
Využití molekulární genetiky v diferenciální diagnostice nádorů ledvin
The use of molecular genetics in the differential diagnosis of renal tumors
Renal tumors are a highly heterogenous group of tumors which makes their diagnosis increasingly complicated. The use of molecular and genetic techniques has resulted not only in the improvement of classification and differential diagnosis, but has also led to a better understanding of the biological behavior of these tumors. The increasing use of targeted therapy makes precise diagnosis of each lesion important and in the future it may become a crucial tool for the selection of proper treatment.
Key words:
kidney, renal cell carcinoma, oncocytoma, translocation renal cell carcinoma, morphology, molecular biology, diagnostics, genetics, biological treatment, differential diagnostics.
Authors:
Jindřich Branžovský 1; Petr Martínek 1; Kevin Baulet 1; Ivan Trávníček 2; Petr Stránský 2; Tomáš Vaneček 1; Milan Hora 2; Ondřej Hes 1
Authors‘ workplace:
Šiklův patologicko-anatomický ústav LF UK a FN, Plzeň
1; Urologická klinika LF UK a FN, Plzeň
2
Published in:
Ces Urol 2012; 16(4): 214-221
Category:
Review article
Overview
Nádory ledvin jsou vysoce heterogenní skupinou nádorů, jejichž diagnostika je stále komplikovanější. Zavedení molekulárně-genetických technik vedlo nejen ke zlepšení klasifikace a diferenciální diagnostiky, ale také k mnohem lepšímu porozumění biologického chování těchto nádorů. Vzhledem ke stále častěji využívané cílené léčbě (targeted therapy) je nepochybně důležitá a v budoucnu bude ještě daleko významnější naprosto přesná diagnóza dané léze.
Klíčová slova:
ledvina, renální karcinom, onkocytom, translokační renální karcinom, morfologie, molekulární-biologie, genetika, biologická léčba, diferenciální diagnostika.
ÚVOD
Nádory ledvin jsou vysoce heterogenní skupinou nádorů, jejichž diagnostika je stále komplikovanější. U některých lézí není možné stanovit správnou diagnózu ani pomocí morfologie, histochemie, imunohistochemie či ultrastrukturálního vyšetření. Vzhledem ke stále častěji využívané cílené léčbě (targeted therapy), je nepochybně důležitá a v budoucnu bude ještě daleko významnější naprosto přesná diagnóza dané léze. V posledních letech se stává molekulární diagnostika nedílnou součástí diagnostického vyšetřovacího algoritmu obtížně určitelných renálních tumorů.
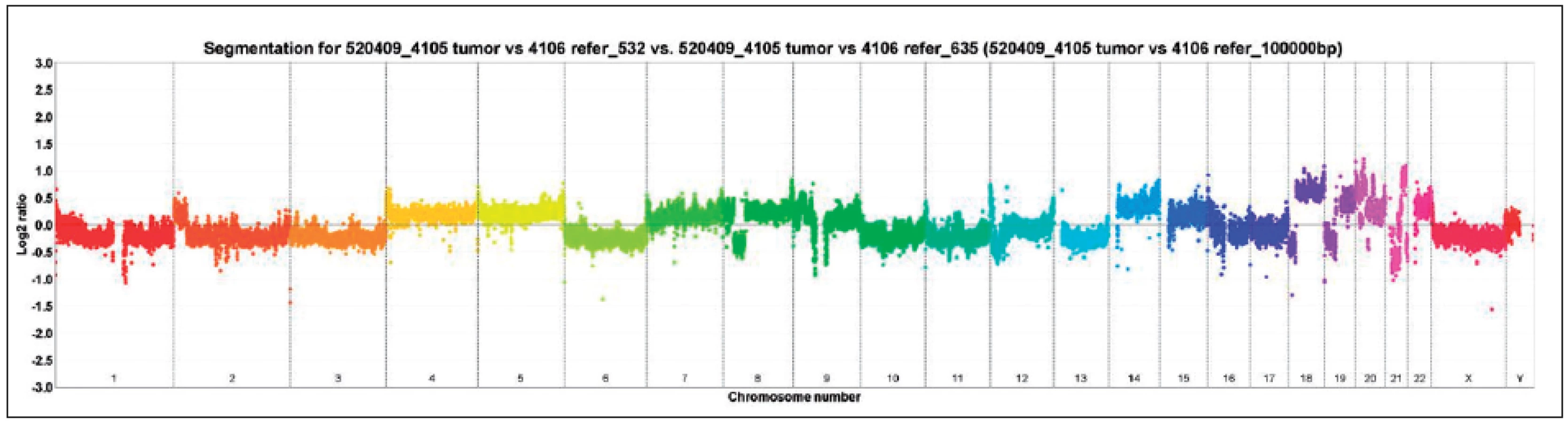
V současné době je k dispozici celá řada technologií, jejichž detailnější popis se vymyká cílům tohoto přehledového článku. Metodiky, které byly dříve dostupné jen pro čerstvou, zamraženou tkáň, je dnes možné aplikovat i na tkáně zpracované rutinní metodikou, tedy na parafinové bloky. Samozřejmě je důležité používat správně koncentrovanou fixaci. Čím je materiál starší, tím obvykle dochází k výraznější degradaci DNA a RNA, ale možnosti využití rutinně zpracovávaného bioptického materiálu jsou stále širší. Při optimální fixaci a dostatečném objemu nádorové tkáně je možné hodnotit i vzorky z „core“ biopsií. Ovšem obecně platí, že „core“ biopsie je vždy významně omezována svoji velikostí a velká část tkáně je většinou spotřebována již při základním histologickém, eventuálně imunohistochemickém vyšetření.
Je nanejvýš pravděpodobné, že ve velmi blízké budoucnosti bude molekulárně genetická analýza využívána rutinně i během plánování další léčby tak, jako se děje u nádorů prsu či kolorektálního traktu, žaludku apod. (HER2-neu, mutace genů K-RAS, BRAF). Z celé řady studií jsou dostupné informace, díky nimž jsme nyní schopni zachytit genetický profil dominující u jednotlivých nádorů ledvin (1, 2).
Cílem tohoto přehledného článku je sumarizovat nejznámější molekulárně genetické rysy a odchylky nejčastěji se vyskytujících nádorů a jejich využití prozatím pouze v histopatologické diferenciální diagnostice. Možné využití genetických vyšetření nádorů ledvin v indikaci biologické léčby je prozatím ve fázi prvotních výzkumů bez naděje k využití v klinické praxi v nejbližší době.
SVĚTLOBUNĚČNÝ RENÁLNÍ KARCINOM
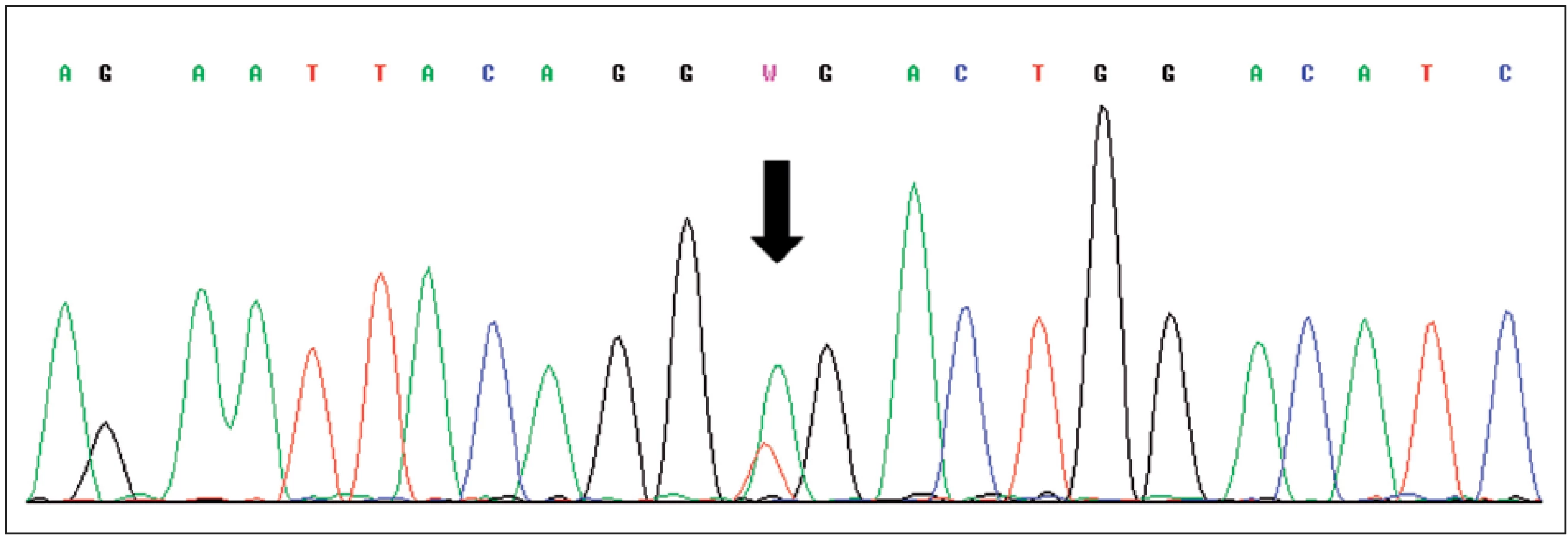
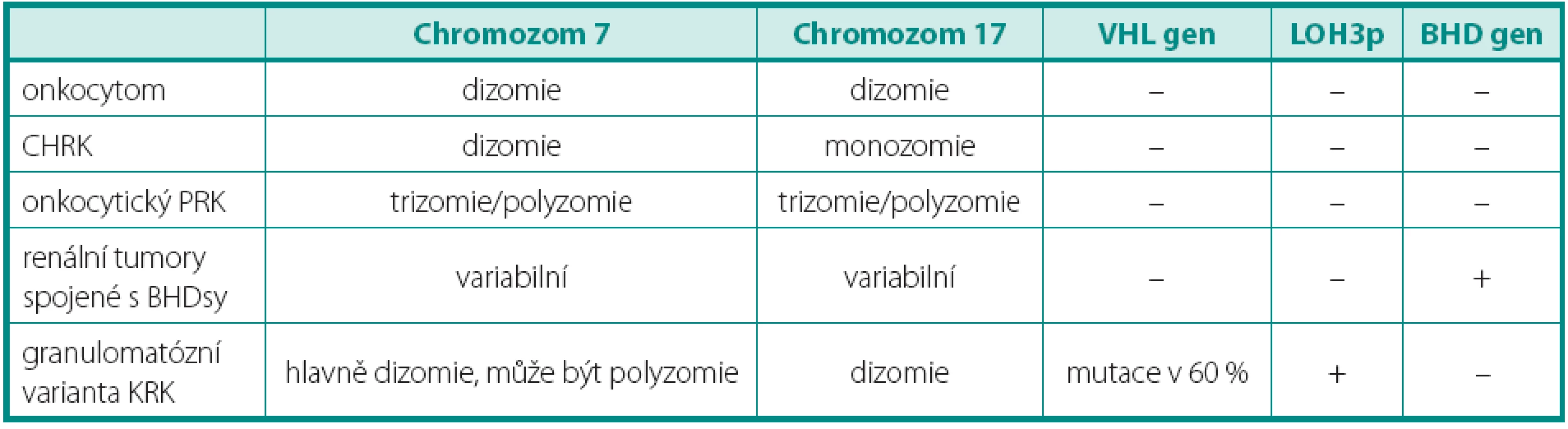
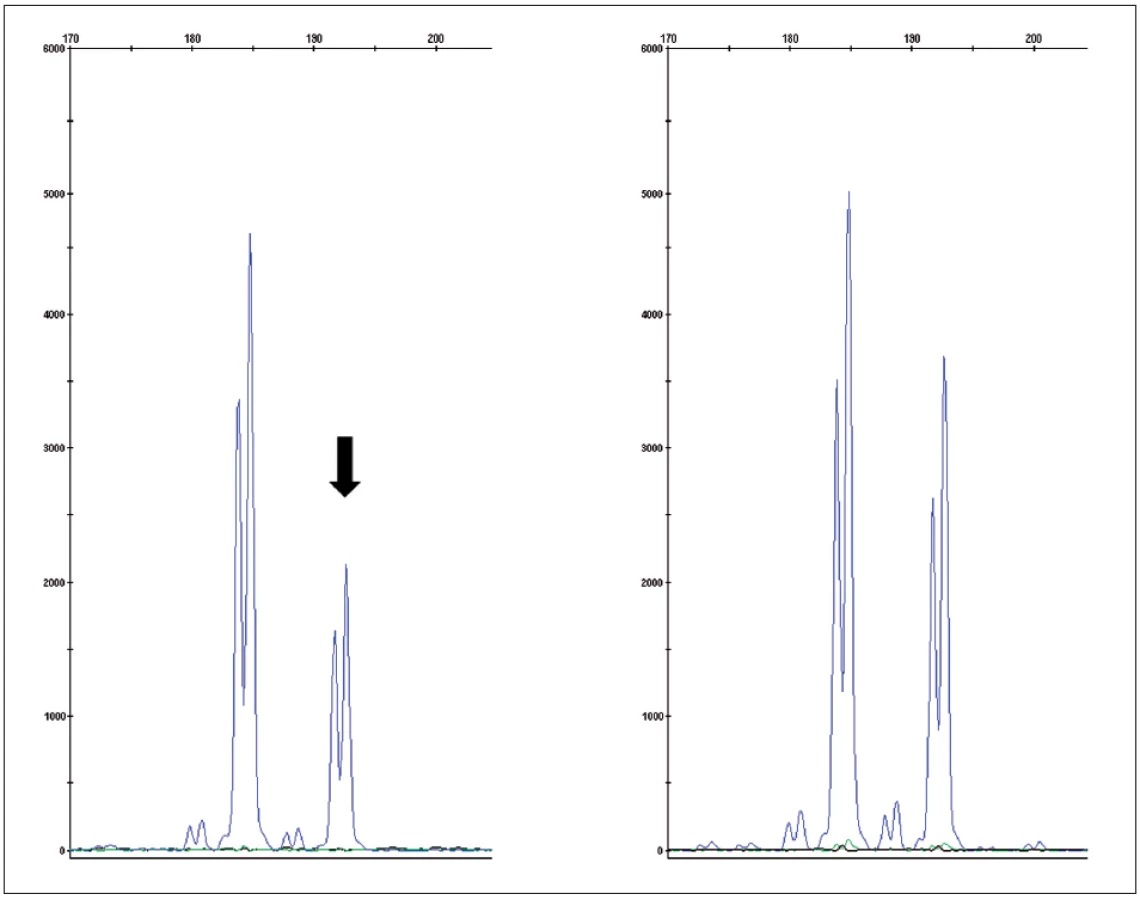
Pro světlobuněčný renální karcinom (SRK) je typická ztráta heterozygozity chromozomu 3p (LOH 3p) u téměř 90 % případů. Jediný doposud známý gen spojený s vývojem SRK je VHL (von Hippel-Lindau) tumor supresorový gen. Ten se nachází v oblasti chromozomu 3p25.3 a bývá mutován jak u sporadických, tak i hereditárních forem nádoru (3). Hlavním diferenciálně diagnostickým problémem u morfologicky a imunohistochemicky nejednoznačných nádorů je odlišení od renálního angioadenomatózního nádoru a světlobuněčného papilárního renálního karcinomu, ať již ve spojení, nebo bez vztahu k end-stage kidney disease (4, 5). Dalšími morfologicky podobnými nádory pak jsou karcinomy spojené s Xp11.2 translokacemi (6) (viz níže), jež jsou rovněž mimo jiné charakterizovány buňkami se světlou cytoplazmou uspořádané do hnízd a pseudopapilárních struktur. Zlatým diagnostickým standardem je průkaz proteinu TFE3, který je výsledným produktem výše zmíněných translokací. Bohužel, imunohistochemický průkaz nebývá vždy jednoznačný. Navíc nádory tvořené světlobuněčnými elementy, hlavně uspořádanými papilárně, se morfologicky a imunohistochemicky překrývají s následující jednotkou papilárního renálního karcinomu (PRK) (tab. 1).
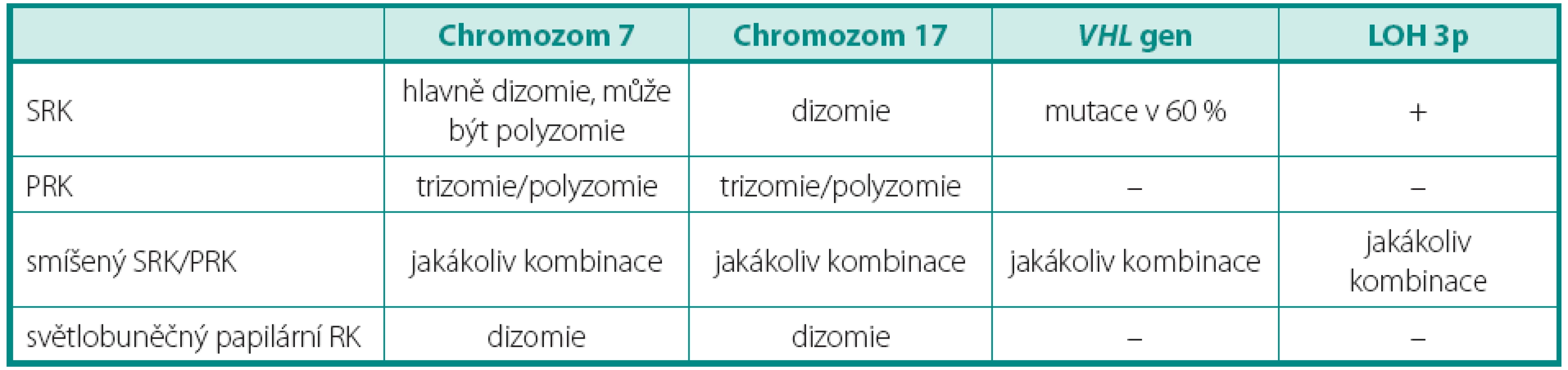
PAPILÁRNÍ RENÁLNÍ KARCINOM
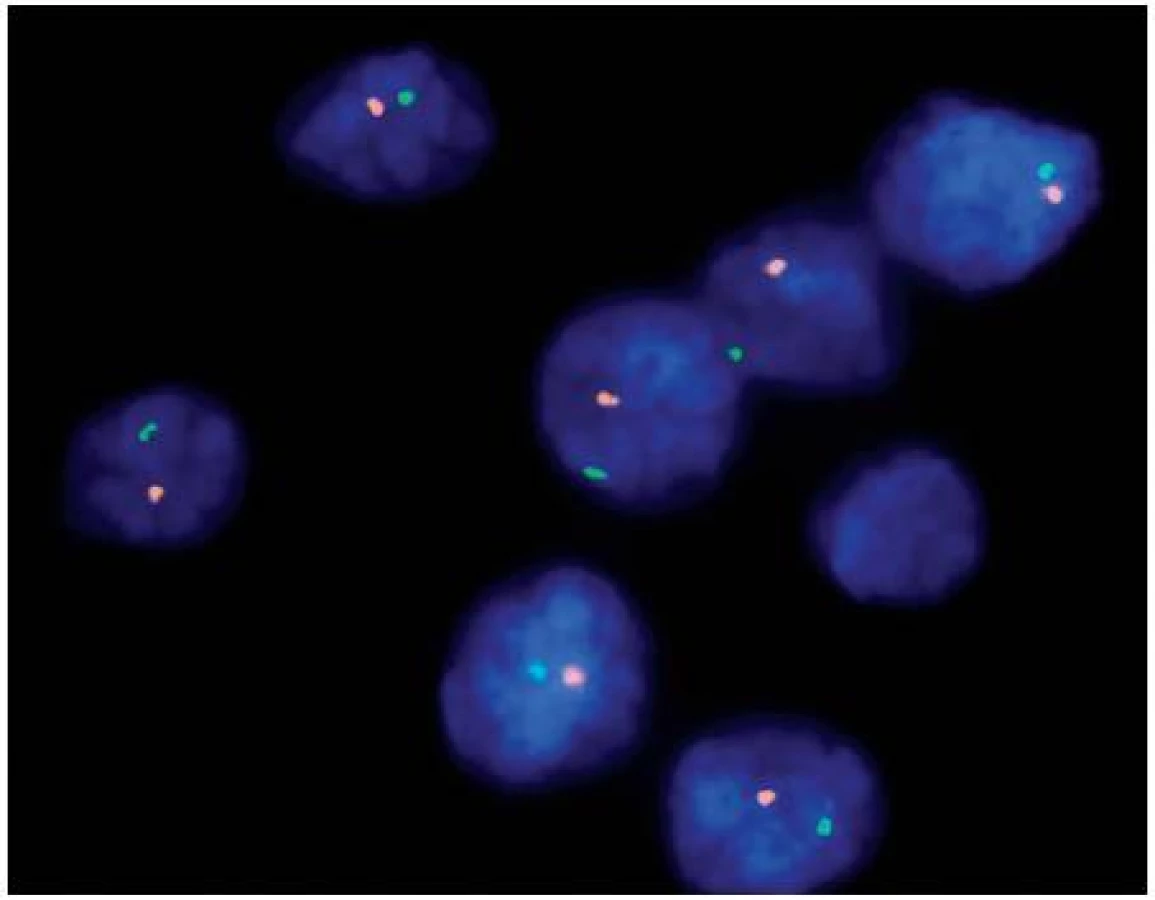
Papilární renální karcinom je charakteristický chromozomálními polyzomiemi, jako typická je udávána trizomie (polyzomie) chromozomů 7 a 17, méně 12, 16 a 20. Často se vyskytujícím znakem je také ztráta chromozomu Y u mužské části pacientů. Vztah mezi PRK a papilárním adenomem lze vysledovat i v přítomnosti chromozomálních aberacích. Papilární adenom vykazuje také ztrátu Y a často kombinovanou trizomii 7 a 17, předpokládá se ale přechod papilárního adenomu v PRK ziskem (gain) chromozomů, nejčastěji 12, 16 a 20, což může napomoci v jejich odlišení (7). Praktický význam však podobná analýza postrádá, vzhledem k definici obou jednotek. Papilární růst se světlobuněčnou komponentou může znamenat značný diagnostický problém, avšak právě cytogenetický profil je zde velmi nápomocný, neboť LOH 3p ukazuje na SRK, kdežto trizomie chromozomů 7, 17 a ztráta Y zase na PRK (tab. 1) (4, 7). Stejný problém nastává u nově popsaného světlobuněčného papilárního renálního karcinomu. Ten nevykazuje žádnou chromozomální aberaci typickou pro PRK (5). Tyto změny nebyly popsány ani u další skupiny, karcinomů spojených s Xp11.2 translokacemi. Ty sice mají podobný morfologický obraz papilární stavby, důležitým diferenciálně diagnostickým znakem je však chromozomální translokace t(X;1)(p11.2;q21) (viz níže) (3). Někdy není jednoduché odlišit mucinózní tubulární a vřetenobuněčný karcinom (MTSRK), který vykazuje mnohotné genetické abnormality, hlavně ztráty chromozomů 1, 4, 6, 8, 9, 10, 12q, 13, 14, 15, 16q, 17, 20q a 22 od PRK (8). Avšak již byly popsány i polyzomie chromozomů 7 a 17 (9). Další komplikovanou jednotkou je tzv. onkocytický papilární renální karcinom. Má stejné chromozomální aberace jako klasický PRK, tedy zejména trizomii či polyzomii chromozomů 7, 17 a ztrátu Y. Morfologicky je však někdy neodlišitelný od renálního onkocytomu (10, 11). U některých případů je diferenciální diagnostika mezi těmito dvěma jednotkami bez využití analýzy chromozomálních aberací nemožná.
CHROMOFOBNÍ RENÁLNÍ KARCINOM
Chromofobní renální karcinom (CHRK) má poměrně pestrý cytogenetický profil. Pro všechny varianty CHRK jsou charakteristické mnohotné změny v počtu chromozómů, zejména ztráty chromozomů Y, 1, 2, 6, 10, 13q, 17 a 21. U některých onkocytomů byla popsána ztráta pouze chromozomů Y, 1 a 14 (12–15), což lze v omezeném počtu případů využít v diferenciálně diagnostické úvaze. Sarkomatoidni CHRK je agresivní forma, která vykazuje polyzomii chromozomů 1, 2, 6, 10 a 17 namísto jejich ztráty (15).
RENÁLNÍ ONKOCYTOM
Renální onkocytom může být provázen některými genetickými změnami, mezi které lze zařadit ztrátu chromozomů 1, 14, Y a translokaci 11q13. U lézí v rámci onkocytózy/onkocytomatózy, byla dále prokázána i ztráta chromozomů 21 a Y. Diferenciální diagnostika onkocytických lézí zahrnuje několik biologickým chováním dramaticky se odlišujících lézí (tab. 2). Předně je nutné od renálního onkocytomu odlišit CHRK. Ten je charakterizován mnohotnými ztrátami chromozomů předně 1, 2, 6, 10, 17 a 21 (12, 16). Renální onkocytomy v rámci BHD (Birt-Hogg-Dube) syndromu na rozdíl od sporadické formy vykazují mutaci genu FLCN (dříve BHD) (17). Genetický profil typický pro SRK odlišuje granulomatózní variantu SRK, tumor též snadno zaměnitelný s renálním onkocytomem. Onkocytický PRCC a jeho charakteristika byl již popsán výše.
MUCINÓZNÍ TUBULÁRNÍ A VŘETENOBUNĚČNÝ KARCINOM
Mucinózní tubulární a vřetenobuněčný renální karcinom (MTSRK) je nádorem, jehož některé morfologické a imunohistochemické charakteristiky jsou podobné, někdy až shodné s PRK. Původně bylo udáváno, že MTSRK nevykazuje polyzomie chromozomů 7 a 17 ani ztrátu chromozomu Y, což jsou typické znaky PRK (8), dnes je jasné, že i MTSRCC může mít řadu numerických chromozomálních aberací včetně polyzomie chromozomů 7 a 17. Mezi takové abnormality patří jak úplná či částečná ztráta chromozomů 1, 4, 6, 8, 9, 13, 14, 15 a 22, tak zisk částí či celých chromozomů 7, 11, 16 a 17 (9).
„MiTF/TFE FAMILY“ TRANSLOKAČNÍ KARCINOMY
„MiTF/TFE family“ translokační karcinomy jsou poměrně nově vytvořenou skupinou, jež zahrnuje nádory spojené s translokacemi některých oblastí chromozomů, čímž dochází k novým genovým fúzím (splynutím) a tvorbě nových proteinů.
První skupina nádorů řazená do této rodiny zahrnuje karcinomy spojené s Xp11.2 translokacemi/TFE3 genovou fúzí. Popsanými mutacemi a geny jsou: t(X;1)(p11.2;q21)/(TFE3-PRCC), t(X;17)(p11.2;q25)/(TFE3-ASPL), t(X;1)(p11.2;p34)/(TFE3-PSF), t(X;17)(p11.2;q23)/(TFE3-CLTG) a inv(X)(p11;q12)/(TFE3-NONO (p54nrb)) (3, 6, 18, 19).
Další skupinu tvoří nádory s t(6;11)/TFEB fúzí. Nejznámější je t(6;11)(p21;q12)/(Alpha-TFEB) (20). Nově je do této skupiny řazen i tzv. „rosette forming t(6;11), HMB45 pozitivní translokační karcinom“ (21, 22).
FAMILIÁRNÍ VÝSKYT LEDVINNÝCH NÁDORŮ
VHL světlobuněčný renální karcinom je familiárně se vyskytující forma SRK. Tumor má stejné charakteristiky morfologické i cytogenetické jako sporadická forma SRK. U obou je (mimo jiné) porucha ve VHL tumor supresorovém genu nacházejícím se v oblasti 3p25.3 (3).
Hereditární papilární renální karcinom je většinou bilaterálně a multifokálně se vyskytující PRK (histologický typ 1). Opět se jedná o familiárně se vyskytující formu, tentokrát PRK, která opět sdílí morfologické a cytogenetické profily se sporadickými nádory. Dominuje zde trizomie (polyzomie) chromozomu 7 a 17, ztráta Y. V oblasti 7q31.1 byla zjištěna genetická alterace proto-onkogenu MET (povrchový receptor pro hepatocytární růstový faktor). Tato mutace se rovněž může vyskytnout u sporadické formy (3).
Syndrom hereditární leiomyomatózy a renálního karcinomu je autozomálně dominantní familiárně se vyskytující syndrom, pro nějž je typická tvorba leiomyomů na kůži a v děloze společně se vznikem renálních tumorů, konkrétně PRK (histologický typ 2). U tohoto syndromu byla identifikována mutace genu pro fumarát hydratázu (FH, 1q42.3-q43) (23).
Birt-Hogg-Dube syndrom je autozomálně dominantní syndrom charakterizovaný vznikem kožních benigních tumorů (fibrofolikulomy, trichodiskomy, akrochordony), plicními cystami, spontánními pneumothoraxy a ledvinnými nádory, které jsou reprezentovány hlavně onkocytomy, CHRK nebo hybridními (onkocytickými/chromofobními) nádory (24). FLCN gen (dříve BHD) je zodpovědný za rozvoj tohoto onemocnění a tento gen byl nalezen na chromozomu 17p11.2 (17).
Konstitucionální translokace chromozomu 3 je familiární syndrom charakterizovaný vznikem mnohotných, bilaterálních či naopak solitárních renálních tumorů, které mají histologické charakteristiky odpovídající SRK. Bohužel, translokace na chromozomu 3 prozatím nelze využít v rutinní diagnostice, jelikož se překrývají s obdobnými změnami u sporadických případů (25–27) Nádory této skupiny jsou však velmi vzácné.
Tuberózní skleróza je geneticky podmíněné onemocnění, které je mimo jiné charakteristické vznikem nízce agresivních či benigních nádorových a pseudonádorových lézí v CNS, kůži a dalších orgánech. V ledvinách je spojena s výskytem angiomyolipomů. Tzv. komplex tuberózní sklerózy je podmíněn mutacemi v genech TSC1 (chromozom 9q) a TSC2 (chromozom 16p), které regulují produkci proteinů tuberin a hamartin. V současné době je bohužel analýza genů TSC1 a TSC2 natolik komplikovaná, že je pro rutinní využití naprosto nevhodná (28).
ZÁVĚR
Z výše uvedených faktů je patrné, že diagnostika renálních tumorů je v současné době komplexní a poměrně složitou disciplínou. Samozřejmě stále platí, že morfologická diagnostika tvoří základní pilíř vyšetřovacího algoritmu. Zejména v konzultační praxi se patologové setkávají s nádory, které však nelze pomocí těchto základních metodik zařadit (často ani do základní skupiny benigní x maligní) a zde nastupují metody molekulární biologie jako integrální součást vyšetřování. Jsme si naprosto jistí, že během několika let budou tyto metodiky hrát v diagnostice a zejména v plánování další onkologické léčby roli mnohem zásadnější.
Došlo: 4. 8. 2012.
Přijato: 22. 11. 2012.
Kontaktní adresa
prof. MUDr. Ondřej Hes, Ph.D.
Oddělení speciální diagnostiky ŠPAÚ FN a LF UK
Alej Svobody 80, 304 60 Plzeň
e-mail: hes@medima.cz
Střet zájmů: žádný.
Podpořeno projektem SVV 264805/2012 Lékařské fakulty v Plzni, Univerzita Karlova v Praze a projektem Ministerstva zdravotnictví ČR koncepčního rozvoje výzkumné organizace 00669806 – FN Plzeň.
Sources
1. Eble JN, Sauter G, Epstein JI, Sesterhenn IA. World Health Organization classification of tumours. Pathology and genetics. Tumours of the urinary system and male genital organs. Lyon: IARC Press 2004.
2. Allory Y, Bazille C, Vieillefond A, Molinié V, Cochand-Priollet B, Cussenot O, Callard P, Sibony M. Profiling and classification tree applied to renal epithelial tumours. Histopathology 2008; 52(2): 158–166 (Epub 2007 Nov 22).
3. Lopez-Beltran A, Montironi R, Egevad L, Caballero-Vargas MT, Scarpelli M, Kirkali Z, Cheng L. Genetic profiles in renal tumors. Int J Urol 2010; 17 : 6–19.
4. Gobbo S, Eble JN, MacLennan GT, et al. Renal cell carcinomas with papillary architecture and clear cell components: the utility of immunohistochemical and cytogenetical analyses in differential diagnosis. Am J Surg Pathol 2008; 32 : 1780–1786.
5. Gobbo S, Eble JN, Grignon DJ, et al. Clear cell papillary renal cell carcinoma: a distinct histopathologic and molecular genetic entity. Am J Surg Pathol 2008; 32 : 1239–1245.
6. Hora M, Hes O, Eret V, Ürge T, Klečka J, Ferda J, Chudáček Z, Vaněček T, Michal M. Translokační renální karcinom Xp11.2 typu ASPL/TFE3 (Kidney carcinoma associated with Xp11.2 translocation type ASPL/TFE3). Ces Urol 2008; 12(3): 186–193.
7. Brunelli M, Eble JN, Zhang S, Martignoni G, Cheng L. Gains of chromosomes 7, 17, 12, 16, and 20 and loss of Y occur early in the evolution of papillary renal cell neoplasia: a fluorescent in situ hybridization study. Mod Pathol 2003; 16 : 1053–1059.
8. Cossu-Rocca P, Eble JN, Delahunt B, et al. Renal mucinous tubular and spindle carcinoma lacks the gains of chromosomes 7 and 17 and losses of chromosome Y that are prevalent in papillary renal cell carcinoma. Mod Pathol 2006; 19 : 488–493.
9. Brandal P, Lie AK, Bassarova A, Svindland A, Risberg B, Danielsen H, Heim S. Genomic aberrations in mucinous tubular and spindle cell renal cell carcinomas. Mod Pathol 2006; 19 : 186–194.
10. Hes O, Brunelli M, Michal M, et al. Oncocytic papillary renal cell carcinoma: a clinicopathologic, immunohistochemical, ultrastructural, and interphase cytogenetic study of 12 cases. Ann Diagn Pathol 2006; 10 : 133–139.
11. Urge T, Hes O, Ferda J, Chudáček Z, Eret V, Michal M, Brunelli M, Martignoni G, Hora M. Clinical characteristics of oncocytic papillary renal cell carcinoma, World J Urol 2010; 28(4): 513–517.
12. Brunelli M, Eble JN, Zhang S, Martignoni G, Delahunt B, Cheng L. Eosinophilic and classic chromophobe renal cell carcinomas have similar frequent losses of multiple chromosomes from among chromosomes 1, 2, 6, 10, and 17, and this pattern of genetic abnormality is not present in renal oncocytoma. Mod Pathol 2005; 18 : 161–169.
13. Crotty TB, Lawrence KM, Moertel CA, et al. Cytogenetic Analysis of six renal oncocytomas and a chromophobe cell renal carcinoma. Evidence that –Y, -1 may be a characteristic anomaly in renal oncocytomas. Cancer Gen Cytogenet 1992; 61 : 61–66.
14. Brimo F, Epstein JI. Selected common diagnostic problems in urologic pathology. Perspectives from a large consult service in genitourinary pathology. Arch Pathol Lab Med 2012; 136 : 360–371.
15. Brunelli M, Gobbo S, Cossu-Rocca P, Cheng L, Hes O, Delahunt B, Pea M, Bonetti F, Mina MM, Ficarra V, Chilosi M, Eble JN, Menestrina F, Martignoni G. Chromosomal gains in the sarcomatoid transformation of chromophobe renal cell carcinoma. Mod Pathol 2007; 20 : 303–309.
16. Dvorakova M, Dhir R, Bastacky SI, Cieply KM, Acquafondata MB, Sherer CR, Mercuri TL, Parwani AV. Renal oncocytoma: a comparative clinicopathologic study and fluorescent in-situ hybridization analysis of 73 cases with long-term follow-up. Diagn Pathol 2010; 5 : 32.
17. Toro JR, Wei MH, Glenn GM, Weinreich M, Toure O, Vocke C, Turner M, Choyke P, Merino MJ, Pinto PA, Steinberg SM, Schmidt LS, Linehan WM. BHD mutations, clinical and molecular genetic investigations of Birt-Hogg-Dube’ syndrome: a new series of 50 families and a review of published reports. J Med Genet 2008; 45 : 321–331.
18. Mosquera JM, Dal Cin P, Mertz KD, Perner S, Davis IJ, Fisher DE, Rubin MA, Hirsch MS. Validation of a TFE3 break-apart FISH assay for Xp11.2 translocation renal cell carcinomas. Diagn Mod Pathol 2011; 20(3): 129–137.
19. Zhong M, De Angelo P, Osborne L, Keane-Tarchichi M, Goldfischer M, Edelmann L, Yang Y, Linehan WM, Merino MJ, Aisner S, Hameed M. Dual-color, break-apart FISH assay on paraffin-embedded tissues as an adjunct to diagnosis of Xp11 translocation renal cell carcinoma and alveolar soft part sarcoma. Am J Surg Pathol 2010; 34(6): 757–766.
20. Argani P, Lae M, Hutchinson B,Reuter V, Collins MH, Perentesis J, Tomaszewski JE, Brooks JS, Acs G, Bridge JA, Vargas SO, Pavis IJ, Fischer DE, Ladanyi M. Renal carcinomas with the t(6;11)(p21;q12): clinicopathologic features and demonstration of the specific alpha-TFEB gene fusion by immunohistochemistry, RT-PCR, and DNA PCR. Am J Surg Pathol 2005; 29 : 230–240.
21. Hora M, Hes O, Ürge T, Eret V, Klečka J, Michal M. A distinctive translocation carcinoma of the kidney (“rosette-like forming”, t(6;11), HMB45 positive renal tumor). Int Urol Nephrol 2009; 41(3): 553–557.
22. Petersson F, Vanecek T, Michal M, Martignoni G, Brunelli M, Halbhuber Z, Spagnolo D, Kuroda N, Yang X, Cabrero IA, Hora M, Branzovsky J, Trivunic S, Kacerovska D, Steiner P, Hes O. A distinctive translocation carcinoma of the kidney; „rosette forming,“ t(6;11), HMB45-positive renal tumor: a histomorphologic, immunohistochemical, ultrastructural, and molecular genetic study of 4 cases. Hum Pathol 2012; 43 : 726–736.
23. Merino MJ, Torres-Cabala C, Pinto P, Linehan WM. The morphologic spectrum of kidney tumors in hereditary leiomyomatosis and renal cell carcinoma (HLRCC) syndrome. Am J Surg Pathol 2007; 31 : 1578–1585.
24. Hora M, Ürge T, Eret V, Stránský P, Klečka J jr, Kreuzberg B, Ferda J, Minčík I, Schraml J, Študent V, Michal M, Hes O. Hybridní onkocytické/chromofóbní tumory (HOChT) ledviny („Birt-Hogg-Dubé syndrome like“ tumory). Čes Urol 2010; 14(4): 237.
25. Cohen AJ, Li FP, Berg S, Marchetto DJ, Tsai S, Jacobs SC, Brown RS. Hereditary renal - cell carcinoma associated with a chromosomal translocation. N Engl J Med 1979; 301 : 592–595.
26. Kovacs G, Hoene E. Loss of der(3) in renal carcinoma cells of a patient with constitutional t(3;12). Hum Genet 1988; 78 : 148–145.
27. Kovacs G, Brusa P, de Riese W. Tissue-specific expression of a constitutional 3;6 translocation: development of multiple bilateral renal-cell carcinomas. Int J Cancer 1989; 43 : 422–427.
28. Au KS, Rodriguez JA, Rodriguez E Jr, Dobyns WB, Delgado MR, Northrup H. Mutations and polymorphisms in the tuberous sclerosis complex gene on chromosome 16. WHO Hum Mutat 1997; 9(1): 23–29.
Labels
Paediatric urologist Nephrology UrologyArticle was published in
Czech Urology

2012 Issue 4
Most read in this issue
- Myoglobinurie jako projev rabdomyolýzy po extrémní fyzické zátěži
- Zobrazení karcinomu prostaty metodami nukleární medicíny
- Prognóza pacientů s T1G3 uroteliálním karcinomem močového měchýře léčených vakcínou BCG – retrospektivní analýza
- Liposarkom retroperitonea v lokalizaci dolního polu ledviny