
NEFROBLASTOM – SOUČASNÁ DIAGNOSTIKA A LÉČBA
NEPHROBLASTOMA – CURRENT EVALUATION AND TREATMENT
Nephroblastoma (Wilms tumor – WT) is the most common solid renal tumor in children. The patients are treated according to SIOP 2001 protocol. This protocol recommends pre-operative chemotherapy, nephrectomy and postoperative treatment. Blastemal and anaplastic histological subtypes are poor prognostic factors. It is a well treatable disease, with up to 90 % of patients cured.
Key words:
Nephroblastoma, therapy, Wilms´ tumor.
:
Marcela Pýchová 1; Karel Švojgr 2; Josef Mališ 2; Michal Rygl 1; Karel Pýcha 1; Martin Kynčl 3; Roman Kodet 4; Jan Starý 2; Jiří Šnajdauf 1
:
Klinika dětské chirurgie 2. LF UK a FN Motol, Praha
1; Klinika dětské hematoonkologie 2. LF UK a FN Motol, Praha
2; Klinika zobrazovacích metod 2. LF UK a FN Motol, Praha
3; Ústav patologie a molekulární medicíny 2. LF UK a FN Motol, Praha
4
:
Ces Urol 2016; 20(2): 113-122
:
Review article
Nefroblastom (Wilmsův tumor – WT) je nejčastějším solidním nádorem ledvin u dětí. V České republice probíhá léčba podle protokolu SIOP 2001. Zahrnuje primárně onkologickou léčbu a poté následuje léčba chirurgická. Další léčba závisí na klinickém stadiu onemocnění a na histopatologickém nálezu. Problémem zůstává léčba blastémového a anaplastického typu WT. Přesto je toto onemocnění velmi dobře léčitelné a přežití se udává až 90 %.
Klíčová slova:
Nefroblastom, terapie, Wilmsův tumor.
ÚVOD
Nefroblastom (Wilmsův nádor – WT) je nejčastější nádor ledvin dětského věku, tvoří 6–7 % všech zhoubných nádorů dětí a dospívajících (1). Prevalence je přibližně 1 : 10 000 dětí. 90 % nádorů se manifestuje ve věku 1–7 let, nejčastější věk diagnózy je mezi 3–4 lety (2).
Poprvé byl popsán německým patologem a chirurgem Maxem Wilmsem v roce 1899. Zpočátku byla léčba jen chirurgická a výsledky nebyly dobré. Významný posun v léčbě znamenal objev rentgenového záření na přelomu století. V roce 1905 použil ozařování k léčbě maligního nádoru ledviny Frielinger v Mnichově. Později se tyto dvě metody, chirurgická léčba s předoperační terapií, začaly používat v kombinaci a znamenalo to významný posun v léčbě. Chemoterapie se začala používat v léčbě Wilmsova nádoru ve 40. letech 20. století, nejlepší účinnost vykazoval aktinomycin, později doplněný vinkristinem a antracykliny (1).
V roce 1969 byla v USA založena National Wilms Tumor Study – NWTS a v Evropě nadnárodní skupina SIOP – International Society of Pediatric Oncology. Tyto dvě organizace si stanovily za cíl hledání optimálního léčebného postupu, nejvhodnější kombinace cytostatik, stanovení rizikových faktorů. Zásadním rozdílem obou protokolů je zařazení nefrektomie v léčebném postupu. NWTS zahajuje léčbu primární nefrektomií a dále pokračuje chemoterapií a radioterapií podle stadia a typu nádoru. SIOP protokol řadí chirurgickou léčbu až po předoperační chemoterapii. Předoperační chemoterapie zmenšuje nádorovou masu a snižuje riziko peroperační ruptury tumoru, její nevýhodou je ovlivnění nádorové tkáně z hlediska dalšího genetického vyšetření.
Díky dlouholeté spolupráci těchto dvou institucí se přežití dětí s nefroblastomem významně zlepšilo, změnily se vyšetřovací postupy, snížila toxicita léčby u příznivých typů nádoru a naopak se zintenzivnila léčba u tumorů s nepříznivou prognózou.
GENETIKA A RIZIKOVÉ FAKTORY
Wilmsův nádor se samostatně většinou vyskytuje u zdravých dětí. Asi v 8 % případů se udává přítomnost dalších přidružených vad. Nejčastěji jsou spojeny se syndromem hypertrofie a hemihypertrofie. Typickým příkladem je Beckwith-Wiedemannův syndrom (nadměrný růst, makroglosie, exomfalus, nefromegalie, hepatomegalie, hemihypertrofie), Perlmanův syndrom (visceromegalie, makrosomie, polyhydramnion), Sotosův syndrom (makrocefalie, opožděný vývoj). Další syndromy nejsou spojené s nadměrným růstem např. WAGR (Wilmsův nádor, aniridie, anomálie genitálu, mentální retardace) a Denys-Drash syndrom (poruchy gonadální diferenciace a glomeruloskleróza).
WT1 protein – gen byl poprvé izolován a klonován v r. 1990 u pacienta s Wilmsovým nádorem. Tento gen je lokalizován na dlouhém raménku 11. chromozomu a má významnou funkci v růstu a diferenciaci buněk (3, 4). U pacientů s Wilmsovým nádorem je mutovaný asi u 20 % nádorů a v některých pracích je mutace WT1 genu spojována s horší prognózou onemocnění. Mutace WT1 genu se nachází i u jiných typů nádorů např. u neuroblastomu, skeletálních nádorů, u akutní leukemie. Germinální mutace na tomto genu způsobuje Denys-Drash syndrom, u těchto pacientů se rozvíjí selhání ledvin a mají velmi vysoké riziko vzniku Wilmsova nádoru. WT2 protein – gen na 11. chromozomu 11p15 je zodpovědný za zvýšenou produkci inzulinu a nadměrný růst (5, 6). V posledních letech studie Pritchard-Jonesové ve Velké Británii vytipovala gen na 1q chromozomu, jako další prognosticky rizikový faktor (5).
Rizikovým faktorem pro vznik Wilmsova nádoru je nefroblastomatóza, která je považována za prekancerózu. Nefroblastomatóza sama o sobě není indikována k léčbě, ale vyžaduje pečlivou dispenzarizaci. Indikací k léčbě je náhlý nadměrný růst ložisek, což může být známkou maligního zvratu. Většina ložisek ale vymizí, nebo se přemění v benigní útvary.
NWTS dále rutinně vyšetřuje nádorovou tkáň na přítomnost ztráty heterozygozity 1p nebo 16q chromozomu, což znamená horší prognózu (7).
PATOLOGICKÁ ANATOMIE
Klasický nefroblastom obsahuje tři buněčné složky: epiteliální, stromální a blastémovou, které jsou v nádoru zastoupeny v různé míře. Pokud jsou v nádoru zastoupeny všechny složky, nazýváme jej trifázický. Dále mohou být v nádoru ložiska anaplazie.
Podle histologie a regrese po předoperační terapii dělí SIOP nefroblastomy na tumory nízkého, průměrného a vysokého rizika. Mezi nefroblastomy nízkého rizika řadíme cysticky diferencovaný nefroblastom a zcela nekrotický tumor po předchozí léčbě. Mezi nádory středního rizika patří epiteliální, stromální, smíšený regresivní po léčbě a fokálně anaplastický. Mezi nádory vysokého rizika konečně patří blastémový typ a difuzní anaplazie. Změnou oproti minulému protokolu SIOP93–2001 je zařazení blastémového typu (BT-WT) mezi nádory vysokého stupně rizika, zatímco dříve byl řazen mezi nádory středního rizika (2, 8).
Pro klasifikaci nádoru a tedy i adekvátní léčbu je proto zásadní správné zhodnocení preparátu patologem. Obecně stále platí, že pokud jsou méně než 2/3 tumoru nekrotické, určuje se typ nádoru podle typu převažujících buněk. Pokud jsou 2/3 z vitální tkáně nádoru blastémové, je nádor klasifikován jako blastémový nefroblastom (2, 8).
KLINICKÉ PROJEVY A DIAGNOSTIKA
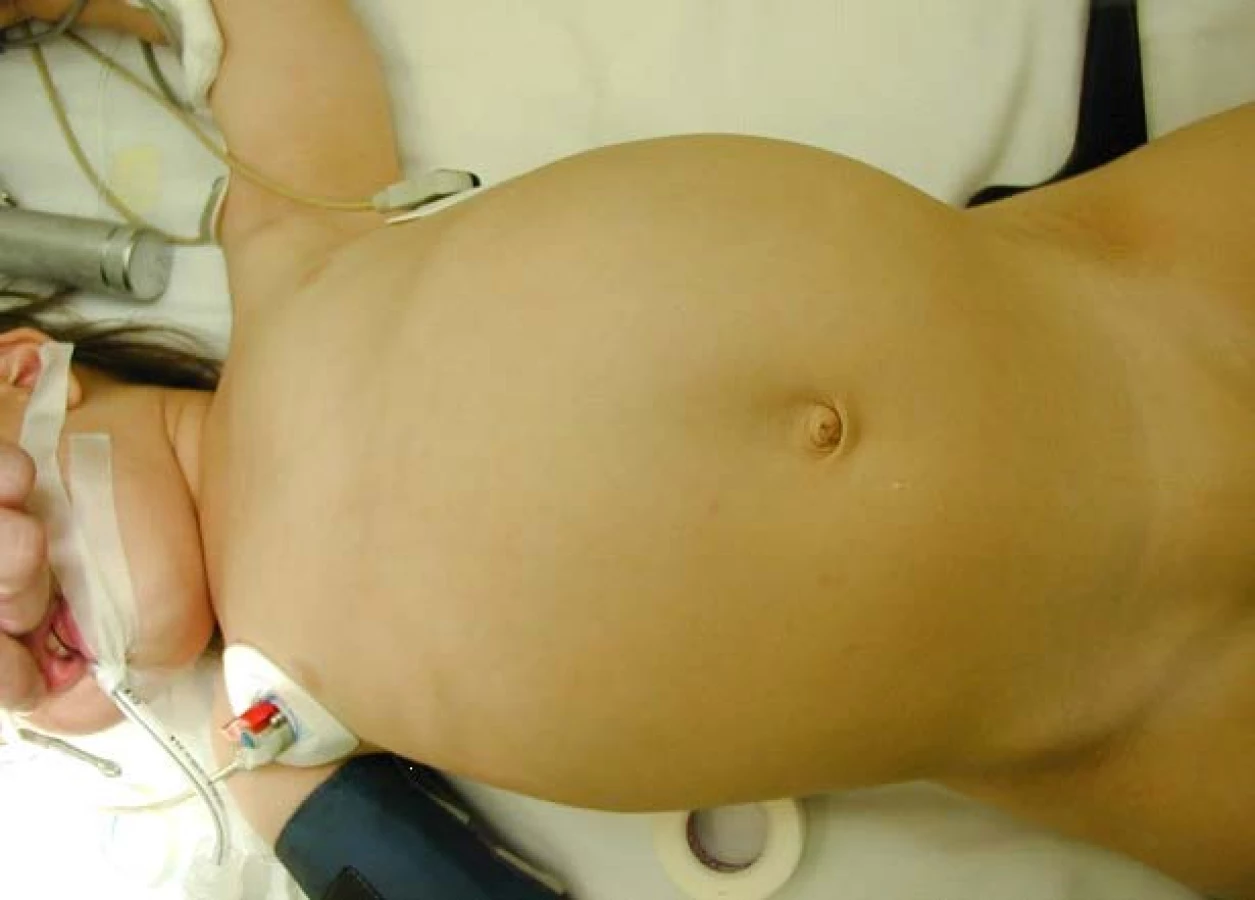
Nejčastějším klinickým projevem je nález hmatné rezistence v dutině břišní, dalšími projevy mohou být bolest břicha, hematurie, horečky.
Mezi základní vyšetření patří fyzikální vyšetření pacienta. Zjišťujeme stranu a velikost hmatného tumoru, velikost jater, měříme krevní tlak (hypertenzi nalézáme až u 50 % pacientů s nefroblastomem), vyšetřujeme regionální lymfatické uzliny a pátráme po kongenitálních anomáliích (aniridie, hemihypertrofie, urogenitální malformace). Vyšetřujeme i genitál, pokročilý tumor se může manifestovat varikokélou, jsou popisované i metastázy lokalizované v testikulární a paratestikulární tkáni (9).
Dále vyšetřujeme laboratorně krevní obraz (počet leukocytů, diferenciál, hodnotu hemoglobinu a hematokritu), sérovou hladinu kreatininu, moč biochemicky, pátráme po proteinurii, hematurii, leukocyturii, přítomnosti katecholaminů.

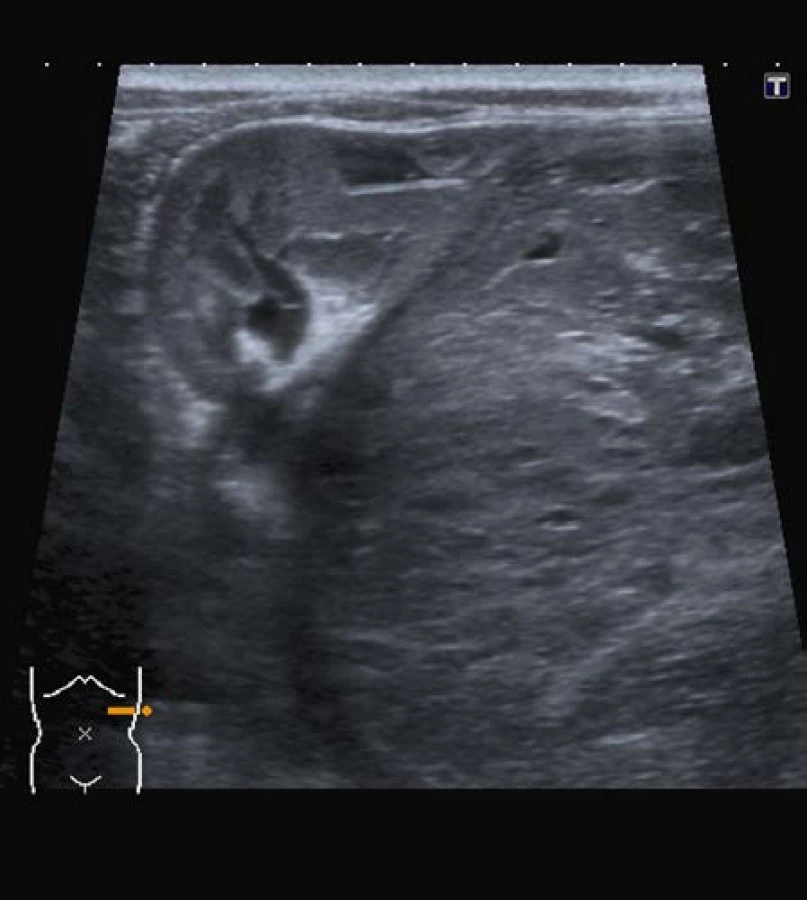
Základní zobrazovací metodou je ultrasonografické vyšetření břicha, které zobrazí typický obraz nádorem změněné ledviny. Vyšetření není invazivní, detekuje i malé tumory v druhostranné ledvině, je schopné rozpoznat tromby v renální a dolní duté žíle, jaterní a abdominální metastázy. Velikost nádoru je měřena ve třech kolmých rovinách a lze odhadnout přibližný objem nádoru.
V případě diagnostických nejasností indikujeme další vyšetření. Magnetická rezonance (MRI) nebo počítačová tomografie (CT) upřesní velikost nádoru, vztah k okolním tkáním a strukturám, přítomnost kalcifikací, cystického podílu v nádoru, infiltraci lymfatických uzlin, infiltraci extrarenálních žil. MRI dnes u dětí, pokud je to možné, upřednostňujeme před CT vyšetřením, z důvodu absence ionizujícího záření. Speciální sekvence diffusion weight (DW-MRI) navíc dokáže rozpoznat vysoce buněčné části nádoru – ložiska blastému (10, 11).




K vyloučení metastatického procesu plic dostačuje předozadní a boční rentgenový snímek hrudníku. Neprokázalo se, že pacienti s metastatickým postižením plic diagnostikovaným jen na CT vyšetření by měli horší prognózu (12). Za metastatický rozsev do plic považujeme jen nález metastáz na rentgenovém snímku plic.
Na základě fyzikálního vyšetření, zobrazovacích metod a peroperačního nálezu je stanoveno klinické stadium nádoru.

LÉČBA
a) Předoperační léčba
Podle protokolu SIOP 2001 je u jednostranného tumoru léčba zahájena předoperační terapií po dobu čtyři týdnů, spočívající v podání vinkristinu v dávce 1,5 mg/m2 v kombinaci s aktinomycinem D 45 mg/kg 1. a 3. týden. Pokud jsou přítomny metastázy, je k této kombinaci podán navíc doxorubicin ve dvou dávkách 50 mg/m2, celková léčba trvá šest týdnů. U dětí s hmotností menší než 12 kg se dávka snižuje na 2/3 vypočtené dávky (2, 8).
b) Chirurgická léčba
Následuje po chemoterapii. SIOP 2001 doporučuje klasický chirurgický postup, popsán Laddem a Grossem v roce 1953, spočívající v transabdominálním přístupu a časné ligaci renálních cév, která snižuje riziko pooperační ruptury nádoru (13). Součástí operace je revize hilových a paraaortálních uzlin a jejich odebrání i v případě, kdy se nám nejeví makroskopicky infiltrované. Zpravidla odebíráme 2–3 uzliny a odstraňujeme pararenální tuk. Při jednostranném postižení není nutné vyšetření druhostranné ledviny, negativní nález na zobrazovacích vyšetřeních považujeme za dostatečný. Dále jsou k chirurgické léčbě indikovány reziduální metastázy v plicích nebo játrech.
V případě bilaterálního postižení, pokud to nález umožňuje, provádíme parciální resekci obou ledvin. Volíme širokou příčnou laparotomii, aby dutina břišní byla co nejvíce přehledná a aby byla možná bezpečná revize obou ledvin, lymfatických uzlin a případných metastáz. Pokud by nebyla jasná hranice nádoru, je možné během operace provést ultrazvukové vyšetření k jejímu upřesnění (14). Je doporučeno resekovat nádor 1 cm do zdravé tkáně, pokud to nález umožňuje. Resekovaný parenchym uzavíráme jednotlivými stehy. Jestliže dojde k otevření dutého systému, provádí se jeho sutura vstřebatelným materiálem a odvodný systém můžeme podle nálezu buď ponechat zcela bez drenáže, nebo zavést nefrostomii či drénovat pomocí zavedeného J-J stentu (15). Popisován je i retroperitoneální přístup u oboustranných nádorů, ale běžně se neužívá. Snižuje riziko postižení střeva a umožňuje časnější obnovení perorálního příjmu (16).
Neméně důležitá je i kvalita života po léčbě. Podstatná ztráta nefronů je spojena s rizikem rozvoje renální hypertenze a následně renálního selhání (17). Pacienti po bilaterální parciální nefrektomii mají zachován větší objem funkčního parenchymu než pacienti s nefrektomií a druhostrannou parciální resekcí a jsou i méně zatíženi vznikem renální hypertenze.
Primární nefrektomii volíme v případě diagnostických nejasností, mezi které patří neobvyklý věk při diagnóze (pod ½ roku a nad 5 let), ruptura tumoru, krvácení do tumoru, masivní hematurie. Většina dětí je operována plánovaně, jen výjimečně musí být operace indikována urgentně, a to zejména v případě krvácení do tumoru a anemizaci. Operace plánovaně během operačního programu snižuje riziko peroperačních komplikací (18).
Poškození pouzdra nádoru, ruptura nádoru nebo nádorový rozsev krevní či lymfatickou cestou může významně zhoršit prognózu pacienta (19). Počet pooperačních ruptur udává NWTS kolem 20 %, SIOP 6 % (1). Podle studie NWTS je větší riziko ruptury u nádorů lokalizovaných na pravé straně a u nádorů větších než 12 cm (19).
Ačkoliv parciální resekce (nephron sparing surgery – NSS) i laparoskopický přístup nejsou stávajícím protokolem doporučovány, některá centra se těmito přístupy zabývají. V rámci SIOP a NWTS probíhají studie, které mají za cíl shromáždit data a zhodnotit výsledky léčby. Podle jejich výsledků nezhoršuje NSS i laparoskopický přístup přežití pacientů (20, 21).
Laparoskopie je oproti klasickému přístupu znevýhodněna absencí palpačního zhodnocení tkáně, navíc vzhledem k malému počtu pacientů, kteří podstoupili miniinvazivní nefrektomii, chybí zkušenosti a data. Indikace pacientů k laparoskopické operaci je omezena hlavně velikostí nádorů. Z multicentrické studie pro SIOP vyplývá, že medián velikosti nádorů operovaných laparoskopicky byl 5 cm (2,8–12) a medián hmotnosti byl 134 g (47–730). Jedná se tedy převážně o velmi malé nádory (20).
c) Pooperační léčba
Léčba 1.–3. klinického stadia
Délka a intenzita pooperační léčby se řídí stagingem: peroperačním nálezem a výsledkem histologie.
- Nefroblastomy nízkého rizika – kompletně nekrotický tumor – nevyžadují v 1. KS další léčbu, ve II. a III. stadiu jsou léčeny kombinací vinkristin, aktinomycin (AV) po dobu 27 týdnů.
- Nádory středního rizika jsou léčeny v I. stadiu AV 4 týdny, ve II. stadiu kombinací AV 27 týdnů, ve III. stadiu AV + radioterapie 27 týdnů.
- Nádory vysokého rizika jsou v 1. klinickém stadiu nádoru léčeny kombinací vinkristin/ aktinomycin/doxorubicin po dobu 26 týdnů. U pacientů II. a III. stadia BT-WT následuje léčba v kombinaci cyklofosfamid, etoposid, carboplatina, doxorubicin (300 mg/m2). Lokální radioterapie je indikovaná u II. i III. stadia. Radioterapie celé břišní dutiny je indikovaná v případě ruptury nádoru (děti 1 rok 20 Gy), jinak se volí radioterapie lůžka tumoru a případných metastáz v lymfatických uzlinách.
Rozdíl oproti minulému protokolu je v léčbě nádorů středního rizika, kdy v I. stadiu se zkrátila léčba a ve II. a III. stadiu se upustilo od podání etoposidu. Zároveň se blastémový typ nefroblastomu zařadil z nádorů středního rizika mezi nádory vysokého rizika a je léčen agresivněji.

Léčba 4. klinického stadia
Po operaci u středního rizika, pokud je lokálně nádor jen v ledvině a jsou přítomny reziduální metastázy, léčíme kombinací AVD po dobu 27 týdnů bez radioterapie, pokud je lokálně nádor ve 3. stadiu pak i s lokální radioterapií. V případě neoperovaných nebo po CHT nemizejících metastáz je indikována léčba CCED – při lokálním stadiu 1 a 2 bez záření, v lokálním stadiu 3 se zářením.
U nádorů vysokého rizika všichni pacienti podstoupí léčbu kombinací CCED, všichni mají ozáření plic, pokud se jedná lokálně o stadium 1, pak bez ozáření břicha, ve stadiu 2 a 3 i s ozářením břicha (8).

Léčba 5. klinického stadia
V případě bilaterálního postižení předoperační chemoterapie trvá tak dlouho, dokud se nádor zmenšuje a poté následuje operace. Cílem operační léčby je zachovat co nejvíce funkčního parenchymu. Nejčastěji provádíme nefrektomii na straně objemnějšího nádoru a parciální resekci na druhé straně. Pokud jsou oba nádory malé s příznivou lokalizací, lze provést i oboustrannou parciální resekci. Dále následuje pooperační léčba podle histopatologického typu nádoru, stejně jako u předchozích stadií.
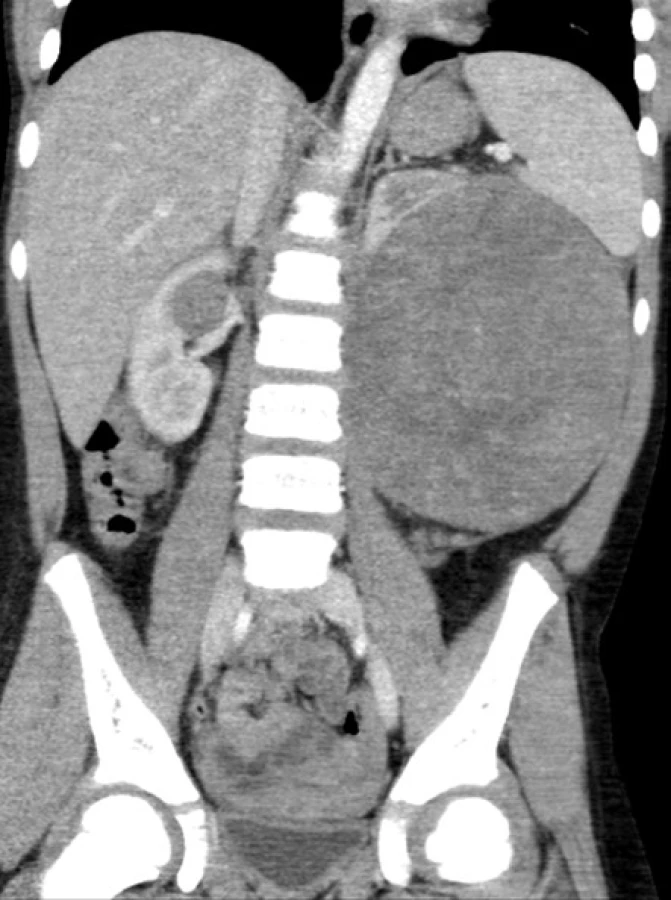


DISKUZE
Wilmsův nádor je velmi dobře léčitelné onemocnění. Zvláště lokalizované tumory nízkého a středního rizika mají přežití více jak 90 %. Častější relapsy a nižší přežití mají histologicky nepříznivé typy nádoru, anaplastický a blastémový. Pokud při histologickém vyšetření přetrvává blastémová složka po dokončení předoperační chemoterapie, blastémový typ (BT-WT), tak je WT řazen mezi nádory vysokého rizika a je léčen intenzivněji než v předchozích protokolech, kombinací čtyři cytostatik (cyklofosfamid, karboplatina, doxorubicin, etoposid) a radioterapie. Výsledky hodnotila multicentrická studie srovnávající předchozí protokol SIOP93–01 a současný SIOP2001 (2). U BT-WT za použití více intenzivní chemoterapie se snížil výskyt relapsů z 33 % na 16 %, pětileté přežití bez příhod (EFS) se zvýšilo z 67 % na 80 % a celkové přežití z 84 % na 88 %. Nejvýznamnější posun byl zaznamenán u BT - -WT v I. klinickém stadiu, kdy EFS podle protokolu SIOP93–01 bylo 71 % (OS 90%) vs. 96 % (OS 100 %) podle protokolu SIOP2001.
V průběhu let na základě výsledků studií upravovaly NWTS a SIOP své protokoly léčby, postupně redukovaly intenzitu chemoterapie, dávku ozáření a došlo až k úplnému vyřazení chemoterapie u nádoru nízkého rizika v I. klinickém stadiu. Hlavním rozdílem mezi SIOP a NWTS je řazení primární chemoterapie a nefrektomie, kdy skupina NWTS primárně indikuje pacienty k nefrektomii, zatímco skupina SIOP primárně indikuje všechny pacienty s nádorem vycházejícím z ledviny k podání předoperační chemoterapie a nefrektomie se provádí odloženě. Výhodou primární nefrektomie je, že poskytuje chemoterapií neovlivněnou tkáň, což umožňuje přesnější vyšetření molekulárně biologickými metodami. Předoperační chemoterapie nádor dokáže zmenšit a významně snížit riziko peroperační ruptury. Poškození pouzdra nádoru, ruptura nádoru nebo nádorový rozsev krevní či lymfatickou cestou je udáváno jako horší prognostický faktor (11). Procento komplikací je podle studiích udávaných SIOP 6 % a až 20 % podle studií NWTS. Přesto se výsledky přežití SIOP i NWTS významně neliší. Také dlouhodobé výsledky našich pacientů, kteří prodělali předoperační chemoterapii nebo kteří byli primárně operováni, byly prakticky stejné (1).
Z výsledků studií protokolu SIOP 2001 vychází i příprava nového protokolu. Jedním z cílů je zlepšit časnou diagnostiku blastému, využití DW-MRI, dále se větší důraz klade na rutinní genetické vyšetření tkáně, přesnější odběr materiálu na histologické vyšetření apod. Přísnější bude i klasifikace blastému patologem, rozhodující bude absolutní množství blastému, bez ohledu na to, jak nádor zareagoval na chemoterapii. Protokol dále umožní laparoskopické operace či parciální resekce u jednostranných nádorů a stanoví kritéria i kontraindikace těchto výkonů. Parciální resekce u jednostranného nádoru bude možná v případě periferně uloženého nádoru, bez porušení pouzdra, bez ruptury, bez propagace do cév, kalichů a pánvičky, s dostatečným lemem a za předpokladu, že bude zachován dostatek funkčního parenchymu. Laparoskopická nebo laparoskopicky asistovaná nefrektomie je možná u malých nádorů s dostatečným lemem zdravé tkáně ledviny, za předpokladu odebrání lymfatických uzlin.
ZÁVĚR
Wilmsův nádor je nádor s dobrou odezvou na léčbu a s velmi dobrou prognózou. Úspěšná léčba a dlouhodobé přežití jsou založeny na úzké multioborové spolupráci. Role chirurga spočívá ve zhodnocení klinického stadia, bezpečném odstranění nádorové tkáně přístupem, který minimalizuje riziko peroperačního rozsevu nádoru a odebrání okolní tkáně a lymfatických uzlin k určení stadia onemocnění. Cílem onkologické léčby je snížit toxicitu léčby u histologicky příznivých typů a nepříznivé typy léčit agresivněji.
Došlo: 18. 1. 2016
Přijato: 6. 4. 2016
Kontaktní adresa:
MUDr. Marcela Pýchová
Klinika dětské chirurgie 2. LF UK a FN Motol
V Úvalu 84, 150 06 Praha
marcela.pychova@fnmotol.cz
Střet zájmů: žádný
Prohlášení o podpoře: Podpořeno projektem MZ ČR – RVO, FN v Motole 00064203.
Sources
1. Mališ J, Švojgr K, Pýcha K, Jeřábková V,Cyprová S, et al. Nephroblastoma-30-years period of treatement in the University Hospital Motol,Klin.Onkol. 2013; 26(5): 336–342. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24107156
2. Van den Heuvel-Eibrink MM, van Tinteren H, Bergeron C, et al. Outcome of localised blastemal-type Wilms tumour patients treated according to intensified treatment in the SIOP WT 2001 protocol, a report of the SIOP Renal Tumour Study Group (SIOP-RTSG). Eur J Cancer [Internet]. Elsevier Ltd; 2015; 51(4): 498–506.
Available from: http://linkinghub.elsevier.com/retrieve/pii/S095980491401171X
3. Parenti R, Salvatorelli L, Musumeci G, et al. Wilms’ tumor 1 (WT1) protein expression in human developing tissues. Acta Histochem [Internet]. Elsevier GmbH.; 2015; 1 : 1–11. Available from: http://linkinghub. elsevier.com/retrieve/pii/S0065128115000574
4. Salvatorelli L, Parenti R, Leone G, Musumeci G, Vasquez E, Magro G. Wilms tumor 1 (WT1) protein: Diagnostic utility in pediatric tumors [Internet]. Acta Histochemica. Elsevier GmbH.; 2015. 1–12 p.
Available from: http://linkinghub.elsevier.com/retrieve/pii/S0065128115000586
5. Freier K, Knoepfle K, Flechtenmacher C, et al. Recurrent copy number gain of transcription factor SOX2 and corresponding high protein expression in oral squamous cell carcinoma. Genes Chromosomes Cancer. 2010; 49(1): 9–16.
6. Dumoucel S, Gauthier-Villars M, Stoppa-Lyonnet D, et al. Malformations, genetic abnormalities, and Wilms tumor.Pediatr Blood Cancer. 2014 Jan, 61(1): 140–144. doi:10.1002/pbc.24709. Epub 2013 Aug 23.
7. Gratias EJ, Jennings LJ, Anderson JR, Dome JS, Grundy P, Perlman EJ. Gain of 1q is associated with inferior event-free and overall survival in patients with favorable histology Wilms tumor: A report from the Children’s Oncology Group. Cancer. 2013; 119(21): 3887–3894.
8. Protocol SIOP 2001Wilm’s tumor.
9. Ansari S, MIiri AB, Rakhshani N. Bilateral Wilms’ Tumor Metastasis to Right Spermatic Cord. Int J Hematol Oncol Stem Cell Res. 2013; 7(4): 45–48.
10. Umbrella Protocol Siop 2014.
11. Dumba M, Jawad N, McHugh K. Neuroblastoma and nephroblastoma: a radiological review. Cancer Imaging [Internet]. 2015; 15(1). Available from: http://www.cancerimagingjournal.com/content/15/1/5
12. Smets AMJB, Tinteren H Van, Bergeron C, et al. The contribution of chest CT-scan at diagnosis in children with unilateral Wilms’ tumour. Results of the SIOP 2001 study. Eur J Cancer [Internet]. Elsevier Ltd; 2012; 48(7): 1060–1065.
Available from: http://dx.doi.org/10.1016/j.ejca.2011.05.025
13. Godzinski J, Graf N, Audry G. Current concepts in surgery for wilms tumor – the risk and function - -adapted strategy. 2014;
14. Harel M, Makari JH, Ferrer FA. Oncology: The role of partial nephrectomy in wilms tumor. Curr Urol Rep. 2013; 14(4): 350–358.
15. Kieran K, Davidoff AM. Nephron-sparing surgery for bilateral Wilms tumor. Pediatr Surg Int [Internet]. 2015; 31(3): 229–236. Available from: http://link.springer.com/10.1007/s00383-015-3668-1
16. Isabel I, Joshua PL, Elizabeth NH, et al. Experience with Retroperitoneal Partial Nephrectomy in Bilateral Wilms Tumor. 2015;
17. Hubertus J, Günther B, Becker K, Graf N, Furtwängler R, Ferrari R, et al. Development of hypertension is less frequent after bilateral nephron sparing surgery for bilateral wilms tumor in a long-term survey. J Urol [Internet]. 2015; 193(1): 262–267.
Available from: http://linkinghub.elsevier.com/retrieve/pii/S0022534714041585
18. Forbes C, Butterworth SA. Perioperative outcomes of primary renal tumour resections: comparison of in-hours to out-of-hours surgery. Pediatr Surg Int [Internet]. 2014; 30(10): 1003–1007.
Available from: http://link.springer.com/10.1007/s00383–014–3560–4
19. Gow KW, Barnhart DC, Hamilton TE, et al. Primary nephrectomy and intraoperative tumor spill: Report from the Children’s Oncology Group (COG) renal tumors committee. J Pediatr Surg [Internet]. Elsevier Inc.; 2013; 48(1): 34–88.
Available from: http://dx.doi.org/10.1016/j.jpedsurg.2012.10.015
20. Warmann SW, Godzinski J, van Tinteren H, et al. Minimally invasive nephrectomy for Wilms tumors in children – data from SIOP 2001. J Pediatr Surg [Internet]. Elsevier Inc.; 2014; 49(11): 1544–1548. Available
from: http://linkinghub.elsevier.com/retrieve/pii/S0022346814003935
21. Kieran K, Williams MA, McGregor LM, Dome JS, Krasin MJ, Davidoff AM. Repeat nephron-sparing surgery for children with bilateral Wilms tumor. J Pediatr Surg [Internet]. Elsevier Inc.; 2014; 49(1): 149–153.
Available from: http://dx.doi.org/10.1016/j.jpedsurg.2013.09.048
Labels
Paediatric urologist Nephrology UrologyArticle was published in
Czech Urology

2016 Issue 2
Most read in this issue
- THE CONSEQUENCES OF PENILE SUBCUTANEOUS APPLICATIONS OF THE FOREIGN MATERIALS
- NEPHROBLASTOMA – CURRENT EVALUATION AND TREATMENT
- BLACK ADENOMA OF THE ADRENAL GLAND
- KEY UROLOGICAL SURGICAL PROCEDURES IN THE CZECH REPUBLIC IN PERIOD 2009–2014