
Myokarditidy a kardiomyopatie
Myocarditis and cardiomyopathy
The review article summarizes the most important information about myocarditis and cardiomyopathies in children. It focuses on their aetiology, clinical manifestations, diagnosis and treatment and may be used as a background material for preparation for the qualification exam in paediatrics.
Keywords:
paediatrics – myocarditis – heart failure – cardiomyopathy
Authors:
Pavlíček Jan 1; Rücklová Kristina 2; David Jan 3
Authors‘ workplace:
Klinika dětského lékařství, Lékařská fakulta Ostravské univerzity a Fakultní nemocnice Ostrava
1; Klinika dětí a dorostu, 3. lékařská fakulta a Fakultní nemocnice Královské Vinohrady, Praha
2; Pediatrická klinika, 2. lékařská fakulta a Fakultní nemocnice v Motole, Praha
3
Published in:
Čes-slov Pediat 2023; 78 (2): 110-121.
Category:
Chapters for Specialization in Pediatrics
doi:
https://doi.org/10.55095/CSPediatrie2023/016
Overview
Přehledový článek shrnuje nejdůležitější informace o myokarditidách a kardiomyopatiích u dětí, věnuje se jejich etiologii, klinickým projevům, diagnostice a léčbě. Slouží jako podkladový materiál k přípravě k atestační zkoušce z pediatrie.
Klíčová slova:
srdeční selhání – pediatrie – myokarditida – kardiomyopatie
1. MYOKARDITIDY
Myokarditida je zánětlivé onemocnění srdce definované histologickým či imunohistochemickým nálezem, případně specifickým obrazem na magnetické rezonanci. Incidence u dětí se pohybuje okolo 1,95 / 100 000 dětí do 15 let za rok s převahou chlapců (77 %).(1) Stanovení diagnózy vždy vyžaduje komplexní vyhodnocení anamnézy, klinických příznaků, laboratorních parametrů, elektrokardiografických (EKG) a echokardiografických nálezů. Vyšetřovací modality je často nutné v kontextu klinického vývoje opakovat a sledovat jejich dynamiku. Výrazným posunem v diagnostice myokarditidy bylo zlepšení technických parametrů a interpretace nálezů srdeční magnetické rezonance (cardiac magnetic resonance, CMR), která umožňuje potvrdit myokarditidu i bez endomyokardiální biopsie (EMB).(2)
Etiologie
Etiologii myokarditidy u dětí lze rozdělit na infekční a neinfekční (tab. 1).(2) Mezi infekčními příčinami v Evropě dominují viry. V minulosti převažovaly adenoviry a enteroviry (Coxsackieviry), v současnosti spíše parvovirus B19 a lidský herpes virus 6, recentně také nový koronavirus (SARS-CoV-2). Myokarditida neinfekční etiologie je většinou součástí vaskulitid, především Kawasakiho nemoci nebo jiných autoimunitních onemocnění.(3) Revmatická horečka je i v současné době příčinou nejčastějšího získaného onemocnění srdce s myokarditidou v rozvojových zemích.(4) Multisystémový zánět u dětí po prodělaném covidu-19, tzv. PIMS – TS (pediatric inflammatory multisystem syndrome temporally associated with SARS-CoV-2), může být asociován s myokarditidou, dle CMR nálezů přibližně v 18 % případů.( 5)

Patogeneze
Předpokládá se, že vznik myokarditidy je zpravidla výsledkem kombinace působení vnějšího faktoru u geneticky predisponovaného jedince a autoimunitní odpovědi.(6) U virové myokarditidy vstupuje virus díky virus-specifickým receptorům do kardiomyocytů, které poškozuje. Napadené kardiomyocyty aktivují systém vrozené a získané imunity, převážně CD4 T lymfocyty. CD4 T lymfocyty produkují prozánětlivé mediátory, které stimulují srdeční fibroblasty k produkci GM-CSF (granulocyte macrophage colony-stimulating factor). GM-CSF aktivuje kostní dřeň, ze které se uvolňují další zánětlivé buňky, především aktivované monocyty. Ty pak infiltrují srdce, poškozují ho a významně se podílí na progresi do dilatační kardiomyopatie.(7)
Klinický obraz
Myokarditida může probíhat jako fulminantní, akutní nebo chronická forma. Diagnostiku komplikuje variabilní klinický obraz s nespecifickými příznaky, které mohou imitovat respirační nebo gastrointestinální onemocnění. Průběh onemocnění může být rozmanitý, od nenápadné tachykardie při virové infekci až po známky srdečního selhávání, kardiogenní šok, nebo dokonce náhlé úmrtí (tab. 2).(2,8) Klinické projevy rovněž závisí na věku dítěte. U novorozenců a kojenců jsou to nespecifické příznaky infekce (subfebrilní a febrilní teploty, apatie, potíže s krmením, zvýšené pocení). U větších dětí může na myokarditidu upozornit bolest na hrudníku, jinak nevysvětlitelná tachykardie nebo nově vzniklá arytmie.
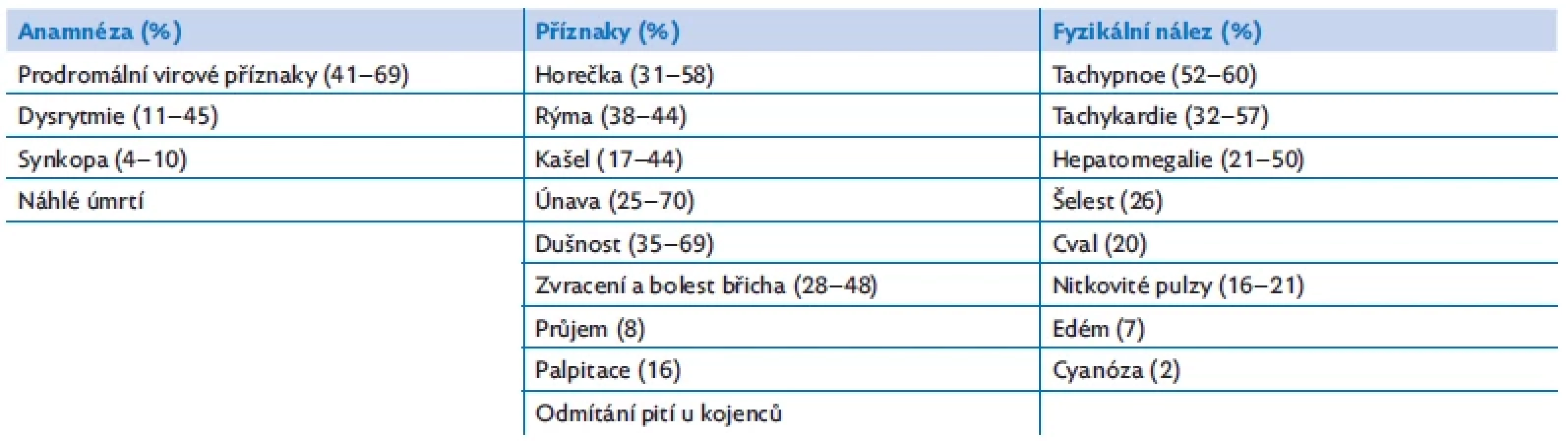
Diagnostika
U myokarditid zpravidla dochází k elevaci kardiálních enzymů. V současné době běžně používaný a citlivý je tzv. vysoce senzitivní (high sensitive, hs) troponin. Jeho zvýšení není specifické, stoupá u jakéhokoliv poškození myokardu, nejen zánětem, ale i při kontuzi nebo ischemii srdce. Jeho hladina nekoreluje s prognózou myokarditidy.(2) Dalším často využívaným parametrem je N-terminální fragment mozkového natriuretického peptidu (NT-proBNP), který koreluje se závažností srdečního selhání a odráží objemovou zátěž srdečních oddílů. Při systémových zánětech typu Kawasakiho nemoci nebo PIMS-TS ale stoupá i při zachované myokardiální funkci. Při podezření na myokarditidu dále vyšetřujeme parametry zánětu, renální a jaterní funkce. Dle dostupnosti vyšetření je při podezření na virovou myokarditidu snahou odhalit infekční agens metodou polymerázové řetězové reakce (PCR) z nazofaryngeálních stěrů, z krve nebo ze stolice. Vyšetření imunologických parametrů je indikováno cíleně při klinickém podezření na konkrétní autoimunitní systémové onemocnění.(2)
Při myokarditidě mohou být přítomny nejrůznější změny na EKG. Typicky se popisuje nízká voltáž, patologické Q vlny, změny ST-T úseků (obr. 1A), nově vzniklé tachyarytmie, komorové a síňové ektopie, nitrokomorové nebo atrioventrikulární (AV) blokády, případně sinusová tachykardie. EKG se v průběhu onemocnění mění a je třeba ho provádět opakovaně.

Echokardiograficky můžeme zobrazit ztluštění stěny myokardu edémem. Může být patrná systolická nebo diastolická dysfunkce komor, případně regionální porucha kinetiky myokardu. Dalším možným nálezem u myokarditidy je perikardiální výpotek, nově vzniklá mitrální nedomykavost či intrakardiální trombus.
CMR je schopna detekovat poškození tkáně ve smyslu edému, hyperemie a fibróz. Ke kvalitnímu posouzení přítomnosti myokarditidy a eventuálnímu odlišení od nezánětlivé kardiomyopatie je potřeba použít kombinaci různých sekvencí (obr. 2). Časným sycením gadoliniem (early gadolinium enhancement, EGE) v T1 vážených sekvencích lze detekovat intracelulární edém, hyperemii a kapilární únik, zatímco pozdní sycení gadoliniem (late gadolinium enhancement, LGE) identifikuje změny ireverzibilní. Velikost srdce dětí a rychlá srdeční frekvence jsou faktory, které ovlivňují kvalitu snímků. Navíc EGE a LGE vyžadují podání kontrastní látky a u menších dětí může být k provedení CMR nezbytná celková anestezie.(8,9) Na rozdíl od dospělých pacientů bývá u dětí akutní koronární syndrom raritní, proto koronarografii či CT koronarografii indikujeme u bolestí na hrudi u dětí zřídka. Toto vyšetření je možné indikovat u pacientů s infarkt-like myokarditidou a při bezprostřední nedostupnosti MR srdce.

Endomyokardiální biopsie je invazivní metoda, která je spojena s rizikem poranění srdce, proto je indikována pouze tehdy, když její výsledek může ovlivnit terapii a prognózu dítěte, a vždy je nutné balancovat očekáváný přínos proti potenciálním rizikům u každého pacienta. Součástí vyšetření bioptického vzorku musí být nejen histologie (Dallaská nebo Marburská kritéria), ale i imunohistochemické vyšetření a detekce virového genomu metodou PCR. Jednoznačnou indikací k EMB je podezření na eozinofilní či obrovskobuněčnou myokarditidu na základě klinického průběhu či laboratorních nálezů.(8)
Terapie
Léčba myokarditidy je zaměřena na zvládnutí komplikací, především srdečního selhání a arytmií. Přijetí k hospitalizaci je vždy indikováno, pacient s myokarditidou by měl ležet na lůžku s možností trvalé monitorace vitálních funkcí.
V rámci terapie srdečního selhání sledujeme bilanci tekutin, při myokardiální dysfunkci se zpravidla snažíme o jejich restrikci. U pacientů v klinicky dobrém stavu a s echokardiograficky mírně sníženou ejekční frakcí je indikována léčba inhibitory angiotensin konvertujícího enzymu (ACEI), případně betablokátory nebo digoxinem. Při závažnějším srdečním selhání s výraznější klinickou symptomatologií volíme převážně intravenózně podané léky a kompletní invazivní zajištění pacienta. Při známkách městnání (otoky, hepatomegalie, snížená kolapsibilita dolní duté žíly, přítomnost výpotků) podáváme diuretika, nejčastěji furosemid se suplementací draslíku. Při nízkém srdečním výdeji (chladná periferie, prodloužený kapilární návrat, hypotenze, oligurie) je často nutná inotropní podpora katecholaminy a tekutinová resuscitace. Inhibitor fosfodiesterázy milrinon má inotropní a periferně vazodilatační efekt vedoucí ke zvýšení tepového objemu a srdečního výdeje a současně ke snížení plnicích tlaků v plicním i systémovém řečišti. Kvůli jeho převažujícímu vazodilatačnímu efektu ho však nemusí akutně selhávající pacient tlakově tolerovat. Před jeho podáním je také nutno doplnit pacientovi cirkulující objem. Kalciový senzitizér levosimendan používáme u nejtěžších případů. Pacienti s myokarditidou a fulminantním průběhem mohou vyžadovat mechanickou podporu. Po odeznění akutní fáze onemocnění a při trvání myokardiální dysfunkce je indikována perorální léčba srdečního selhání, především ACEI, betablokátory, v případě městnání diuretika.(2,8) Obecně platí, že je nevhodné u pacientů s akutní myokarditidou podávat ibuprofen, který může potencovat myokardiální nekrózu.(10)
Tachyarytmie působící nestabilitu oběhu (supraventrikulární či komorová tachykardie nebo fibrilace) musí být vyřešena urgentně elektrickou kardioverzí nebo defibrilací. Při recidivující komorové tachykardii nebo závažné komorové ektopii je indikována antiarytmická léčba amiodaronem nitrožilně. K dalším možným lékům u arytmií při myokarditidě patří například betablokátory nebo lidokain. Zahájení antiarytmické léčby by mělo být vždy s maximální opatrností a po konzultaci s kardiologem. Většina antiarytmik působí na myokard negativně inotropně a mohou způsobit nestabilitu oběhu až jeho zhroucení. V případě výskytu závažné bradyarytmie, většinou na podkladě nově vzniklé pokročilé atrioventrikulární blokády v zánětlivém terénu, je indikována stimulace transvenózně dočasně zavedenou elektrodou.
Antivirová léčba je doporučena při známkách probíhající virové infekce a pozitivitě PCR. Antivirotika jsou k dispozici pouze pro několik typů konkrétních virů. Podání intravenózních imunoglobulinů (IVIG) je často indikováno u dětí s akutní myokarditidou pro příznivý efekt na zlepšení srdeční funkce.(2,8) Doporučení pro jejich použití ale nejsou jednotná. Kortikosteroidy jako imunosupresivní terapie jsou jednoznačně indikovány u dětí s obrovskobuněčnou nebo eosinofilní myokarditidou a v rámci léčby systémových autoimunitních onemocnění.(2) Další možné použití imunosuprese je u lymfocytární myokarditidy, výsledky jsou ale nejednoznačné a zahájení této léčby by mělo být podmíněno průkazem zánětu v EMB a zároveň negativním virovém PCR. Kortikoidy mohou být použity v kombinaci s jinými imunosupresivy (cyklosporin A, azathioprin).
Návrh vyšetření a diagnostický a terapeutický algoritmus s ohledem na závažnost klinického stavu shrnují tabulka 3 a obrázek 3.


Sledování a prognóza
Přežití u dětí s myokarditidou se pohybuje mezi 70–100 %, dvě třetiny dětí se zcela uzdraví, nejvyšší mortalitu mají novorozenci.( 1,2) Fulminantní nebo akutní forma ohrožuje pacienta na životě v úvodu onemocnění, při dobrém průběhu a přežití akutní fáze zánět rychle ustupuje a dlouhodobá prognóza je dobrá. Naopak chronický zánět v mírnější formě může být dlouhodobě asymptomatický, je ale možnou příčinou remodelace myokardu a rizikem progrese do chronického srdečního selhání.
Děti po prodělané myokarditidě zpravidla sledujeme do normalizace srdeční funkce a vymizení arytmií, minimálně však jeden rok po akutním onemocnění. Neomezený sport je povolen nejdříve za tři až šest měsíců za předpokladu normálního echokardiografického nálezu, nízkých kardiomarkerů a absence závažných arytmií v klidu i v zátěži.
2. KARDIOMYOPATIE
Kardiomyopatie (KMP) jsou u dětí vzácné, přinášejí ale významné riziko morbidity a mortality.(11) KMP jsou heterogenní skupina onemocnění asociovaná s poruchou struktury a funkce srdečního svalu při absenci koronárního onemocnění, systémové hypertenze nebo vrozené srdeční vady. Morfologická a funkční klasifikace dělí KMP na dilatační, hypertrofické, restriktivní, arytmogenní, nekompaktní a neklasifikované. U dětí se nezřídka vyskytuje i takzvaný smíšený fenotyp s rysy dvou různých KMP (např. kombinace hypertrofické a nekompaktní). Incidence KMP se pohybuje okolo 1,3 případu na 100 000 dětí a rok, vyšší záchyt je v kojeneckém období. Nejčastější je forma dilatační (50 %) a hypertrofická (35–50 %), zatímco ostatní subtypy jsou mnohem vzácnější, např. restriktivní tvoří méně než 5 % případů.( 12)
Stanovení diagnózy kardiomyopatie a posouzení její závažnosti je založeno na zhodnocení klinických projevů, echokardiografie s posouzením srdeční struktury, systolické a diastolické funkce a EKG, případně pozdních komorových potenciálů. V poslední době se také stále více využívá CMR. Srdeční katetrizace je u dětí indikována především k posouzení plicní hypertenze před ev. transplantací, výjimečně k vyloučení koronární anomálie jako příčiny dilatace a dysfunkce myokardu.
V souvislosti s větší dostupností a kvalitou genetického testování je v dnešní době v mnoha případech odhalena genetická příčina KMP. Testování většinou probíhá metodou NGS (next generation sequencing) s použitím panelu genů asociovaných s KMP. KMP jsou typické neúplnou expresivitou a penetrací, kdy ne všichni nositelé stejné patogenní varianty onemocní anebo se u nich onemocnění projeví s odlišnou závažností.(13) Každý z uvedených typů KMP může být způsoben patogenními variantami v mnoha genech kódujících součásti srdečního svalu. Zároveň porucha jednoho genu může vézt k obrazu různých forem KMP. Kromě genetických příčin se může etiologicky uplatnit i negenetická etiologie. Zároveň ale genetická predispozice může fenotyp i u převážně negenetických příčin modifikovat.
Specifikum kardiomyopatií u dětí oproti dospělým spočívá především v tom, že jsou relativně často součástí systémových onemocnění, např. vrozených poruch metabolismu, neuromuskulárních onemocnění nebo malformačních syndromů. Mohou se tedy projevit i extrakardiálními příznaky a v některých případech existuje specifická léčba základního onemocnění. Z tohoto hlediska je včasné odhalení příčiny u dětí enormně důležité.
DILATAČNÍ KARDIOMYOPATIE
Dilatační kardiomyopatie (DKMP) je definována přítomností dilatované levé komory (obr. 4) nebo obou komor se sníženou systolickou funkcí při absenci hemodynamické příčiny (srdeční vady, ischemie, sepse).(14) Pro DKMP existuje široká škála příčin a asociací, které lze rozdělit na genetické a negenetické (tab. 4).(12,14)


Etiologie a patogeneze
Genetické DKMP
Na většině DKMP se do značné míry podílí genetická predispozice. Familiární výskyt (postižení několika členů rodiny) lze odhalit asi ve 30–50 % případů, dědičnost je nejčastěji autozomálně dominantní. Mezi příčinné geny patří geny kódující součásti cytoskeletu, Z-disk, sarkomerické proteiny (nejčastěji gen pro titin, TTN) a další.(15) V některých případech nás mohou určité fenotypické znaky přivést k podezření na konkrétní genetickou příčinu. Např. DKMP způsobená patogenní variantou v genu pro lamin (LMNA, laminopatie) se typicky projevuje dilatací srdce v kombinaci s poruchami elektrického vedení, závažnými arytmiemi a vyšším rizikem náhlé srdeční smrti. DKMP mohou existovat izolovaně, ale i jako součást systémových onemocnění, např. neuromuskulárních. Typickým příkladem je Duchenneova/Beckerova muskulární dystrofie, způsobená patogenní variantou genu pro dystrofin (DMD). Onemocnění významně postihuje kosterní svalstvo. Dědičnost je X-vázaná, a proto jsou postiženi především chlapci. DKMP je hlavní příčinou úmrtí těchto dětí. DKMP může být asociována i s většinou dalších dědičných svalových dystrofií a myopatií. Jako součást péče o děti s nervosvalovými onemocněními jsou indikovány i pravidelné kardiologické kontroly.
DKMP může být součástí vrozených poruch metabolismu, především poruch 𝛃-oxidace mastných kyselin s dlouhým řetězcem. Tyto se vyznačují autozomálně recesivní dědičností. Kromě kardiomyopatie se projevují např. retinopatií, periferní polyneuropatií, hepatopatií, opakovanými atakami rhabdomyolýzy anebo život ohrožujícími Reye-like epizodami s hyperamonemií a poruchou vědomí. Dalším příkladem mohou být mitochondriální choroby, v některých případech způsobené poruchami mitochondriální DNA s maternální dědičností. Postižení srdce u mitochondriálních poruch může probíhat pod obrazem jak dilatační, tak hypertrofické kardiomyopatie, charakteristické jsou poruchy nitrokomorového vedení a preexcitace na EKG.(16)
Negenetické DKMP
Mezi negenetické příčiny DKMP patří prodělaná myokarditida, vzácně deficit některých vitaminů a stopových prvků (vitamin B1, selen), vliv toxinů (některá cytostatika, především anthracykliny), ev. dlouhodobá tachykardie (tachycardia - induced cardiomyopathy) (tab. 4).
Klinický obraz a diagnostika
Příznaky jsou důsledkem vývoje srdečního selhávání, méně často arytmií. Menší děti se projevují potížemi při krmení, špatným prospíváním, u starších jedinců dochází k rozvoji horší tolerance námahy, dušnosti, únavnosti, palpitací. Mezi základní klinické příznaky patří tachypnoe, tachykardie, bledost, alterace periferních pulzací. S progresí selhávání může dojít k vývoji cyanózy, šoku, hypotenzi, hepatomegalii, otokům. Srdeční šelesty můžeme detekovat při regurgitacích chlopní, může být přítomný cval. Chrůpky na plicích slyšíme při vývoji plicního edému. V důsledku pomalé progrese srdeční dysfunkce a značné adaptace organismu je někdy těžká systolická dysfunkce paradoxně dlouho klinicky velmi dobře tolerována.
EKG je většinou nespecifické, nejčastěji se změnami ST úseků (obr. 1B), rentgen popisuje dilataci srdečního stínu a plicní městnání. Echokardiografie identifikuje a kvantifikuje dilataci srdečních komor, síní a poruchu systolické funkce. Dopplerovská měření zobrazí případnou regurgitaci mitrální chlopně. Speciální echokardiografické techniky, např. strain, mohou v některých případech odhalit dyssynchronii srdeční kontrakce. Srdeční katetrizace se u dětí indikuje především k posouzení plicní hypertenze před ev. transplantací srdce a endomyokardiální biopsie k rozlišení myokarditidy od kardiomyopatie. V současné době lze k tomuto účelu využít méně invazivní CMR.
Terapie a prognóza
U genetických forem je terapie většinou symptomatická. Léčebně ovlivňujeme srdeční selhávání (katecholaminy, kardiotonika, diuretika, ACE inhibitory, betablokátory). Standardní moderní léčbu chronického srdečního selhávání u dětí s dávkami základních léků uvádí tabulka 5. U významné dysfunkce je pro riziko trombóz a embolizací možno doplnit antiagregaci (anopyrin). Při výskytu nebo riziku významných arytmií (komorová tachykardie a fibrilace) indikujeme elektrofyziologické vyšetření a implantaci kardioverteru - defibrilátoru, dysfunkci levé komory je možno v případě přítomnosti dyssynchronie ovlivnit resynchronizační terapií. Při refrakterním těžkém srdečním selhávání je možnost zavedení mechanické podpory jako přemostění k transplantaci srdce.

Pro léčbu kardiálního postižení v rámci neuromuskulárních onemocnění jsou k dispozici doporučení Americké kardiologické společnosti.(17) Specifická terapie přichází v úvahu např. u Duchenneovy muskulární dystrofie, kdy je u pacientů s bodovými nonsense mutacemi k dispozici ataluren, měnící genetickou informaci. Progresi onemocnění dále zpomalují kortikoidy. Z hlediska prevence nebo zpomalení progrese kardiálního postižení je doporučeno od 10 let preventivní podávání ACE inhibitorů. U poruch β-oxidace mastných kyselin s dlouhým řetězcem připadá v úvahu dietoterapie s omezením tuků s dlouhým řetězcem, suplementace karbohydrátů a tuků se středně dlouhým řetězcem.
Prognóza DKMP u dětí není dobrá. Roční přežívání je do 70 %, pětileté přežívání do 60 %.(18) Vyšší mortalita je v kojeneckém a batolecím věku. Nejčastější příčinou úmrtí je náhlá smrt. Vyšší mortalitu mají děti se symptomatickým srdečním selháním při stanovení diagnózy DKMP.
HYPERTROFICKÁ KARDIOMYOPATIE
Hypertrofická kardiomyopatie (HKMP) je definována přítomností hypertrofovaného myokardu levé a/nebo pravé komory (obr. 4B) při absenci hemodynamické příčiny (srdeční vada, systémová hypertenze, fyzická zátěž – atletické srdce).(12) HKMP je také velmi heterogenní skupina onemocnění, genetická příčina se předpokládá v naprosté většině případů. HKMP u dětí se asi ve 25 % spojuje s vrozenými poruchami metabolismu, nervosvalovými poruchami a malformačními syndromy.(19) Hlavní příčiny uvádí tabulka 6.(20)

Etiologie a patogeneze
Genetické HKMP s izolovaným postižením srdce
Patogenní varianty v genech pro sarkomerické proteiny jsou dominantní příčinou HKMP s izolovaným postižením srdce.(21) Dědičnost je převážně autozomálně dominantní, u poloviny případů je identifikován familiární výskyt, u ostatních jde pravděpodobně o patogenní varianty de novo. Nejčastější kauzální geny kódují těžký řetězec β-myosinu (MYH7), myosin-binding protein C (MYBPC3), srdeční troponin T (TNNT2), srdeční troponin I (TNNI3), α-tropomyosin (TPM1), případně proteiny Z-disku a proteiny ovlivňující vápníkový metabolismus. Výsledkem patogenních variant v uvedených genech je dezorganizace myofibril, zvětšení kardiomyocytů a vývoj fibrózy, dále kompenzatorní dilatace koronárních arterií. Relativní koronární insuficience při významné myokardiální hypertrofii vede k ischemickým klinickým projevům. Pro sarkomerické HKMP je typická asymetrická lokální hypertrofie komorového septa.
Genetické HKMP v rámci multisystémového onemocnění
U těchto onemocnění je HKMP jedním z příznaků základního onemocnění. Jedná se o některé dědičné poruchy metabolismu, malformační syndromy (RASopatie) a neuromuskulární onemocnění typu Friedreichovy ataxie.
Mezi dědičné poruchy metabolismu typicky asociované s HKMP patří střádavé choroby. Známým příkladem je autozomálně recesivní glykogenóza typu II (Pompeho choroba) charakterizovaná hypotonií a těžkou hypertrofickou kardiomyopatií s manifestací v kojeneckém věku, dále X-vázaná Danonova choroba asociovaná s rychle progredující HKMP, která často přechází do dilatace se systolickou dysfunkcí. Těžká HKMP je také součástí patogenních variant v genu PRKAG2, které vedou k akumulaci glykogenu a amylopektinu v kardiomyocytech. Akumulace glykosfingolipidů v lysozomech kardiomyocytů, chlopenních fibroblastů, neuronů a endotelu je příčinou X-vázané Fabryho nemoci. Onemocnění se manifestuje většinou v adolescenci pálivou bolestí nohou a rukou, kožními angiokeratomy, zákalem rohovky, bolestmi hlavy, v rané dospělosti se pak objevuje hypertrofie myokardu, postižení mitrální chlopně a někdy předčasné koronární příhody. Pro všechna uvedená střádavá onemocnění je kromě HKMP typická preexcitace/pseudo-preexcitace nebo jiné poruchy nitrokomorového vedení na EKG. Podobnou kombinaci HKMP a poruch vedení na EKG vídáme i u mitochondriálních onemocnění včetně MELAS, MERRF a deficitu proteinu TMEM 70. Mukopolysacharidózy způsobují v důsledku hromadění glykosaminoglykanů také ztluštění myokardu a výrazně postihují i aortální a mitrální chlopeň s následnou insuficiencí.(16)
Hypertrofická kardiomyopatie u pacientů s RASopatiemi může být velmi závažná, manifestuje se už v kojeneckém věku a rychle progreduje. Pouze 22 % těchto pacientů přežívá první rok života. Typickým příkladem je syndrom Noonanové, který se klasicky kromě kardiomyopatie projevuje stenózou plicnice, poruchou lymfatických cév, poruchami koagulace, dysmorfickými rysy a malým vzrůstem.( 14)
Negenetické HKMP
Mezi negenetické HKMP můžeme zařadit HKMP v rámci některých endokrinopatií (tab. 6).
Klinický obraz a diagnostika
Klinický obraz závisí na etiologii HKMP. Metabolické nebo syndromické HKMP jsou diagnostikovány většinou v časném dětství, prvním projevem může být srdeční šelest nebo i projevy městnání při srdečním selhávání (tachypnoe, dyspnoe, potíže při krmení, pocení, neprospívání). U starších dětí bývají prvními symptomy bolest na hrudi, palpitace, arytmie, synkopy a netolerance fyzické námahy při diastolické dysfunkci.(20) HKMP může být diagnostikována náhodně pro šelest nebo EKG abnormality při preventivních vyšetřeních provedených z jiných důvodů. Prvním projevem HKMP může být i náhlá smrt.
EKG je u HKMP velmi citlivé a specifické, detekujeme známky hypertrofie levé komory (vysoké voltáže nad levou komorou), známky její zátěže (negativní T vlny v levostranných hrudních svodech) a prohloubené kmity Q (obr. 1C). Z důvodu rizika závažných arytmií je stěžejní pravidelné EKG holterovské monitorování. Echokardiografie stanoví diagnózu, identifikuje velikost a lokalizaci hypertrofie a případně obstrukci výtokového traktu levé komory. Systolická funkce většinou není alterována, kontraktilita může být zvýšená. Patologická je zpravidla poddajnost levé komory a její diastolická funkce.
U HKMP vždy navrhujeme genetické došetření, v klinické rozvaze je nutno posoudit rodinnou historii (typ dědičnosti) a přítomnost možných fenotypových projevů jednotlivých genetických syndromů. Dále se zaměřujeme na neuromuskulární symptomatologii (ataxie, myotonie, hypotonie). Také zvažujeme přítomnost příznaků dědičných poruch metabolismu (encefalopatie, hepatosplenomegalie, metabolická acidóza s hyperlaktacidemií, hypoglykemie, poruchy vedení typu preexcitace, ev. pseudo-preexcitace apod.) a indikujeme patřičná metabolická vyšetření.(22)
Terapie a prognóza
Stěžejní v péči o pacienty s HKMP je vyhodnocení rizika závažných arytmií, a tedy náhlé srdeční smrti a její včasná prevence. V současné době existují dva kalkulátory rizika náhlé smrti pro děti s HKMP. Oba berou v úvahu především přítomnost hlavních rizikových faktorů, jako jsou synkopy, tloušťka stěny levé komory, velikost levé síně, míra obstrukce výtokového traktu levé komory, přítomnost nesetrvalé komorové tachykardie na EKG holteru a v případě Primacy kalkulátoru i genotyp.(23,24) Základem léčby jsou betablokátory (zpomalení frekvence a prodloužení diastolického plnění) a blokátory kalciových kanálů u starších pacientů (zlepšení plnění komory a snížení její obstrukce). Tyto jsou indikovány v případě symptomů a/nebo přítomnosti obstrukce výtokového traktu levé komory. Chirurgická terapie (myektomie hypertrofické septální svaloviny) se využívá u dětí zřídka v případě významné obstrukce nebo trvání příznaků i přes farmakoterapii. Implantace defibrilátoru je indikována jako sekundární prevence náhlé smrti po proběhlé fibrilaci komor nebo jako primární prevence na základě pečlivého zvážení individuálního rizika náhlé smrti daného pacienta. Kromě výpočtu rizika pomocí dostupných kalkulátorů hraje roli i rizikovost některých genotypů (vysoce rizikové jsou např. desminopatie, laminopatie nebo Kaernsův – Sayreův syndrom), zkušenost kardiocentra a postoj rodiny. U pacientů s významnou HKMP nedoporučujeme větší fyzickou zátěž a závodní sport. Ve vybraných případech existuje specifická léčba základního onemocnění. V České republice je dostupná např. enzymatická substituční terapie pro Pompeho onemocnění, Fabryho chorobu a některé mukopolysacharidózy.
Pětileté přežití se pro dětské pacienty velmi liší, od 40 % u dětí s HKMP s vrozenou poruchou metabolismu až po 95 % u dětí se sarkomerickou HKMP.(12) Pro celou skupinu HKMP platí, že nejhorší prognóza je při stanovení diagnózy v prvních dvou letech věku.
RESTRIKTIVNÍ KARDIOMYOPATIE
Restriktivní kardiomyopatie (RKMP) je definována poruchou diastolické funkce při absenci hypertrofie myokardu. Systolická funkce bývá normální nebo lehce snížená.(25)
Etiologie a patogeneze
Etiologicky se uplatňují některé sarkomerické geny (MYBPC3, MYH7, TNNT2, TNNI3), gen pro α-kardiální aktin, desmin nebo filamin C. Patofyziologickým podkladem poruchy diastolické funkce je intersticiální infiltrace nebo fibróza.
Nejčastější příčina RKMP u dospělých, amyloidóza, je u dětí zcela raritní. Z lysozomálních chorob ze střádání může RKMP způsobit vzácná Andersonova–Fabryho choroba, postižení srdce se přidává ve druhém a třetím decenniu, tato choroba může být asociována i s HKMP. Další etiologií je přetížení železem u pacientů s hemochromatózou a po opakovaných krevních transfuzích. Endomyokardiální fibrózu mohou způsobit některé parazitární infekce, autoimunitní choroby nebo malignity s hypereozinofilí.
Klinický obraz a diagnostika
Pro patologickou funkci většinou levé komory jsou hlavními projevy příznaky systémového a plicního městnání (dušnost námahová i klidová, hepatomegalie). Možné jsou bolesti na hrudníku, synkopy. Rtg identifikuje kardiomegalii a projevy plicního městnání, na EKG popisujeme známky zvětšení síní, změny ST úseků. Echokardiografie je většinou schopna identifikovat tento typ onemocnění, důležitý je popis diastolické dysfunkce a dilatace síní.
Terapie a prognóza
Terapie je symptomatická (diuretika při známkách městnání), farmakologické možnosti ovlivnění diastolické funkce jsou minimální. Pro časté trombotické komplikace doplňujeme antikoagulancia. Prognóza je závažná, RKMP má pro děti nejhorší výsledky z celé skupiny kardiomyopatií.(26) Již samo stanovení této diagnózy může být indikací k zařazení na čekací listinu pro srdeční transplantaci.
DALŠÍ TYPY KARDIOMYOPATIÍ DŮLEŽITÉ V PEDIATRII
Nonkompaktní myokard je výsledkem patologického vývoje primární embryonální tkáně v definitivní myokard. Může být izolovaný nebo asociovaný s vrozenými srdečními vadami. Většinou tento obraz zřetelných trabekul a lakun popisujeme v levé komoře. Komplikací jsou komorové arytmie, trombózy a embolie, vývoj oběhového selhávání. V zahraniční literatuře se používá termín LVNC (left ventricular noncompaction cardiomyopathy). I u těchto pacientů jsou postupně objevovány sarkomerické mutace, případně mutace v genu pro ZASP (hlavní součást Z disku) nebo pro α-aktinitin - 2. Obraz nekompaktní kardiomyopatie je typický pro X-vázaný Barthův syndrom (porucha acetylace kardiolipinu s poruchou mitochondriální membrány). Klinickými projevy jsou postižení srdce, neutropenie, myopatie s hypotonií a růstová retardace.
Arytmogenní KMP je charakterizována patologickou fibrózní a tukovou přestavbou myokardu. Klinickým důsledkem jsou komorové arytmie s palpitacemi a synkopami. Nezřídka bývá první manifestací náhlé úmrtí. Geneticky byly identifikovány mutace genů kódující mezibuněčná spojení, např. pro desmozomy (DSC2, DSG2, DSP, JUP a další), výsledkem mutací je chybná adheze mezi buňkami a následná funkční i elektrická porucha. V současnosti jsou popsány i jiné typy než mutace desmozomů a arytmogenní KPM je považována za heterogenní skupinu onemocnění.(27) Významnou roli v diagnostice hraje CMR, signálově průměrované EKG s detekcí pozdních potenciálů a genetické vyšetření v rámci kaskádového rodinného screeningu. Vzhledem k vysokému riziku fatálních arytmií a progrese onemocnění v důsledku fyzické zátěže je v případě arytmogenní KMP i samotný genotyp indikací k zákazu sportu. Implantace defibrilátoru (ICD) je předmětem společného rozhodnutí rodiny a lékaře, v případě proběhlé komorové fibrilace, setrvalých komorových arytmií, synkop na podkladě susp. arytmie nebo rizikového genotypu (geny pro fosfolamban, lamin A/C, filamin C) je ICD indikován.(28)
SOUHRN OBOU KAPITOL A ZÁVĚRY
Myokarditida u dětí je vzácné a zároveň potenciálně závažné onemocnění. Etiologicky se v našich podmínkách setkáváme téměř výhradně s viry nebo s myokarditidami v rámci Kawasakiho nemoci či PIMS-TS. Myokarditida často představuje diagnostické a terapeutické dilema. Při klinickém podezření je nutná hospitalizace s možností monitorace pacienta, laboratoř včetně hs-troponinu, EKG a echokardiografii provádíme vždy opakovaně. Smyslem je včasné odhalení arytmií nebo srdeční dysfunkce. V posledních letech stoupá diagnostický význam magnetické rezonance. Terapie je cílena na oběhovou stabilizaci a léčbu arytmií. V terapeutických postupech je často opomíjeno použití antivirotik. V případě jejich dostupnosti, například pro chřipku, herpetické viry apod., se přikláníme k jejich podání za předpokladu známek akutní infekce, a to i při průkazu viru pouze z periferních tkání. Doporučení pro podání IVIG nejsou jednoznačná, většinou jsou indikovány při komplikované myokarditidě, tedy při poruše funkce nebo nově vzniklých dysrytmiích.
Kardiomyopatie jsou v dětském věku vzácné. Jedná se o heterogenní skupinu diagnóz, u kterých je v důsledku větší dostupnosti a dokonalosti genetického testování stále častěji odhalena genetická příčina. Pro dětský věk je typické, že KMP mohou být součástí systémových onemocnění, především dědičných poruch metabolismu, malformačních syndromů nebo neuromuskulárních onemocnění. Pro některá z nich existuje specifická léčba. V diferenciálně diagnostické rozvaze u dětí s KMP je stěžejní vytvoření rodokmenu s odhalením typu dědičnosti, dále popis echokardiografického i EKG fenotypu. Symetrická hypertrofie celého srdce a preexcitace na EKG jsou typické např. pro střádavé choroby, zatímco asymetrická hypertrofie septa spíše pro sarkomerické HKMP. U dětí pátráme po extrakardiálních příznacích včetně regresu psychomotorického vývoje, malého vzrůstu, dysmorfických rysů, hypotonie, přítomnosti hepatomegalie, kostních změn. Laboratorní biochemické, metabolické a genetické vyšetření je vždy indikováno. Léčba je většinou cílena na podporu srdeční funkce a léčbu nebo prevenci závažných arytmií a náhlé smrti. Ve vybraných případech však existuje specifická léčba základního onemocnění, proto je nutné děti vyšetřovat komplexně a danou diagnózu včas odhalit.
Korespondující autor:
doc. MUDr. Jan Pavlíček, Ph.D.
Klinika dětského lékařství LF OU
a FN Ostrava
17. listopadu 1789
708 52 Ostrava-Poruba
jan.pavlicek@fno.cz
Ces-slov Pediat 2023; 78(2): 110–121
Sources
1. Arola A, Pikkarainen E, Sipilä JO, et al. Occurrence and features of childhood myocarditis: a nationwide study in Finland. J Am Heart Assoc 2017; 6(11): e005306.
2. Law YM, Lal AK, Chen S, et al. Diagnosis and management of myocarditis in children: a scientific statement from the American Heart Association. Circulation 2021; 144(6): e123–e135.
3. McCrindle BW, Rowley AH, Newburger JW, et al. Diagnosis, treatment, and long-term management of Kawasaki disease: a scientific statement for health professionals from the American Heart Association. Circulation 2017; 135(17): e927–e999.
4. de Loizaga SR, Beaton AZ. Rheumatic fever and rheumatic heart disease in the United States. Pediatr Ann 2021; 50(3): e98–e104.
5. Aeschlimann FA, Misra N, Hussein T, et al. Myocardial involvement in children with post-COVID multisystem inflammatory syndrome: a cardiovascular magnetic resonance based multicenter international study-the CARDOVID registry. J Cardiovasc Magn Reson 2021; 23(1): 140.
6. Arbustini E, Narula N, Giuliani L, et al. Genetic basis of myocarditis: myth or reality? Myocarditis 2020; 45–89.
7. Bracamonte-Baran W, Čiháková D. Cardiac autoimmunity: myocarditis. Adv Exp Med Biol 2017; 1003 : 187–221.
8. Ammirati E, Veronese G, Bottiroli M, et al. Update on acute myocarditis. Trends Cardiovasc Med 2021; 31(6): 370–379.
9. Friedrich MG, Sechtem U, Schulz-Menger J, et al. Cardiovascular magnetic resonance in myocarditis: a JACC White Paper. J Am Coll Cardiol 2009; 53 : 1475–1487.
10. Meune C, Spaulding C, Mahé I, et al. Risks versus benefits of NSAIDs including aspirin in myocarditis: a review of the evidence from animal studies. Drug Saf 2003; 26 : 975–981.
11. Lipshultz SE, Sleeper LA, Towbin JA, et al. The incidence of pediatric cardiomyopathy in two regions of the United States. N Engl J Med 2003; 348 : 1647–1655.
12. Lipshultz SE, Law YM, Asante-Korang A, et al.; American Heart Association Council on Cardiovascular Disease in the Young; Council on Clinical Cardiology; and Council on Genomic and Precision Medicine. Cardiomyopathy in children: classification and diagnosis: a scientific statement from the American Heart Association. Circulation 2019; 140(1): e9–e68.
13. Tomašov P. Genetika kardiomyopatií. Cor Vasa 2010; 52 : 399–402.
14. Lee TM, Hsu DT, Kantor P, et al. Pediatric cardiomyopathies. Circ Res 2017; 121 : 855–873.
15. Braunwald E. Cardiomyopathies: an overview. Circ Res 2017; 121 : 711 – 721
16. Towbin JA, Jefferies JL. Cardiomyopathies due to left ventricular noncompaction, mitochondrial and storage diseases, and inborn errors of metabolism. Circ Res 2017; 121(7): 838–854.
17. Feingold B, Mahle WT, Auerbach S, et al.; American Heart Association Pediatric Heart Failure Committee of the Council on Cardiovascular Disease in the Young; Council on Clinical Cardiology; Council on Cardiovascular Radiology and Intervention; Council on Functional Genomics and Translational Biology; and Stroke Council. Management of cardiac involvement associated with neuromuscular diseases: a scientific statement from the American Heart Association. Circulation 2017; 136(13): e200–e231.
18. Alvarez JA, Orav EJ, Wilkinson JD, et al.; Pediatric Cardiomyopathy Registry Investigators. Competing risks for death and cardiac transplantation in children with dilated cardiomyopathy: results from the Pediatric Cardiomyopathy Registry. Circulation 2011; 124 : 814–823.
19. Monda E, Rubino M, Lioncino M, et al. Hypertrophic cardiomyopathy in children: pathophysiology, diagnosis, and treatment of non-sarcomeric causes. Front Pediatr 2021; 9 : 94.
20. Moak JP, Kaski JP. Hypertrophic cardiomyopathy in children. Heart 2012; 98(14 : 1044–1054.
21. Ziółkowska L, Turska-Kmieć A, Petryka J, Kawalec W. Predictors of long - -term outcome in children with hypertrophic cardiomyopathy. Pediatr Cardiol 2016; 37(3): 448–458.
22. Colan SD, Lipshultz SE, Lowe AM, et al. Epidemiology and cause-specific outcome of hypertrophic cardiomyopathy in children: findings from the Pediatric Cardiomyopathy Registry. Circulation 2007; 115 : 773–781.
23. Miron A, Lafreniere-Roula M, Steve Fan CP, et al. A validated model for sudden cardiac death risk prediction in pediatric hypertrophic cardiomyopathy. Circulation 2020; 142(3): 217–229.
24. Norrish G, Ding T, Field E, et al. Development of a novel risk prediction model for sudden cardiac death in childhood hypertrophic cardiomyopathy (HCM Risk-Kids). JAMA Cardiol. 2019; 4(9): 918-927.
25. Ditaranto R, Caponetti AG, Ferrara, V, et al. Pediatric restrictive cardiomyopathies. Front Pediatr 2021; 9 : 745365.
26. Anderson HN, Cetta F, Driscoll DJ, et al. Idiopathic restrictive cardiomyopathy in children and young adults. Am J Cardiol 2018; 121(10): 1266–1270.
27. Chen L, Song J, Chen X, et al. A novel genotype-based clinicopathology classification of arrhythmogenic cardiomyopathy provides novel insights into disease progression. Eur Heart J 2019; 40(21): 1690–1703.
28. T owbin JA, McKenna WJ, Abrams DJ, et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm 2019; 16(11): e301–e372.
Labels
Neonatology Paediatrics General practitioner for children and adolescentsArticle was published in
Czech-Slovak Pediatrics

2023 Issue 2
- What Effect Can Be Expected from Limosilactobacillus reuteri in Mucositis and Peri-Implantitis?
- The Importance of Limosilactobacillus reuteri in Administration to Diabetics with Gingivitis
Most read in this issue
- Myocarditis and cardiomyopathy
- Prenatal diagnosis of ovarian cysts, management and pregnancy outcomes
- Preparation of the child for the magnetic resonance examination
- Current pharmacotherapy options in pediatric obesity