
VÝVOJ A KLINICKÉ HODNOCENÍ NOVÝCH LÉČIV
DEVELOPMENT AND CLINICAL EVALUATION OF NEW DRUGS
The discovery and development of a drug is a process of many steps. The purpose of drug discovery is to identify chemicals that have a great chance of providing therapeutic benefit. Experiments designed to discover pharmacologic effect in patients have their fundamentals in the experimental data obtained from animals. Clinical experiments in humans classically are divided into at least three phases. Phase I presents first in man administration of study drugs. Phase II shows the response to treatment in patients with target disease state. The keystone to any drug development program and the gold standard of experimentation with drugs in humans is the randomized, controlled clinical trial. Th ese trials (phase III) are designed to test whether a drug is efficacious in comparison to current standard therapy. Phase IV trial is also known as Post Marketing Surveillance Trial. Phase IV trials involve the safety surveillance (pharmacovigilance) of drug aft er it receives permission to be sold. In this paper are discussed some basic terms relating to drug development, pre clinical and clinical experiments.
Key words:
drug discovery, clinical experiments, randomisation, blinding, design, bioequivalency
Authors:
David Suchý 1; Milan Hora 2; Jindřich Fínek 3
Authors‘ workplace:
Oddělení klinické farmakologie FN, Plzeň
1; Urologická klinika LF UK a FN, Plzeň
2; Onkologické a radioterapeutické oddělení FN, Plzeň
3
Published in:
Ces Urol 2009; 13(2): 141-148
Category:
Review article
Overview
Objev a vývoj nového léku je proces o mnoha krocích. Testování molekul začíná dnes v laboratorních podmínkách mj. na tkáňových kulturách. Cílem je identifikace chemické substance, která má šanci vykazovat terapeutický benefit. Následující rozsáhlé experimenty na zvířatech. Experimenty zaměřené na farmakologický efekt nové substance u člověka jsou opřeny o data získaná z těchto pokusů na zvířatech. Klinické experimenty u člověka jsou rozdělovány minimálně do tří fází. Fáze I představuje první podání člověku, fáze II zjišťuje, jak na nový terapeutický postup reaguje skupina nemocných. Základem každého vývojového programu je randomizovaná klinická studie. Tyto studie jsou ve fázi III zaměřeny na průkaz účinnosti nového léku ve srovnání se současnou standardní terapií. Studie fáze IV jsou označované jako studie postmarketingového sledování. Tyto studie jsou zaměřeny na sledování bezpečnosti (farmakovigilance) léčiva po jeho uvedení na trh. V této práci autoři diskutují základní pojmy z oblasti vývoje léků, preklinických a klinických experimentů.
Klíčová slova:
objev léku, klinický experiment, randomizace, zaslepení, uspořádání, bioekvivalence.
ÚVOD
Vývoj nových léčiv prošel v posledních šedesáti letech dramatickým rozvojem. Bylo objeveno obrovské množství nových účinných látek i celá řada nových farmakologických skupin. Na druhou stranu, vývoj nového léku představuje stále náročnější proces s rostoucími ekonomickými a technologickými nároky. K vývoji nového léku je třeba shromáždit týmy vědeckých pracovníků s vysokou kvalifikací a mezioborovým přesahem. Roste rozsah nutných preklinických testů, je propracována náročná metodologie klinického zkoušení, což konečně vede i k významnému nárůstu nákladů spojených s vývojem nového léčiva (1–3). Klinické zkoušení léků a jeho výsledky jsou neodmyslitelnou součástí současné lékařské praxe. Výsledky klinických studií a jejich metaanalýz jsou základní oporou tzv. medicíny založené na důkazu (EBM – Evidence Based Medicine) (4).
VÝVOJ NOVÉHO LÉKU
Inovativní lék má přinášet možnosti vyléčení dosud neléčitelné choroby nebo kvalitativní nové možnosti v terapii, očekává se zpravidla také větší bezpečnost léčby a pozitivní ovlivnění kvality života. Jeho použití by mělo být přínosem i z hlediska farmakoekonomického (4). V současnosti existuje několik základních strategií ve vývoji potencionálně inovativní substance:
- modifikace struktury již existujících léků
- vyhledáváním nových účinků již používaných léčiv
- systematický „screening“ souboru náhodně vybraných sloučenin s určitou biologickou aktivitou
- náhodné výsledky
- racionální projektování na základě pochopení molekulární struktury
- proteonomika, genomika
Dnes vzniká většina léků na základě racionálních postupů, při kterých hraje důležitou roli projektování léčiv za použití počítačové techniky (CADD – Computer Assisted Drug Design) (4).
Další vývojový program léku spočívá v hledání důkazu o účinnosti a bezpečnosti léku v podmínkách experimentálních a následně i klinických. Tím jsou vymezeny dvě základní etapy: etapy předklinických studií a etapa klinických testů, jejichž stručnou charakteristikou se zabývá následující text.
PŘEDKLINICKÉ ZKOUŠENÍ LÉČIV
Je charakterizováno hledáním důkazů o účinnosti nové substance (farmakodynamický „screening“) a riziku (toxikologický „screening“). Tato etapa probíhá na různých úrovních experimentu na zvířatech a v modelových pokusech na izolovaných buňkách a tkáních (1, 3–5). Významným pokrokem je použití geneticky modifikovaných zvířat. U slibných látek jsou prováděny farmakokinetické studie.
Po analýze a zhodnocení získaných výsledků je rozhodnuto, v jakých dávkách a jakým způsobem bude léčivo poprvé podáno lidem (4, 6). To je již náplní etapy klinických testů.
Cílem klinického hodnocení (KH) je podání objektivních důkazů o účinnosti a bezpečnosti léku u člověka. KH je problematikou mezioborovou, časově a finančně náročnou (6). Účinek potenciálních léčiv se vyhodnocuje během několika fází KH v podmínkách kontrolované randomizované klinické studie na základě předdefinovaných kritérií – cílů (endpointů) buď přímo (např. uzdravení), nebo nepřímo za pomoci biomarkerů (surrogate point, např. snížení krevního tlaku při hypertenzi, pokles C-reaktivního proteinu u zánětlivých onemocnění, změny glykémie u diabetes mellitus). Míru objektivity posouzení účinku léčiva ztěžuje řada faktorů, jejichž vliv má být během KH použitím vhodné metodologie minimalizován (4, 7).
Mezi takové faktory například patří:
- přirozená variabilita onemocnění, individualita pacienta, vliv pohlaví (6)
- farmakogenetické rozdíly v metabolismu léčiv
- tendence některých nemocí ke spontánnímu vyléčení
- přítomnost dalších onemocnění, rizikových faktorů, lékové interakce
- neúmyslná předpojatost pacienta i lékaře (bias) při očekávaném pozitivním/negativním efektu terapie: přecenění nebo podcenění léčebného efektu, placebo efekt
Klinické hodnocení probíhá ve třech fázích před registrací léčiva, čtvrtá fáze je již poregistrační. Každé klinické hodnocení musí probíhat ve smyslu zásad Správné klinické praxe (SKP, GCP – Good Clinical Practice), což je mezinárodně přijatý soubor pravidel pro navrhování, provádění, zpracování a vyhodnocování dokumentace klinického hodnocení léčiv (4, 5). SKP je navrhována tak, aby získané údaje a závěry z KH byly přesné a věrohodné. Klinické studie uskutečňují přímo originální farmaceutické společnosti nebo v převážné míře specializované agentury (CRO – Contract Research Organisation), orientované na jednotlivé fáze vývoje léku. Ty pracují na objednávku farmaceutických firem, zaměřených na vývoj (RD – Research and Development Company) (4).
Charakteristika jednotlivých fází KH
Tradičně je používáno dělení KH do čtyř fází. V současné době je již částečně zastaralé a zcela nevyhovuje, protože se jednotlivé fáze velmi často prolínají. V současné době je nahrazováno rozdělením KH podle záměru nebo uspořádání.
Fáze I.
Představuje první podání člověku. Používá dávku odvozené z preklinického výzkumu, která je většinou zlomkem dávky léčiva, která byla pokusnými zvířaty tolerována bez toxických projevů.
Cíle fáze I jsou zejména:
- vyhodnocení bezpečnosti a tolerance farmakoterapie novým léčivem
- farmakodynamický rozsah dávkování
- základní farmakokinetické parametry
- terapeutický záměr není primárním cílem hodnocení
V této fázi je lék podáván zpravidla malému množství pečlivě vybraných zletilých zdravých dobrovolníků nebo malé skupině nemocných. Zcela vyloučeno je zpravidla zařazení vulnerabilní skupiny osob (těhotné ženy, geriatričtí nemocní).
S ohledem na toxicitu nebo charakter některých léčivých přípravků se některé léčivé přípravky na dobrovolnících nezkouší – např. cytostatika (1, 4–6).
Fáze II, terapeutická
Představovaná úvodní (pilotní) klinickou studií, označováno jako fáze IIa. Cílem této fáze je ověření účinnosti a bezpečnosti léčivého přípravku u menší skupiny (cca 200 pacientů) s cílovým onemocněním, stanovení rozsahu farmakoterapeutických dávek a terapeutického rozmezí a vyhodnocení závislosti účinku na dávce. Zahrnuje opět doplňující měření farmakokinetických údajů, studium metabolitů, vliv patologických podmínek na farmakokinetiku léčiva, četnost a závažnost nežádoucích účinků i v závislosti na dávce (2, 4, 8).
Fáze IIb (pivotal trial) – dokazuje účinek léčivého přípravku na větším počtu pacientů, hodnocení účinku v závislosti na dávce. Tyto studie se nejprve uskutečňují na menším množství pacientů, při pozitivním výsledku pilotní studie se počty pacientů rozšiřují, design studií se přibližuje fázi III (randomizace, zaslepení atd.) (2). Jak již bylo řečeno, dělení jednotlivých fází již není v posledních letech takto rigidní vzhledem k překryvu jednotlivých fází.
Fáze III. Klinický kontrolovaný pokus, rozšířená klinická studie
Cílem této fáze je stanovení bezpečnosti a účinnosti léčby na mnohem větším počtu pacientů (až tisíce) ve srovnání s placebem nebo aktuálně používanou léčbou. Studie této fáze má tedy prokázat srovnatelnost nové léčby z hlediska bezpečnosti a účinnosti se soudobým standardem.
Klinické pokusy jsou navrhovány tak, aby byla minimalizována systematická chyba studie nebo zkreslení (bias). Systematická chyba je definována jako cokoliv, co chybně ovlivňuje závěry a zkresluje porovnání ramen klinického hodnocení. Při zařazování pacientů do jednotlivých ramen studie je zpravidla použita randomizace (2, 8).
Randomizace je proces, při kterém je subjekt zařazován do kontrolní skupiny nebo skupiny léčené zkoumaným lékem tak, aby měl každý jedinec stejnou šanci, že bude vybrán do jednoho z obou souborů. Randomizace se provádí na základě předem určeného plánu, který formou náhodného nebo pseudonáhodného rozdělování přiřazuje subjekty do jednotlivých skupin.
Randomizace tedy teoreticky zamezuje subjektivnímu a selektivnímu zařazování subjektů do jednotlivých ramen, zajišťuje požadovaný počet subjektů ve srovnávaných ramenech, umožňuje rovnoměrné rozložení prognosticky významných faktorů (souběžná léčba, onemocnění atd.) i rušivých faktorů (confounders). Používání randomizace je dnes standardem především při provádění srovnávacích klinických studií fáze III (1, 2, 8). V současnosti je řada dříve používaných randomizačních technik opuštěna.
Například se již nepoužívá randomizace podle pořadového čísla vstupu do studie nebo randomizace podle iniciál pacienta. V případě zaslepených studií tyto techniky neumožňují vytvořit skupiny pacientů s homogenně rozloženými prognostickými faktory ani požadovaný počet subjektů v jednotlivých skupinách. Za méně vhodnou je považována i kompletní randomizace, kdy je o přiřazení pacienta rozhodnuto pouze náhodně (jako při hodu mincí). Mezi doporučované randomizační techniky patří dnes stratifikační permutační bloková randomizace a adaptivní randomizace, které lépe vyhovují požadavku zajištění kontroly distribuce prognostických faktorů v ramenech studie (2, 8).
Možnost kontroly rovnoměrného rozložením prognostických faktorů je zásadní pro zajištění srovnatelnosti porovnávaných ramen KH. Jedná se o prognostické faktory ve vztahu k účinnosti a bezpečnosti studijní léčby, tedy například věk, pohlaví, anamnestická data, stadium onemocnění, souběžná léčba atd.).
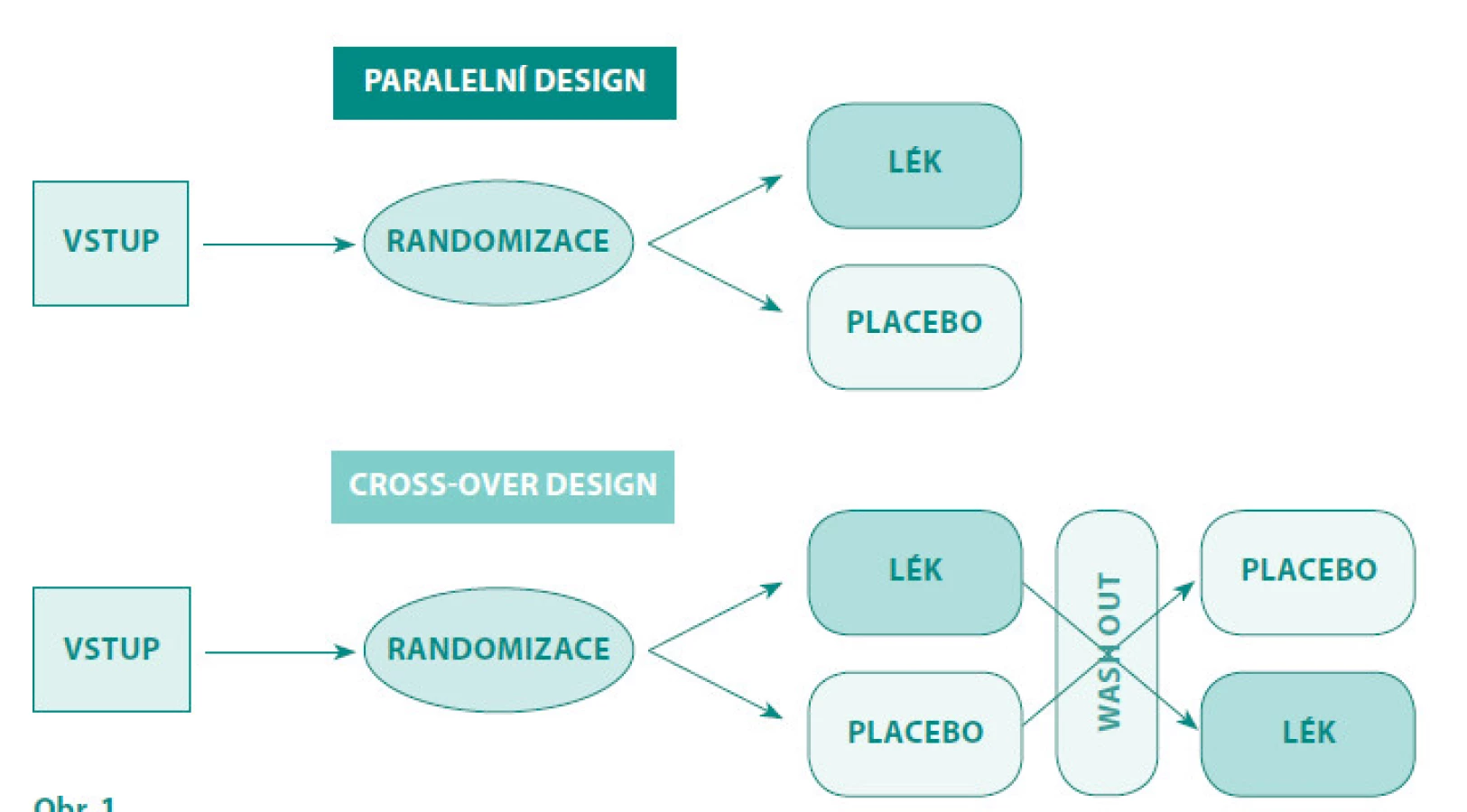
V uspořádání (designu) těchto studií je možno použít různá uspořádání, často se setkáváme s paralelním nebo „cross-over“ uspořádáním studie.
Paralelní uspořádání: Subjekty hodnocení jsou randomizovaně zařazeni do dvou nebo více ramen s odlišným způsobem léčby, která se v celém průběhu klinického hodnocení již nemění (2, 9).
„Cross-over“ uspořádání (obr. 1): Všechny hodnocené subjekty léčby podstoupí všechny srovnávané léčby. V průběhu studie si subjekty v jednotlivých ramenech zkříženě mění léčbu (placebo, aktivní komparátor. U studií s chirurgickou intervencí nebo u onemocnění s rychlým zhoršováním stavu (např. u některých nádorových onemocnění) není toto uspořádáním možné (2, 9).
Faktoriální uspořádání: Umožňuje zkoumání účinku více léků současně. Při tomto uspořádání jsou jednotlivým skupinám pacientů podávány všechny možné kombinace zkoumaných léčebných postupů. Toto uspořádání umožňuje testovat současně účinek dvou a více léků bez zvýšení nároků na počet zařazených pacientů. Podmínkou je, aby bylo léky možno podávat současně, bez interakce (6, 9).
Zaslepení (blinding): Pokud je cílem klinického experimentu srovnání dvou nebo více léčivých přípravků nebo aktivní látky a placeba, je z důvodů zvýšení objektivity experimentu používáno zaslepení.
Zaslepením se rozumí taková úprava lékové formy i balení, která neumožňuje odlišit na první pohled nově zkoušené léčivo od kontrolní účinné látky, eventuálně placeba. Při jednoduchém zaslepení („single blinding“) ví pouze lékař, v jaké skupině je pacient zařazen (2).
Pacient neví, zda je zařazen do kontrolní nebo hodnocené skupiny.
Dvojité zaslepení (double blinding) znamená, že ani lékař ani pacient neví, zda užívá aktivní léčbu nebo kontrolní přípravek, eventuálně placebo (2, 7, 8). Oba jsou zaslepeni. Informaci o zařazení pacienta má zadavatel nebo sponzor KH, který studií řídí. K odslepení medikace dojde při ukončení studie nebo při manifestaci závažných nežádoucích účinků. Existují i vyšší stupně zaslepení studie – trojité zaslepení (triple blinding), kdy je kromě lékaře a pacienta zaslepen i personál spravující data studie a čtyřnásobné zaslepení (quadriple blinding), jestliže jsou osoby podávající studijní medikaci odlišné od osob vyhodnocujících efekt a bezpečnost terapie a tyto osoby jsou rovněž zaslepeny. Zaslepení je zásadním prvkem klinických studií, které zvyšuje objektivitu a validitu získaných dat (8).
Klinické hodnocení fáze IV (poregistrační) Tato klinická hodnocení probíhají až po registraci léčiva (v ČR registraci provádí SÚKL). Cílem je ověření vlastností přípravku v reálném prostředí klinické praxe a u širší populace pacientů. Počet pacientů bývá v této fázi většinou nejvyšší, populace pacientů je oproti předchozím stadiím klinického zkoušení méně selektovaná. Klinická hodnocení jsou často uspořádána obdobně jako experimenty fáze III, někdy využívají odlišné metodiky (Case Control – osoby exponované (léčené) vs. kontrolní skupina neléčených, kohortové – hodnotí i specifický výsledek terapie ve skupinách jedinců, které jsou podobné, ale liší se jistou vlastností, např. kuřáci/nekuřáci). Jelikož se mnoho nežádoucích účinků manifestuje až v této fázi KH, je velmi důležité pečlivé hlášení nežádoucích účinků (6, 8).
V současné době je tendence opouštět tradiční dělení klinických studií na čtyři fáze, neboť se již není považováno za dostatečně odpovídající (2). Stále častěji se objevují charakteristiky klinického zkoušení, které refl ektují více zamyšlený primární cíl hodnocení. (rozdělení klinických studií podle záměru). Takto jsou rozlišovány studie farmakologické, průzkumné (explorační), ověřovací (konfirmační) a terapeutické. Další způsob dělení je možný podle uspořádání na studie otevřené a randomizované. Farmakologické studie procházejí všemi čtyřmi fázemi klinického hodnocení, přičemž logicky převažují u studií fáze I. Zabývají se stanovením základní farmakokinetiky, farmakodynamiky a ověřováním tolerance léčiva. Studie explorační (průzkumné) odpovídají fázi II klinického zkoušení s přesahem do dalších fází. Jejich cílem je stanovení terapeutické účinnosti léčiva a dávkování. Ověřovací (konfirmační studie) ověřují na velkých souborech nemocných terapeutickou účinnost, dávkovací schéma, bezpečnost a nežádoucí účinky. Odpovídají fázi II–III. Studie zaměřené na terapeutické užití léku se provádí po registraci léčiva a odpovídají tradiční IV fázi (tab. 1).

PRŮKAZ BIOEKVIVALENCE LÉČIV
Zcela jiným typem klinického zkoušení je průkaz bioekvivalence dvou léčiv. V tomto případě se nejedná o vývoj inovativního přípravku. Takto koncipované sledování je podmínkou registrace kopie již existujícího léku, tedy tzv. generického přípravku (10).
Bioekvivalenční studie
Jsou jiným typem studií, nicméně jejich význam roste s velkým nárůstem výroby a spotřeby generických léků, které v současné době v Evropě i USA tvoří přibližně 50 % preskripce. Registrace generika nevyžaduje na rozdíl od originálních přípravků náročné klinické zkoušení. Porovnání originálního přípravku a přípravku generického pomocí bioekvivalenční studie je považováno za dostatečně průkazné (10–12).
Dva přípravky jsou bioekvivalentní, jestliže jsou farmaceuticky ekvivalentní nebo farmaceuticky alternativní a jestliže jejich biologická dostupnost je po podání stejné dávky natolik podobná, že lze předpokládat stejnou účinnost i bezpečnost přípravku. Jde tedy o to, aby si množství a rychlost, kterou se účinná látka dostává do místa působení, byly navzájem podobné. Přípravky musí mít kvantitativně i kvalitativně stejný obsah aktivní látky se stejnou koncentraci a jejich léková forma má být shodná nebo podobná (12).
Průkaz bioekvivalence se provádí formou randomizované, překřížené studie relativní biologické dostupnosti u zdravých dobrovolníků. Generický lék je porovnáván s referenčním přípravkem, zpravidla originálním lékem. Sledují se farmakokinetické (opakované odběry ke zjištění plazmatických koncentrací) a farmakodynamické parametry. Míra absorpce je vyjadřována jako plocha pod křivkou plazmatických koncentrací (AUC – Area Under Curve), rychlost lze odhadnout z hodnot maximální plazmatické koncentrace (c max) a času, kdy byla dosažena (t max). Pokud jsou plochy pod křivkou testovaného a referenčního léku podobné v akceptovatelném rozmezí (0,8–1,25), lze léky prohlásit za bioekvivalentní (10, 12).
LEGISLATIVA
V naší legislativě je zakotveno, že jakékoliv klinické hodnocení fází I–III schvaluje Státní ústav pro kontrolu léčiv (SÚKL). Ten se spolu se zadavatelem a příslušnou etickou komisí podílí na dohledu nad prováděním klinického hodnocení (13). Pro KH IV fáze platí pouze oznamovací povinnost. K žádosti o schválení musí být přiložen soubor informací pro zkoušejícího, plán klinického zkoušení označovaný jako protokol a informace pro subjekt hodnocení a informovaný souhlas pacienta. Informovaný souhlas je dobrovolné vyjádření vůle subjektu (popřípadě jeho zástupce) zúčastnit se na klinickém hodnocení. Podpis informovaného souhlasu je získáván až po předchozím informování subjektu o relevantních aspektech klinického hodnocení (2). Tyto informace jsou pacientovu nejčastěji zprostředkovány zkoušejícím lékařem. Informace pacienta mají obsahovat údaje o výzkumném záměru, smyslu a cílech, dále možný přínos pro subjekt klinického hodnocení, předvídatelná a známá rizika léčby. Je třeba rovněž uvést podmínky odškodnění pro případ, že by došlo k poškození zdraví subjektu. Nesmí chybět ani informace o zabezpečení důvěrnosti osobních údajů a souhlas se zpřístupněním údajů vybraným osobám. Po ukončení KH musí zadavatel vypracovat souhrnnou zprávu.
Protokol studie musí být plánován podle zásad již zmíněné správné klinické praxe neboli Good Clinical Practise (GCP). Protokol má standardní části a kapitoly (2, 14). Je to podrobný plán klinického hodnocení, popisuje cíl, uspořádání, metodiku, hodnocené parametry, statistické vyhodnocení a organizaci klinického hodnocení. Je základním dokumentem, který podléhá schválení státními autoritami i etickými komisemi. Protokol zahrnuje v úvodní části základní informace o klinickém zkoušení, v dalších kapitolách jsou popsány cíle klinického hodnocení, plán klinického hodnocení (uveden typ studie, plánované kroky, označení medikace, trvání léčby, opatření pro předčasné ukončení, přerušení atd.), kritéria pro výběr subjektů hodnocení, parametry pro zařazení, resp. nezařazení subjektů, léčby, hodnocení účinnosti a bezpečnosti a část věnovaná statistickému hodnocení (2). Pokud nejsou dodrženy podmínky dané protokolem (odchylky od protokolu), nejsou ve vážnějších případech data tohoto pacienta zahrnuta do závěrečné analýzy (2, 14).
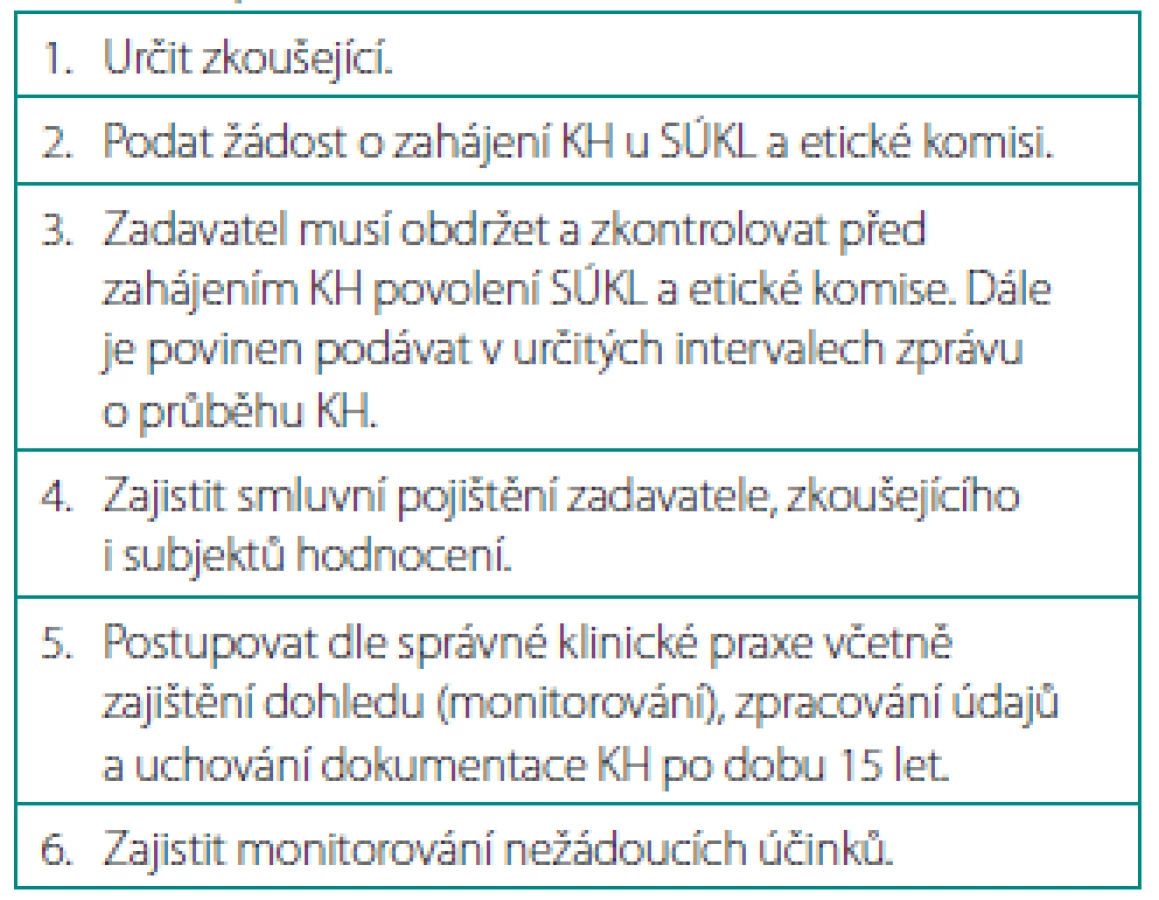
Zadavatel je definován jako fyzická osoba, společnost, instituce nebo organizace, která plně přijímá zodpovědnost za přípravu, provedení, ukončení a případně i financování KH.
Dle naší právní normy má zadavatel řadu povinností – předně žádá již zmíněné schválení KH Státním ústavem pro kontrolu léčiv (SÚKL). Ten informuje i o zahájení, ukončení, výskytu nežádoucích účinků, změnách a dodatcích k protokolu. Dále určuje zkoušející, zajišťuje pojištění účastníků KH, dohled nad prováděním studie podle pravidel správné klinické praxe atd. (tab. 2).
ETICKÉ OTÁZKY
Farmaceutické společnosti jako zadavatelé zřizují pro vývoj a výzkum samostatná oddělení anebo si najímají na jednotlivé činnosti výzkumné organizace (CRO – Contract Research Organization) (5).
Na základě Helsinské deklarace byly vytvořeny komise, které mají nezávisle posoudit etické aspekty KH. Tyto komise mají být nezávislé na státních orgánech, zdravotnickém zařízení, zadavateli a zkoušejících. Jedná se o Etické komise (EK), které tvoří skupina osob, kterou tvoří zdravotničtí profesionálové i nezdravotníci. Etické komisi nepřísluší odborné hodnocení preklinických a klinických údajů, k protokolu se vyjadřuje z hlediska etických otázek spojených s metodikou klinického hodnocení, očekávaných přínosů a rizik pro subjekty hodnocení. Dále se vyjadřuje k vhodnosti zdravotnického zařízení, morální a odborné způsobilosti zkoušejících, způsobu získání a formě dobrovolného prohlášení o účasti ve studii, pojištění subjektů hodnocení a zkoušejícího/zadavatele, kompenzace pro subjekt hodnocení (13). V České republice jsou dva typy etických komisí. Jsou to EK pro multicentrická klinická hodnocení (MEK) u studií, které probíhají podle stejného protokolu na několika místech, a místní (lokální) etické komise (LEK), které se vyjadřují ke klinickému hodnocení, které probíhá pouze v jejich zdravotnickém zařízení. Lokální EK se rovněž vyjadřují i k multicentrickým studiím, kde je již známo stanovisko MEK. V našich podmínkách předkládá žádost Etické komisi většinou zadavatel, ale je přípustné, aby s lokální etickou komisí jednal i zkoušející.
Etické komisi je třeba předložit:
- protokol studie
- text informovaného souhlasu
- postupy náboru, inzerce
- soubor informací pro zkoušejícího
- informace o kompenzacích výdajů a odměnách pro subjekty hodnocení
- životopis zkoušejícího, jeho kvalifikaci pro klinické hodnocení
- pojistné smlouvy: subjekt hodnocení, zadavatel, zkoušející
- další materiály mohou být vyžádány lokálně
ZÁVĚR
Výzkum a vývoj léků je složitý multidisciplinární proces, jehož náročnost stále roste. Úspěšné klinické hodnocení završuje aplikovaný farmaceuticko-medicínský výzkum daného léku a umožňuje registraci léku. Čas potřebný k dokončení vývoje inovativního přípravku přesahuje zpravidla 10 let a náklady se pohybují na úrovni miliardy dolarů. Originální lék je po určitou dobu chráněn patentem, jehož má vlastník patentu exkluzivní právo na prodej léku (3). Základní patent zahrnuje obvykle 20 let, přičemž se ale část této doby spotřebovává již v období klinických zkoušek. Po vypršení platnosti patentu může být lék vyráběn a prodáván jinou farmaceutickou společností, která tedy uvede na trh kopii originálního léku, tzv. generický lék.
Došlo: 18. 2. 2009
Přijato: 23. 3. 2009.
Kontaktní adresa
MUDr. David Suchý
Oddělení klinické farmakologie FN
E. Beneše 13, 305 99 Plzeň
e-mail: suchyd@fnplzen.cz
Sources
1. Švihovec J. Vývoj a registrace nových léčiv. In: Lincová D, Farghali H. Základní a aplikovaná farmakologie. 1.ed. Praha: Galén 2002; 47–52.
2. Strnadová V, Svobodník A, Křepelka F. Úvod do metodiky klinického hodnocení léků. 1. vyd. Praha: Grada Publishing 2007; 150 s.
3. Kaztung B. Předklinické a klinické hodnocení léčiv. In: Katzung B. Základní a klinická farmakologie. 8 ed. Praha: H & H 2006; 73–83.
4. Kriška M. Vývoj nového léku. In: Kriška M. a kol. Memorix klinickej farmakologie a liekov. 2.ed. Bratislava: SAP 2006; 55–61.
5. Petr P, Kalová H. Nápady čtenáře klinických studií. Praktické lékárenství 2006; 6 : 270–273.
6. Perlík F. Klinické hodnocení účinků léčiv. In: Perlík F. Základy klinické farmakologie. 1. ed. Praha: Triton 1999; 90–98.
7. Carruthers SG, Hoff man BB., Melmon KL, et al. Drug discovery and development. In: Melmond and Morrellis Clinical Pharmacology. 4 ed. New York: McGraw-Hill 2000; 1289–1306.
8. Svobodník A, Coufal O, Dušek L. Základní pojmy v designu, analýze dat a interpretaci výsledků klinických hodnocení léčiv. Klinická onkologie 2005; 18(Suppl): 238–241.
9. Tomášek J, Kiss I. Design klinické studie. Klinická onkologie 2005; 18(Suppl): 245–249.
10. Vetchý D, Rabišková M, Švarcová M, et al. Porovnání vybraných perorálních originálních a generických léků používaných v kardiologii na základě disoluční studie. Klin Farmakol Farm 2005; 19 : 84–88.
11. Vetchý D, Frýbortová K, Rabišková M, Daněčková H. Bioekvilalenční studie léčivých přípravků v České republice. Čas Lék čes 2007; 146 : 431–433.
12. Perlík F. Biologická dostupnost. In: Perlík F. Základy klinické farmakologie. 1. vyd. Praha: Triton 1999; 28–33.
13. Perlík F. Etická problematika klinického hodnocení léků. In: Perlík F. Základy klinické farmakologie. 1. vyd. Praha: Triton 1999; 101–108.
14. Demlová R. Výběr pacientů v klinických studiích – rizika pro interpretaci v běžné klinické praxi. Klinická onkologie 2005 : 18(Suppl): 250–251.
Labels
Paediatric urologist Nephrology UrologyArticle was published in
Czech Urology

2009 Issue 2
Most read in this issue
- ATYPICKÝ PRŮBĚH TORZE PŘÍVĚSKU VARLETE
- PROGNOSTICKÝ VÝZNAM EXPRESE p53, Ki-67 VE TKÁNI UROTELIÁLNÍHO KARCINOMU A NENÁDOROVÉ SLIZNICI MOČOVÉHO MĚCHÝŘE
- VÝVOJ A KLINICKÉ HODNOCENÍ NOVÝCH LÉČIV
- ČASNÉ ZKUŠENOSTI S ROBOTICKY ASISTOVANOU LAPAROSKOPICKOU RADIKÁLNÍ PROSTATEKTOMIÍ - PRVNÍCH 153 PACIENTŮ