánková R, et al. Problematika včasného stanovení diagnózy těžkého kombinovaného imunodeficitu. Čes-slov Pediat 2007; 62 (1): 5–15.
5. Šedivá A, et al. Závažné primární imunodeficience a možnosti jejich časné diagnostiky, uvedení testu TREC/KREC. Čes-slov Pediat 2019; 74 (3): 182–187.
6. Schuetz C, et al. An immunodeficiency disease with RAG mutations and granulomas. N Engl J Med 2008; 358 : 2030.
7. Heimalla J, Cowanb MJ. Long term outcomes of severe combined immunodeficiency: therapy implications. Expert Rev Clin Immunol 2017; 13 (11): 1029–1040.
8. Wahlstrom JT, et al. Hematopoietic stem cell transplantation for severe combined immunodeficiency. Curr Pediatr Rep 2015; 3 (1): 1–10.
9. Brüggemann M, et al. Standardized next-generation sequencing of immunoglobulin and T-cell receptor gene recombinations for MRD marker identification in acute lymphoblastic leukaemia; a EuroClonality-NGS validation study. Leukemia 2019; 33 (9): 2241–2253.
Labels
Neonatology Paediatrics General practitioner for children and adolescentsArticle was published in
Czech-Slovak Pediatrics

2020 Issue 6
- What Effect Can Be Expected from Limosilactobacillus reuteri in Mucositis and Peri-Implantitis?
- The Importance of Limosilactobacillus reuteri in Administration to Diabetics with Gingivitis
Most read in this issue
- Extrémní hypokalémie u familiární periodické paralýzy – kazuistika
- Škrkavka jako vzácná příčina nočních bolestí břicha u dětí – kazuistika
- Mozkový absces u dětí – souhrnný článek a dvě kazuistiky
- Vaskulitida jako vzácný projev infekce Sarcoptes scabiei – kazuistika