
Novinky WHO klasifikace z roku 2022: klasifikace renálních tumorů
The WHO Classification of Urinary and Male Genital Tumours 2022: renal tumours classification
The 5th edition WHO (World Health Organization) classification of the urogenital tract tumors was published in the middle of last year (so-called blue book). The new WHO classification reflects very dynamic developments in the field of renal neoplasms (caused mainly by the more frequent implementation of molecular genetic testing into the routine diagnosis of kidney tumors), and the new WHO classification shows changes especially in the chapter on renal tumors. The new kidney tumor classification modifies some traditional entities and also redefines and includes some new kidney tumors. This review article aims to briefly outline and comment on the most significant changes in the WHO classification of renal neoplasia.
Keywords:
urogenital tract – Molecular pathology – Renal cell carcinoma – renal tumours – WHO 2022 classification.
Authors:
Adriena Bartoš Veselá 1; Tomáš Pitra 1; Jiří Kolář 1; Josef Skopal 2; Ondřej Fiala 3; Milan Hora 1; Ondřej Hes 2,4; Kristýna Pivovarčíková 2,4; Zesnulý
Authors‘ workplace:
Urologická klinika, Lékařská fakulta Plzeň, Univerzita Karlova a Fakultní nemocnice Plzeň
1; Šiklův ústav patologie, Lékařská fakulta Plzeň, Univerzita Karlova a Fakultní nemocnice Plzeň
2; Onkologická a radioterapeutická klinika, Lékařská fakulta Plzeň, Univerzita Karlova a Fakultní, nemocnice Plzeň a Biomedicínské centrum, Lékařská fakulta Plzeň, Univerzita Karlova
3; Bioptická laboratoř, s. r. o., Plzeň
4
Published in:
Ces Urol 2023; 27(2): 70-77
Category:
Review articles
Overview
V polovině minulého roku vyšlo již 5. vydání klasifikace nádorů urogenitálního traktu Světové zdravotnické organizace (WHO), tzv. modrá kniha. Nová WHO klasifikace přináší především změny v rámci kapitoly renálních tumorů, pouze menší změny se pak dotkly ostatních kapitol. Právě nová klasifikace renálních tumorů odráží velmi dynamický vývoj na poli renálních neoplazií z posledních let, který je z části i odrazem čím dál tím častějšího využití molekulární genetiky při diagnostice renálních tumorů. Nová klasifikace renálních tumorů upravuje některé tradiční jednotky a vymezuje a zařazuje též některé nově vzniklé entity. Toto sdělení si klade za cíl v krátkosti a přehledně nastínit a okomentovat nejvýznamnější změny v rámci klasifikace renálních neoplazií.
Klíčová slova:
urogenitální trakt – renální karcinom – molekulární patologie – renální neoplazie – WHO 2022 klasifikace.
ÚVOD
WHO představila v první polovině minulého roku již 5. vydání (nejprve elektronicky a posléze i v tištěné formě) nové klasifikace nádorů vylučovacího a mužského genitálního traktu (1). Histologické klasifikace v rámci různých patologických odvětví prožívají v současné době velmi dynamický vývoj a výrazné změny, často pod „palbou“ nových molekulárně‑genetických zjištění. Ani genitourinární (GU) trakt pak není výjimkou. Velký nápor přibývajících nových zjištění v GU patologii pak odráží i fakt, že od 4. vydání „modré knihy“ uplynulo pouhých šest let a je tak evidentní, že interval mezi po sobě následujícími vydáními WHO se výrazně zkrátil (3. vydání WHO klasifikace vyšlo v roce 2004 (2), 4. vydání v roce 2016 (3) a 5. vydání v roce 2022 (1)). Nejlépe je dynamika rozvoje patrná zejména na poli renálních neoplazií.
Jak už je zvykem, vlastnímu vydání nové WHO klasifikace předcházela řada diskuzí a „consensus“ konferencí odborných mezinárodních patologických GU společností (Genitourinary Pathology Society/GUPS, International Society of Urological Pathology/ISUP) a publikace doporučených postupů daných společností, ze kterých nové WHO klasifikace z větší části vycházejí (4–6).
NOVINKY WHO 2022
Za hlavní změnu 5. vydání klasifikace renálních neoplazií lze považovat zavedení dvou nových kategorií (skupiny „molekulárně definovaných renálních karcinomů“ a kategorie „ostatní onkocytické tumory“), dále pak začlenění vybraných nových neoplastických entit do klasifikace a místy výraznější i drobné změny nomenklatury některých neoplazií (konkrétně viz níže) (1). Jak už je vzhledem k názvu skupiny „molekulárně definovaných renálních karcinomů“ patrné, diagnostika tumorů zařazených do této skupiny vždy vyžaduje provedení molekulárně‑genetických testů pro stanovení diagnózy, je tedy třeba si uvědomit, že bez genetických vyšetření se dnešní diagnostika renálních tumorů již neobejde.
Jako klasicky WHO popisuje základní diagnostická kritéria renálních neoplazií, jejich morfologii, imunohistochemické a molekulárně genetické vlastnosti, komentuje incidenci, etiologii, biologické chování. Zde prezentovaný text si neklade za cíl do detailu popsat všechny v současnosti rozeznávané renální neoplazie, v textu níže budou komentovány pouze jednotky s menšími či většími změnami oproti předchozí WHO klasifikaci.
Papilární renální karcinom (PRCC) byl historicky a v předchozí WHO klasifikaci 2016 členěn na typ 1 a typ 2 (7). Zároveň předchozí WHO klasifikace připouštěla i existenci tzv. onkocytické varianty PRCC (3). Postupně se však ukázalo, že dělení PRCC na typ 1 a 2 je nedostatečné. PRCC může vykazovat výraznou heterogenitu morfologického vzhledu a jsou relativně časté i PRCC, které do daných kategorií nelze spolehlivě zařadit (8). Zároveň v posledních 10 letech začalo přibývat velké množství prací, popisujících různé další morfologické varianty/ subtypy PRCC (9–13). Nová WHO klasifikace 2022 tak dělení PRCC na typ 1 a typ 2 již nadále nedoporučuje, odlišování „onkocytického PRCC“ jako subtypu PRCC též není ve světle současných znalostí doporučováno (14). V rámci PRCC však WHO radí spíše popisovat jednotlivé histologické subtypy (pokud je to možné) – mezi ně patří: PRCC klasický typ (morfologicky zcela odpovídá dříve popisovanému PRCC typ 1), papilární renální karcinom se světlobuněčnými elementy/světlobuněčnými změnami, bifázický typ se skvamoidními buňkami a emperipolézou, Warthin‑like PRCC, případně PRCC blíže nespecifikovaný (Not Otherwise Specified/NOS) (1).
Nově zavedenou kategorií je diagnostická skupina „ostatní onkocytické tumory ledviny“. Jedná se o kategorii, v níž by měly být klasifikovány eosinofilní/onkocytické tumory (patology často označované jako „růžové“ tumory) morfologicky nesplňující kritéria pro diagnózu renálního onkocytomu, chromofobního renálního karcinomu, ani jiného jasně definovaného „růžového“ renálního tumoru (tj. „růžové“ tumory z „šedé zóny“) (1). Tato kategorie tak představuje „odpadkový koš“, do nějž spadnou různé morfologicky „růžové“ neoplazie, které nelze klasifikovat do žádné ze známých entit. Zavedení této jednotky, která je spíše než opravdovou svébytnou entitou jen diagnostickou heterogenní skupinou nádorů, je celkem racionálním krokem, neboť dříve byly nezařaditelné eosinofilní/ onkocytické renální tumory dle WHO klasifikovány jako renální karcinomy blíže nespecifikované (RCC NOS). Pokud však urolog pracoval s pacientem diagnostikovaným jako RCC NOS, většinou na něj nahlížel jako na pacienta s agresivní malignitou. To však u většiny těchto blíže nespecifikovaných/nezařaditelných eosinofilních/onkocytických renálních tumorů neplatí – prognóza je často dobrá, tumory se většinou chovají neagresivně. Do této skupiny však byly WHO zahrnuty i tumory, které možná do budoucna svébytnou jednotkou opravdu budou a též je v rámci této skupiny upravena nomenklatura týkající se neoplazií vznikajích za specifických klinických okolností. Konkrétně pak do této skupiny spadá např. dobře popsaná jednotka s konstantním morfologickým vzhledem, imunohistochemickým profilem a specifickým genetickým pozadím – low grade onkocytický tumor (LOT) (15). Podobně dobře definovanou jednotkou v současné době spadající do kategorie „jiných onkocytických tumorů“ je i eosinofilní vakuolizovaný tumor (EVT), který ač svou morfologií věrně připomíná high grade tumor (v úvodní práci popisující tento tumor autoři pro tuto jednotku celkem nešťastně zvolili lehce zavádějící název high grade onkocytický tumor/HOT (16)), je svým klinickým chováním indolentní (17). EVT i LOT mají abnormality v genech pro mTOR signální dráhy, mohou vznikat sporadicky, ale mohou být součástí manifestace tuberózní sklerózy (18). Jako „hybridní onkocytický tumor“ (též jednotka v rámci kategorie „ostatní onkocytické tumory“) by v současné době měly být označovány pouze „růžové“ tumory ze šedé zóny vznikající u pacientů s Birt-Hogg-Dubé syndromem (tj. prokázanou mutací genu FLCN), typicky spojené s výskytem mnohočetných renálních tumorů (často bilaterálně) (1). Označení „onkocytická renální neoplazie nízkého maligního potenciálu NOS/oncocytic renal neoplasms of low malignant potential NOS“ je rezervována pouze pro solitární sporadické „růžové“ renální neoplazie šedé zóny (1). Některými autory je též doporučováno používání tohoto indiferentního pojmu při diagnostice z limitovaného materiálu renálních punkcí, neboť heterogenita morfologického vzhledu skupiny „růžových tumorů“ v limitovaném materiálu často činí významné diagnostické obtíže a stanovení definitivní diagnózy z punkce je často nemožné (19).
Změnou nomenklatury prošel i tumor dříve nazývaný světlobuněčný papilární renální karcinom (ve WHO 2016) (3), nově na základě současných znalostí (benigní tumor) označovaný jako světlobuněčný papilární renální tumor (dle WHO 2022) (1). Jedná se o neoplazii s mírným histologickým překryvem s více častým low‑grade světlobuněčným renálním karcinomem (CCRCC), ale i některými vzácnějšími jednotkami. V diferenciálně diagnosticky obtížných případech lze využít molekulárně‑genetických metod pro definitivní stanovení diagnózy této indolentní jednotky (světlobuněčný papilární renální tumor nevykazuje genetické znaky typické pro CCRCC, tedy je pro něj typická absence abnormalit v genu VHL – methylace, mutace; bez ztráty heterozygozity 3p) (20).
Tab. 1. Overview of renal tumors (taken from WHO 2022 (1))
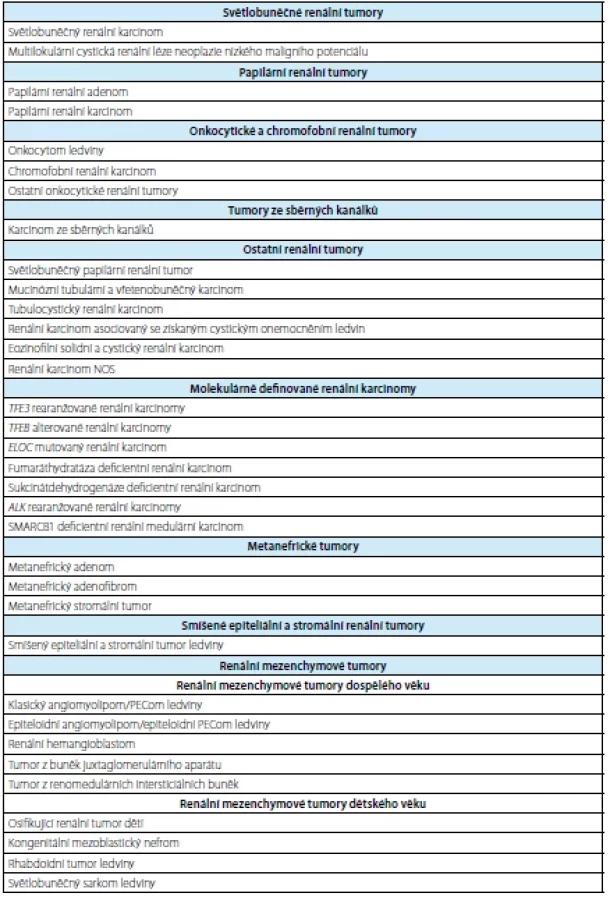
Tab. 1. Overview of renal tumors (taken from WHO 2022 (1))

Eosinofilní solidní a cystický renální karcinom (ESC RCC) je WHO nově definovanou entitou renálních karcinomů. Může se vyskytovat v hereditární formě u tuberózní sklerózy, častěji se však vyskytuje sporadicky, klasickým pacientem je starší žena (21). ESC RCC má typicky popisované abnormality v genech dráhy mTOR (22). Ač bylo v prvních studiích popisujících tuto jednotku polemizováno o indolenci této neoplazie, pozdější práce zdokumentovaly několik metastazujících případů (23).
Zásadní koncepční změnou nové WHO klasifikace je zavedení kategorie/kapitoly „molekulárně definovaných renálních karcinomů“ (1). Do kategorie byly zavzaty nádorové jednotky, jejichž histologický vzhled (a i imunohistochemický profil) často může být sice velmi sugestivní pro danou diagnózu, ale pro definitivní stanovení diagnózy je vždy vyžadováno použití molekulárně‑genetických metod s průkazem pro danou jednotku specifických genetických alterací. Před tím, než budeme komentovat jednotlivé neoplazie v této kategorii, je třeba zmínit, že z části jsou v této kapitole zahrnuty tumory z předchozí WHO klasifikace již známé (s menšími či většími úpravami), najdeme zde však i jednotky ve WHO zcela nové. Původní diagnostická kategorie „MiT rodina translokačních renálních karcinomů“ z WHO 2016 (3) byla rozdělena na samostatné jednotky – TFE3 rearanžované RCC a TFEB alterované RCC. V rámci TFEB alterovaných RCC pak rozlišujeme dále ještě dvě další jednotky TFEB translokované/rearanžované RCC a TFEB amplifikované RCC (1, 6). TFEB amplifikované RCC pak v porovnání s TFEB translokovanými/ rearanžovanými RCC vykazují výrazně agresivnější klinické chování, proto je třeba mezi těmito dvěma modalitami (které mohou pro nezasvěceného pozorovatele působit jako „slovíčkaření“) rozlišovat (24, 25). V rámci TFE3 rearanžovaných RCC nedošlo oproti předchozí WHO klasifikaci k výraznějším změnám.
Změnou nomenklatury a lehkým přehodnocením přístupu prošel i tumor, v nové klasifikaci označený jako „fumarát hydratáza (FH) deficientní renální karcinom“ (1). Toto označení je preferováno oproti původnímu krkolomnému názvu z WHO 2016 „s hereditární leiomyomatázou a renálním karcinomen asociovaný RCC“ (3). Tato jednotka si prošla ve třech WHO klasifikacích výrazným vývojem. Ve WHO z roku 2004 byla uvedena jako hereditární protipól PRCC (2). Ve WHO 2016 se stala samostatnou jednotkou a dostala již výše zmíněný název „s hereditární leiomyomatózou a renálním karcinomen asociovaný RCC“ (26), což odráželo pevnou víru autorů, že tyto agresivní tumory vznikají u pacientů s germinální mutací genu FH a syndromovou manifestací. Postupně však literatura odhalila, že nádory mohou vznikat (a často i vznikají) sporadicky (27), bez syndromové manifestace a tedy přišel čas na další změnu názvu. Nádory se vyznačují patologickou alterací FH genu (mutace, LOH), vedoucí k inaktivaci fumarát hydratázy (enzymu Krebsova cyklu podílejícího se na přeměně fumarátu na L‑malát) a tím mění metabolické pochody.
Další jednotkou „molekulárně definovaných RCC“ je nová entita TCEB1 (dříve ELOC) mutovaný renální karcinom. Ten se do značné míry dokáže podobat CCRCC, odlišuje se však přítomností objemného fibroleiomyomatózního stromatu a genetickým podkladem (kterým je přítomnost mutace TCEB1/ELOC genu) (28, 29). Fibroleiomyomatózní stroma a světlobuněčná morfologie buněk však mohou být markantou i jiných renálních neoplazií (30), které přicházejí v diferenciální diagnóze (tj. světlobuněčný papilární renální tumor, RCC s fibroleiomyomatózním stromatem). Genetické vyšetření by tak mělo být cíleno nejen na mutaci genu ELOC/TCEB1, ale i MTOR, TSC1 a TSC2, popř. VHL a LOH3p. Jedná se o raritní lézi, v současné literatuře nebylo zaznamenáno agresivní chování, avšak právě pro podobnost s jinými renálními neoplaziemi musíme za použití molekulárně genetických metod vyloučit jiné potenciálně agresivní typy nádorů (CCRCC).
Mezi molekulárně definované renální karcinomy patří i renální karcinomy s přestavbou ALK genu. U ALK rearanžovaných RCC byl zaznamenán potenciál agresivního chování a velmi dobrá odpověď na cílenou léčbu ALK inhibitory (31). Jedná se však opět o raritní typ neoplazie s omezenými klinickými daty vyžadující další studie. Ke stanovení diagnózy je nezbytná imunohistochemie a molekulární genetika. Význam této diagnózy tkví zejména v dostupnosti efektivní cílené terapie (32).
Další staronovou entitou je SMARCB1 deficientní renální medulární karcinom (původně medulární RCC). Jedná se o vysoce agresivní nádor, který se v našich podmínkách téměř nevyskytuje – typicky jsou postiženi pacienti Afroameričané se srpkovitou anémií. Diagnostická je nejčastěji inaktivační mutace genu SMARCB1 vedoucí ke ztrátě imunohistochemického barvení SMARCB1 (INI1) (33).
ZÁVĚR
Klasifikace renálních nádorů se dynamicky vyvíjí, a to zejména za přispění molekulárně genetických metod, které se stávají součástí rutinní patologické diagnostické praxe. Genetická vyšetření však nelze indikovat na všechny renální tumory, cena vyšetření je stále vysoká. Je tedy nutné na základě morfologie a imunohistochemického profilu léze pečlivě vybírat tumory, jejichž diagnostiku bez genetických vyšetření provést nelze.
Pro urologa se však management chirurgické léčby pacientů s renálním tumorem příliš nemění. Na základě nových poznatků však do budoucna nejspíše bude možné u některých pacientů aplikovat cílenou léčbu, jako je tomu u jedné z prvních vlaštovek – RCC s přestavbou ALK genu.
Došlo: 21. 2. 2023
Přijato: 20. 3. 2023
Kontaktní adresa:
doc. MUDr. Kristýna Pivovarčíková, Ph.D.
Šiklův ústav patologie LF UK a FN Plzeň
Alej Svobody 80
304 60 Plzeň
e-mail: pivovarcikovak@fnplzen.cz
Střet zájmů: Žádný.
Prohlášení o podpoře: Karlova univerzita Praha, Lékařská fakulta Plzeň (Cooperation Program, SURG), Institucionální výzkum Fakultní nemocnice Plzeň (FNPl 00669806) a SVV 260652.
Ces Urol 2023; 27(2): 70-77
Sources
1. Amin MB, Gill Aj, Hartmann A, et al. Chapter 2: tumours of the kidney. In: WHO Classification of Tumours Editorial Board. Urinary and male genital tumours. 5. ed. Lyon: IARC 2022 : 32–130.
2. Eble JN, Sauter G, Epstein JI, Sesterhenn I. Pathology and Genetics of Tumours of the Urinary System and Male Genital Organs.
3. ed. Lyon: IARC Press; 2004. 3. Moch H, Humphrey P, Ulbright TM, Reuter VE. WHO Classification of tumours of the urinary system and male genital organs. 4. ed. Lyon: IARC 2016.
4. Williamson SR, Gill AJ, Argani P, et al. Report From the International Society of Urological Pathology (ISUP) Consultation Conference on Molecular Pathology of Urogenital Cancers: III: Molecular Pathology of Kidney Cancer. Am J Surg Pathol. 2020; 44(7): e47–65.
5. Trpkov K, Williamson SR, Gill AJ, et al. Novel, emerging and provisional renal entities: The Genitourinary Pathology Society (GUPS) update on renal neoplasia. Mod Pathol. 2021; 34(6): 1167–1184.
6. Trpkov K, Hes O, Williamson SR, et al. New developments in existing WHO entities and evolving molecular concepts: The Genitourinary Pathology Society (GUPS) update on renal neoplasia. Mod Pathol. 2021; 34(7): 1392–1424.
7. Delahunt B, Eble JN. Papillary renal cell carcinoma: a clinicopathologic and immunohistochemical study of 105 tumors. Mod Pathol. 1997; 10(6): 537–544.
8. Pitra T, Pivovarcikova K, Alaghehbandan R, Hes O. Chromosomal numerical aberration pattern in papillary renal cell carcinoma: Review article. Ann Diagn Pathol. 2019; 40 : 189–199.
9. Pivovarcikova K, Peckova K, Martinek P, et al. „Mucin“-secreting papillary renal cell carcinoma: clinicopathological, immunohistochemical, and molecular genetic analysis of seven cases. Virchows Arch. 2016; 469(1): 71–80.
10. Rogala J, Kojima F, Alaghehbandan R, et al. Papillary renal cell carcinoma with prominent spindle cell stroma – tumor mimicking mixed epithelial and stromal tumor of the kidney: Clinicopathologic, morphologic, immunohistochemical and molecular genetic analysis of 6 cases. Ann Diagn Pathol. 2020; 44 : 151441.
11. Ulamec M, Skenderi F, Trpkov K, et al. Solid papillary renal cell carcinoma: clinicopathologic, morphologic, and immunohistochemical analysis of 10 cases and review of the literature. Ann Diagn Pathol. 2016; 23 : 51–57.
12. Skenderi F, Ulamec M, Vanecek T, et al. Warthin‑like papillary renal cell carcinoma: Clinicopathologic, morphologic, immunohistochemical and molecular genetic analysis of 11 cases. Ann Diagn Pathol. 2017; 27 : 48–56.
13. Hes O, Condom Mundo E, Peckova K, et al. Biphasic Squamoid Alveolar Renal Cell Carcinoma: A Distinctive Subtype of Papillary Renal Cell Carcinoma? Am J Surg Pathol. 2016; 40(5): 664–675.
14. Pivovarcikova K, Grossmann P, Hajkova V, et al. Renal cell carcinomas with tubulopapillary architecture and oncocytic cells: Molecular analysis of 39 difficult tumors to classify. Ann Diagn Pathol. 2021; 52 : 151734.
15. Williamson SR, Hes O, Trpkov K, et al. Low‑grade oncocytic tumour of the kidney is characterised by genetic alterations of TSC1, TSC2, MTOR or PIK3CA and consistent GATA3 positivity. Histopathology. 2023; 82(2): 296–304.
16. He H, Trpkov K, Martinek P, et al. „High‑grade oncocytic renal tumor“: morphologic, immunohistochemical, and molecular genetic study of 14 cases. Virchows Arch. 2018; 473(6): 725–738.
17. Farcaş M, Gatalica Z, Trpkov K, et al. Eosinophilic vacuolated tumor (EVT) of kidney demonstrates sporadic TSC/MTOR mutations: next‑generation sequencing multi‑institutional study of 19 cases. Mod Pathol. 2022; 35(3): 344–351.
18. Pivovarcikova K, Alaghehbandan R, Vanecek T, et al. TSC/mTOR Pathway Mutation Associated Eosinophilic/ Oncocytic Renal Neoplasms: A Heterogeneous Group of Tumors with Distinct Morphology, Immunohistochemical Profile, and Similar Genetic Background. Biomedicines. 2022; 10(2): 322.
19. Hora M, Albiges L, Bedke J, et al. European Association of Urology Guidelines Panel on Renal Cell Carcinoma Update on the New World Health Organization Classification of Kidney Tumours 2022: The Urologist’s Point of View. Eur Urol. 2023; 83(2): 97–100.
20. Williamson SR. Clear cell papillary renal cell carcinoma: an update after 15 years. Pathology 2021; 53(1): 109–119.
21. Trpkov K, Hes O, Bonert M, et al. Eosinophilic, Solid, and Cystic Renal Cell Carcinoma: Clinicopathologic Study of 16 Unique, Sporadic Neoplasms Occurring in Women. Am J Surg Pathol. 2016; 40(1): 60–71.
22. Mehra R, Vats P, Cao X, et al. Somatic Bi‑allelic Loss of TSC Genes in Eosinophilic Solid and Cystic Renal Cell Carcinoma. Eur Urol. 2018; 74(4): 483–486.
23. McKenney JK, Przybycin CG, Trpkov K, Magi‑Galluzzi C. Eosinophilic solid and cystic renal cell carcinomas have metastatic potential. Histopathology. 2018; 72(6): 1066–1067.
24. Argani P, Reuter VE, Zhang L, et al. TFEB‑amplified Renal Cell Carcinomas: An Aggressive Molecular Subset Demonstrating Variable Melanocytic Marker Expression and Morphologic Heterogeneity. Am J Surg Pathol. 2016; 40(11): 1484–1495.
25. Peckova K, Vanecek T, Martinek P, et al. Aggressive and nonaggressive translocation t(6;11) renal cell carcinoma: comparative study of 6 cases and review of the literature. Ann Diagn Pathol. 2014; 18(6): 351–357.
26. El‑Zaatari Z, Divatia MK. Hereditary leiomyomatosis and renal cell carcinoma syndrome‑associated renal cell carcinoma: Morphological appraisal with a comprehensive review of differential diagnoses. Indian J Pathol Microbiol. 2020; 63(Supplement): S7–17.
27. Kuroda N, Tsutsui M, Iguchi M, et al. Fumarate hydratase‑deficient renal cell carcinoma: A clinicopathological study of seven cases including hereditary and sporadic forms. Ann Diagn Pathol. 2020; 49 : 151599.
28. Hakimi AA, Tickoo SK, Jacobsen A, et al. TCEB1-mutated renal cell carcinoma: a distinct genomic and morphological subtype. Mod Pathol. 2015; 28(6): 845–853.
29. Wang Y, Zhao P, Wang L, et al. Analysis of clinicopathological and molecular features of ELOC(TCEB1) ‑ -mutant renal cell carcinoma. Pathol Res Pract. 2022; 235 : 153960.
30. Shah RB, Stohr BA, Tu ZJ, et al. „Renal Cell Carcinoma With Leiomyomatous Stroma“ Harbor Somatic Mutations of TSC1, TSC2, MTOR, and/or ELOC (TCEB1): Clinicopathologic and Molecular Characterization of 18 Sporadic Tumors Supports a Distinct Entity. Am J Surg Pathol. 2020; 44(5): 571–581.
31. Iannantuono GM, Riondino S, Sganga S, Roselli M, Torino F. Activity of ALK Inhibitors in Renal Cancer with ALK Alterations: A Systematic Review. Int J Mol Sci. 2022; 23(7): 3995.
32. Pal SK, Bergerot P, Dizman N, et al. Responses to Alectinib in ALK‑rearranged Papillary Renal Cell Carcinoma. Eur Urol. 2018; 74(1): 124–128.
33. Calderaro J, Moroch J, Pierron G, et al. SMARCB1/INI1 inactivation in renal medullary carcinoma. Histopathology. 2012; 61(3): 428–435.
Labels
Paediatric urologist Nephrology UrologyArticle was published in
Czech Urology

2023 Issue 2
Most read in this issue
- Prognostické faktory metastatického karcinomu prostaty
- 5. edice WHO klasifikace karcinomu prostaty z roku 2022: změny a novinky v „Blue Book“
- Časné komplikace po radikální cystektomii před zavedením konceptu ERAS (Enhanced Recovery After Surgery) u pacientů podstupujících radikální cystektomii
- Výsledky zavádění punkční nefrostomie pod ultrazvukovou kontrolou