
Muir-Torre syndrom – fenotypická varianta Lynchova syndromu
Muir-Torre Syndrome – a Phenotypic Variant of Lynch Syndrome
Muir-Torre syndrome (MTS) represents an autosomal dominantly inherited condition and is considered a phenotypic variant of the more common hereditary nonpolyposis colorectal cancer syndrome (HNPCC), or Lynch syndrome. MTS combines at least one cutaneous neoplasm with sebaceous differentiation (e.g. sebaceoma, sebaceous adenoma, and sebaceous carcinoma), and at least one visceral malignancy. MTS is a genetic disorder caused by a germline mutation in one of the DNA mismatch repair (MMR) genes. Tumors in MTS patients are characteristically associated with the loss of MMR protein expression and/or microsatellite instability (70%).
Patients who are suspected to have MTS/Lynch syndrome are often identified by dermatologists, dermatopathologists/pathologists, gastroenterologists and gynecologists. If MTS is suspected on a clinicopathological ground, necessary additional laboratory investigations should be performed only in specialized pathological departments providing immunohistochemistry and molecular biologic analysis service.
Key words:
Muir-Torre syndrome – hereditary nonpolyposis colorectal cancer – Lynch syndrome – mismatch repair (MMR) genes – MMR deficiency – MMR germline mutation – microsatellite instability
Authors:
D. Kacerovská 1,2; D. V. Kazakov 1,2; Karin Černá 1,2
![]() ; L. Hadravský 1; M. Michal Jr. 1; J. Dostál 1
; L. Hadravský 1; M. Michal Jr. 1; J. Dostál 1
![]() ; A. Skálová Jr. 1; M. Michal 1,2
; A. Skálová Jr. 1; M. Michal 1,2
Authors‘ workplace:
Šiklův patologicko-anatomický ústav, Fakultní nemocnice a Lékařská fakulta Univerzity Karlovy, Plzeň
1; Bioptická laboratoř s. r. o., Plzeň
2
Published in:
Čes.-slov. Patol., 46, 2010, No. 4, p. 86-94
Category:
Reviews Article
Overview
Muir-Torre syndrom (MTS) je autozomálně dominantně dědičné onemocnění, které je dnes považováno za fenotypickou variantu mnohem častějšího hereditárního nepolypózního kolorektálního karcinomu neboli Lynchova syndromu. MTS zahrnuje kombinaci nejméně jednoho kožního nádoru se sebaceózní diferenciací (sebaceózní adenom, sebaceom, sebaceózní karcinom) a minimálně jednoho viscerálního tumoru. Příčinou vzniku je většinou autozomálně dominantně dědičná zárodečná mutace v některém z genů zodpovědných za opravy replikačních chyb v DNA, tzv. mismatch repair (MMR) genů. Tumory vznikající u MTS se ve velké části případů vyznačují ztrátou exprese MMR proteinů a v cca 70 % vykazují vysokou mikrosatelitní nestabilitu.
Podezření na tento syndrom bývá nejčastěji vysloveno dermatology, dermatopatology/patology, event. gastroenterology a gynekology. K definitivnímu potvrzení diagnózy jsou však vyžadována další vyšetření, která lze provádět jen na specializovaných patologických pracovištích disponující imunohistochemickými a molekulárně biologickými metodami.
Klíčová slova:
Muir-Torre syndrom – hereditární nepolypózní kolorektální karcinom – Lynchův syndrom – mismatch repair (MMR) geny – MMR deficience – MMR zárodečná mutace – mikrosatelitní nestabilita
Úvod
Muir-Torre syndrom (MTS) je autozomálně dominantně dědičné onemocnění, které bylo poprvé popsáno nezávisle dvěma autory, tj. Muirem a Torrem v roce 1967 a 1968. Tento syndrom zahrnuje kombinaci nejméně jednoho kožního nádoru se sebaceózní diferenciací a minimálně jednoho viscerálního tumoru. Stejně tak jsou kožním příznakem MTS mnohočetné keratoakantomy vyskytující se u mladých osob na místech chráněných před sluncem. Mezi nejčastější interní malignity asociované s tímto syndromem patří karcinomy gastrointestinálního (cca 50 %) a urogenitálního traktu (25 %). Kožní léze ve většině případů předcházejí anebo se vyskytují současně s nádory vnitřních orgánů (1).
MTS je dnes považován za fenotypickou variantu mnohem častějšího hereditárního nepolypózního kolorektálního karcinomu (hereditary nonpolyposis colorectal cancer; HNPPC) neboli Lynchova syndromu, pro který je typický časný výskyt kolorektálních karcinomů lokalizovaných převážně v proximálním úseku tlustého střeva a obvykle asociovaných s dalšími malignitami postihující jiné orgány. Právě „otec“ HNPCC Henry Lynch v roce 1981 poukázal na společnou možnou etiologii mezi MTS a HNPCC poté, co identifikoval pacienty s fenotypem MTS v rodině postižené HNPCC/Lynchovým syndromem (2). Následně bylo prokázáno, že oba syndromy jsou genetická onemocnění způsobená zárodečnou mutací v jedné alele některého z genů zodpovědných za opravy replikačních chyb v DNA, tzv. mismatch repair (MMR) genů, která po inaktivaci druhé alely téhož genu vede ke ztrátě exprese příslušného proteinu, což má za následek mimo jiné tzv. mikrosatelitní nestabilitu (microsatellite instability; MSI).
Kožní nálezy
Kožní léze u MTS zahrnují nádory se sebaceózní diferenciací, a to sebaceózní adenom, sebaceom a extraokulární sebaceózní karcinom. Hyperplazie mazových žlázek může být také přítomna (3). Výskyt periokulárních sebaceózních tumorů u MTS je vzácný. Kožní tumory jsou většinou mnohočetné, ale byly popsány i případy solitárních lézí (4). Klinický obraz se neliší od jejich sporadických protějšků, které jsou statisticky častější. Obecně lze shrnout, že mnohočetné kožní nádory se sebaceózní diferenciací vyskytující se u jedinců před 50. rokem života nebo postihující tělní partie mimo obličej jsou silným indikátorem MTS (5). Stejně tak mnohočetné keratoakantomy vznikající na místech chráněných před sluncem u mladých osob slouží jako vodítko pro správnou diagnózu.
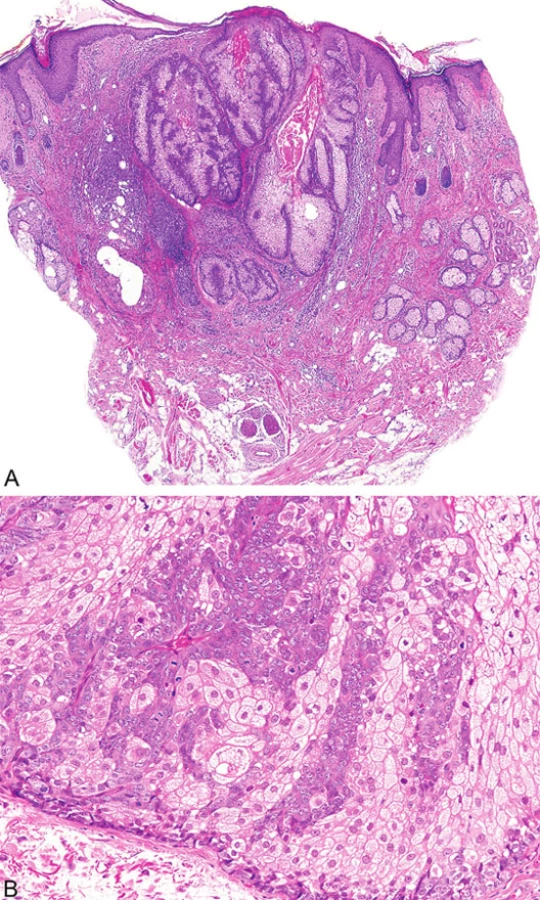
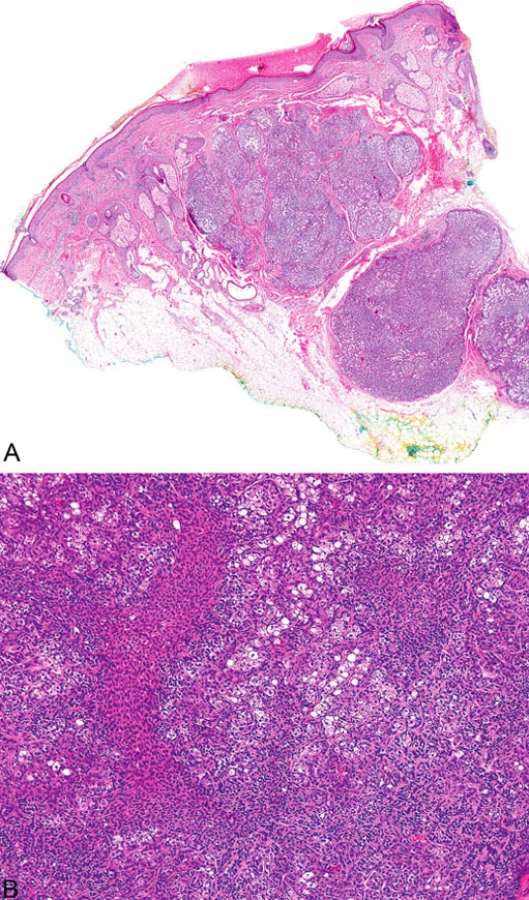
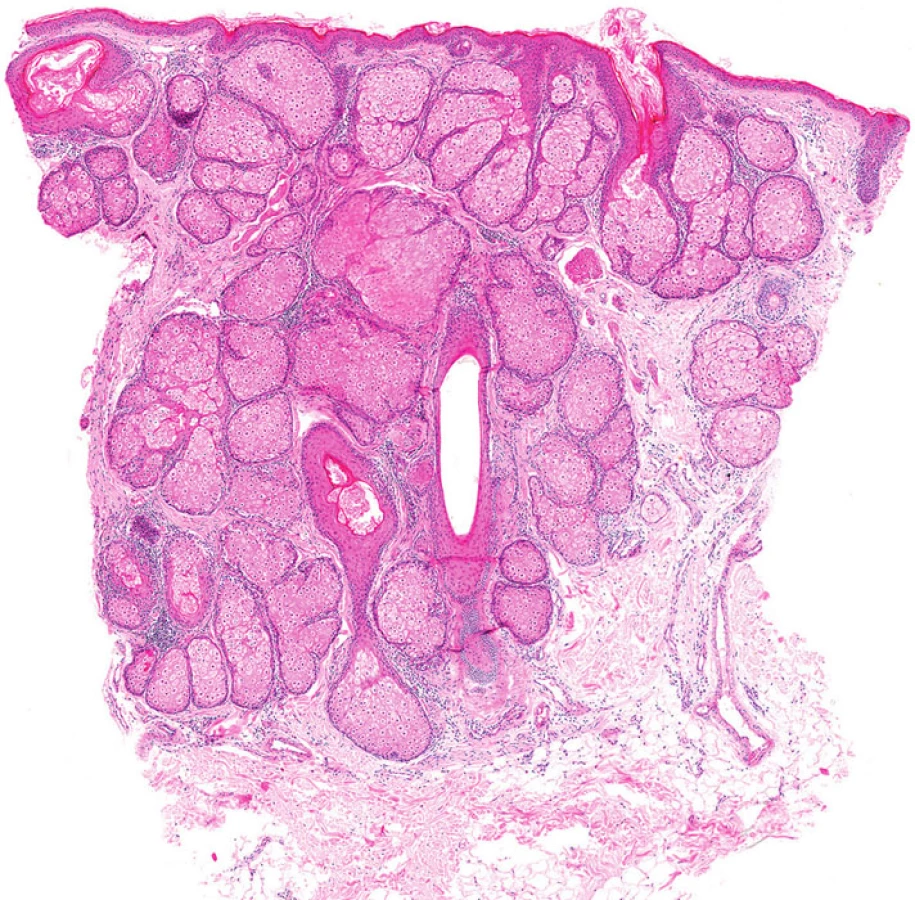
Mikroskopicky jsou sebaceózní adenomy charakterizovány lobulárním uspořádáním a spojením s přilehlou epidermis (obr. 1A). Jednotlivé lobuly jsou složeny z periferní vrstvy tvořené několika řadami malých bazaloidních germinativních buněk a z centrálně uložených zralých sebocytů s objemnou, narůžovělou, pěnitou až jemně vakuolizovanou cytoplazmou a s jádrem hvězdicového tvaru (obr. 1B). Naproti tomu sebaceom je uložen především v dermis, tvořen obvykle několika noduly s velmi vzácným epidermálním spojením (obr. 2A). Jednotlivé noduly bývají kulatého tvaru, avšak různé velikosti, složené z převážně malých, monomorfních, bazaloidních buněk. Zralé sebocyty tvoří méně než 50 % samotného tumoru (obr. 2B). U sebaceózních karcinomů jsou rozhodujícím nálezem cytologické detaily zahrnující buněčný a nukleární pleomorfismus a/nebo atypické mitózy (obr. 3). Může být přítomna i nekróza. Sebaceózní hyperplazie představuje zmnožený počet a větší velikost jinak normálních mazových žlázek, často vázaných na vlasový folikl (obr. 4).
Histopatologicky jsou výše zmíněné sebaceózní kožní nádory prakticky identické s jejich sporadickými analogy, avšak existuje několik morfologických rysů, které mohou u patologa vzbudit podezření na možnou asociaci s MTS. Jedná se o:
- 1. Cystické změny
Některé nádory u MTS vykazují nápadnou cystickou přeměnu. Tzv. cystický sebaceózní tumor (cystic sebaceous neoplasm) zahrnutý v posledním vydání WHO klasifikace je často citován jako nejčastější indikátor MTS (obr. 5) (6).Na druhé straně v některých lézích může být cystická přestavba výsledkem vystupňované holokrinní sekrece, fokální desintegrace buněk a sloučení mikrocyst (7).
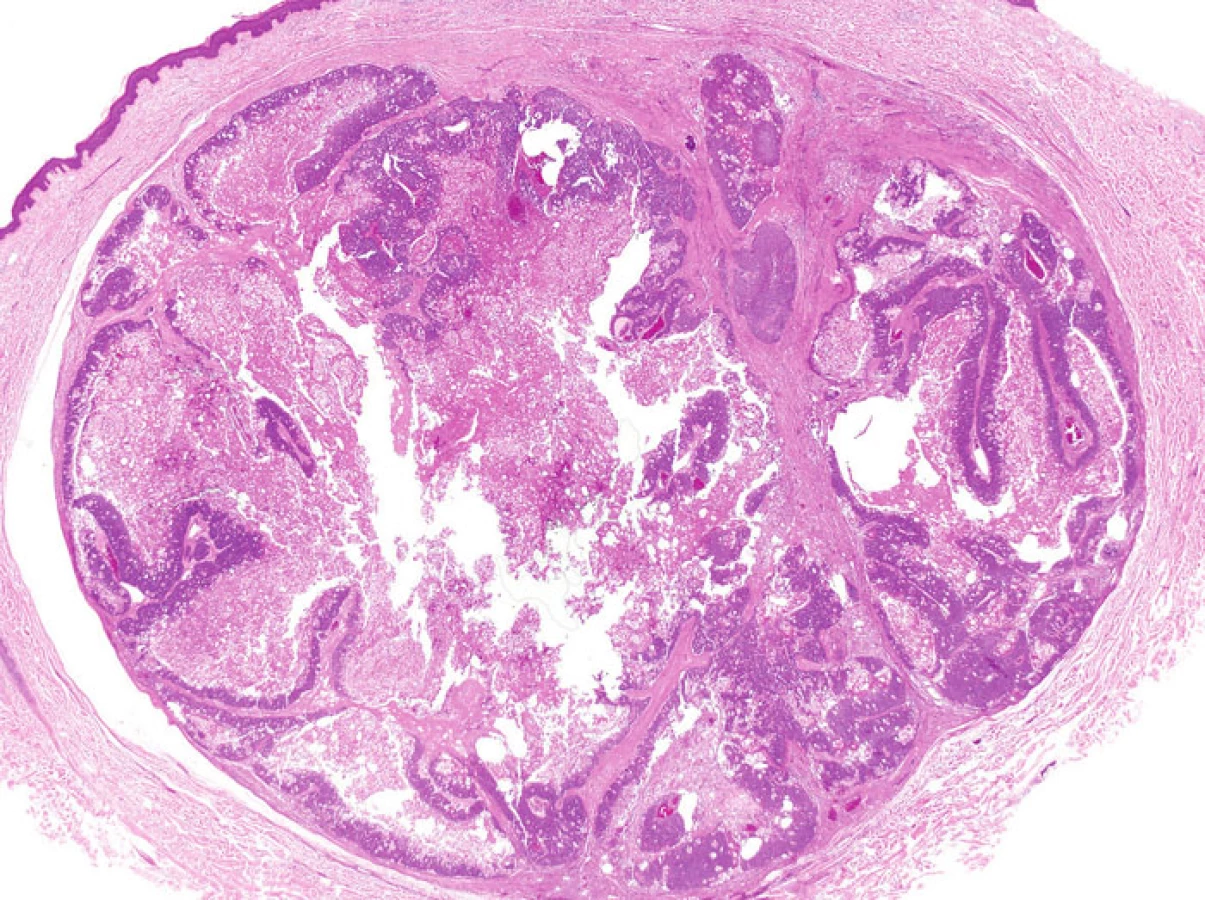
- 2. Architektura napodobující keratoakantom
Tento morfologický rys bývá často přítomen u sebaceózních adenomů. Projevuje se jako centrální, kráteru podobná invaginace (obyčejně mělká), která může být přítomna samostatně anebo v kombinaci s periferním koloretem a dlaždicobuněčnou diferenciací (obr. 6). Podobné léze byly v minulosti nazývány seboakantomy.
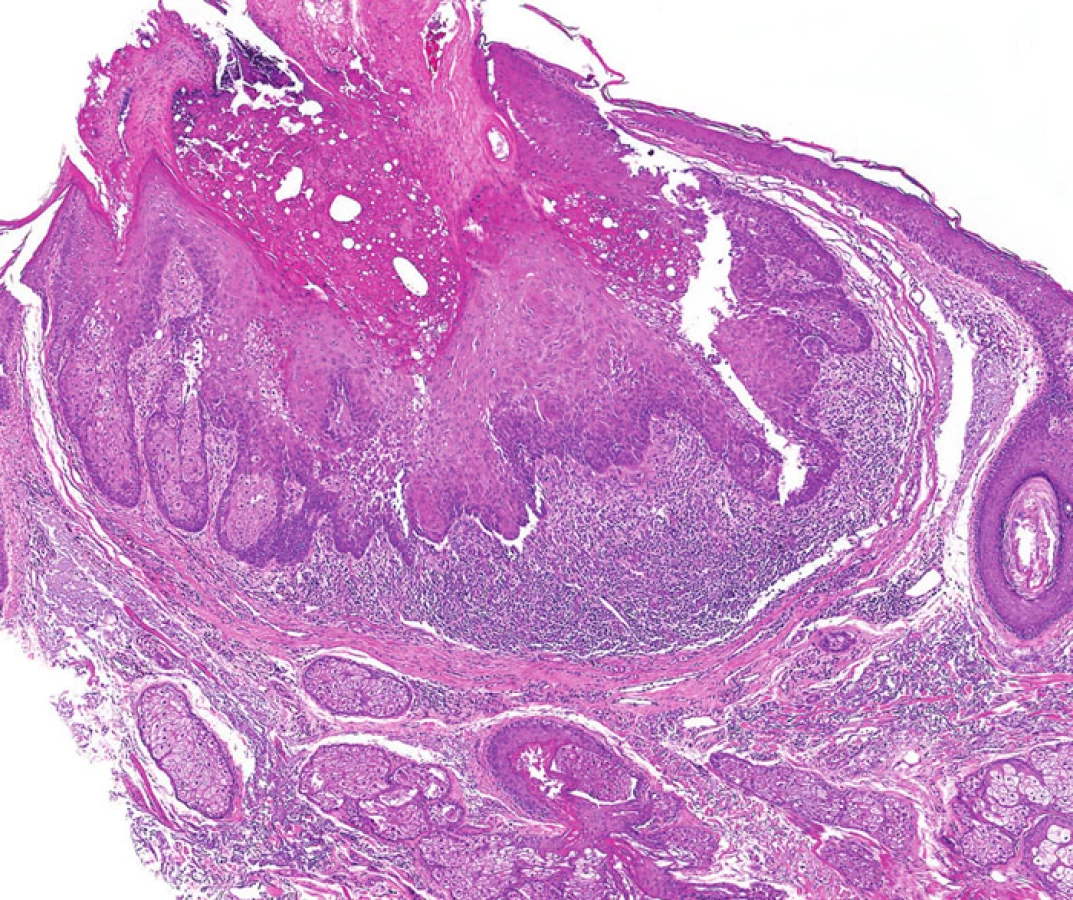
- 3. Intra - a peritumorální lymfocytární infiltrace
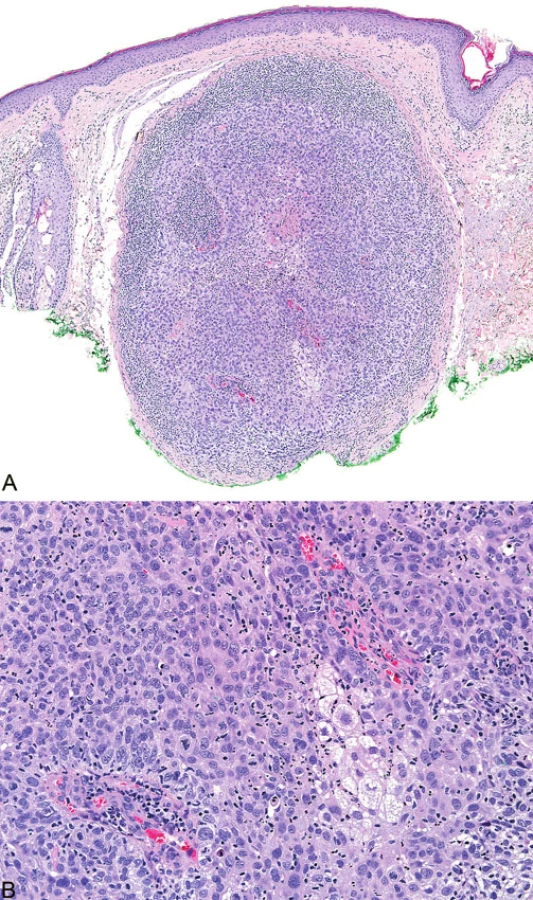
Podobně jako u kolorektálních a endometriálních karcinomů (8) v rámci HNPCC/Lynchova syndromu přítomnost intra - a peritumorálních lymfocytů u sebaceózních kožních neoplazií pravděpodobně koreluje s MMR deficiencí (obr. 7). Existuje však jen několik málo studií zabývajících se touto problematikou (9).
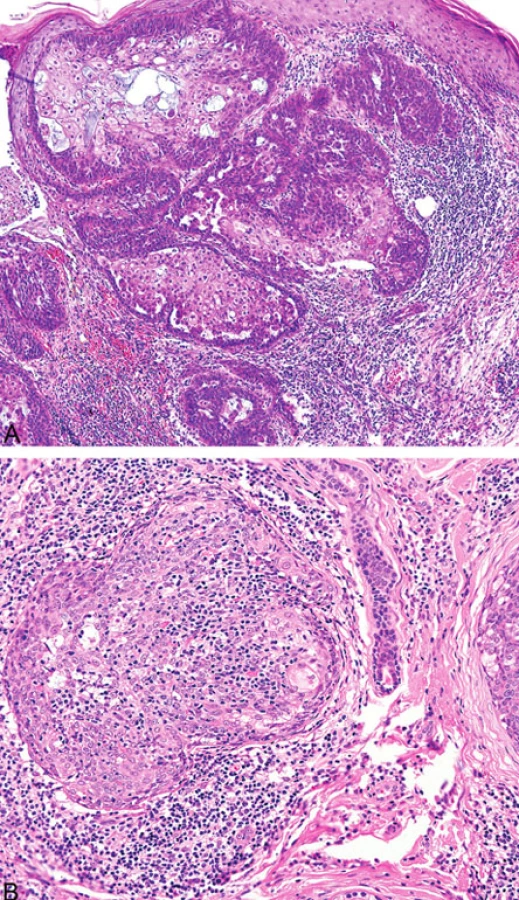
- 4. Mucinóza
Fokální mucinóza je vzácně zmiňována jako jeden z rysů MTS, zejména u sebaceózních adenomů a karcinomů (7).
- 5. Intra - a intertumorální heterogenita
Biopsie odebrané z různých lokalit od jedince s MTS mohou poměrně často vykazovat rozdílný vzhled, a to i tehdy, jedná-li se o stejné histologické léze (např. adenom, karcinom). Dokonce i v rámci samotného nádoru mohou být pozorovány rozdíly, např. ve stupni sebaceózní diferenciace a nukleárního pleomorfismu (10).
Existují i další histopatologické rysy zmiňované u sebaceózních nádorů v kontextu s MTS. Burgdorf a kol. popsali přítomnost kribriformních žlázek v sebaceózní komponentě (7).Stejně tak byl u pacientů s MTS popsán mírný nukleární pleomorfismus u jinak běžných sebaceózních adenomů (11).
Zdá se, že sebaceomy s organoidním uspořádáním (karcinoid napodobující, labyrintní a sinusoidální růst) nejsou pravděpodobně předzvěstí MTS (12). Stejně tak retikulární akantom se sebaceózní diferenciací a nevus sebaceus Jadassohn nejsou spojeny s tímto syndromem (13, 14).
Periokulární sebaceózní léze se vyskytují jen ve vzácných případech. Dosud bylo popsáno cca 40 periokulárních sebaceózních nádorů u pacientů s MTS zahrnujících karcinomy (nejčastější asociace), sebaceomy a sebaceózní adenomy (15, 16). Postižené osoby byly často mladšího věku ve srovnání se sporadickými případy, s průměrným věkem diagnózy 53 let. Horní a dolní víčko bylo postiženo se stejnou častostí. Léze vznikající v oblasti mediálního kantu byly vzácné (15). Podobně jako u jejich extraokulárních analogů byly u periokulárních lézí popsány neobvyklé morfologické rysy.
Je třeba zmínit i jeden z extrémních pohledů na kožní sebaceózní léze vznikající u pacientů s MTS vyslovený Ackermanem, který považoval všechny sebaceózní tumory kůže u těchto jedinců za karcinomy (17). Tento názor však nebyl obecně akceptován.
Mimokožní nálezy
Gastrointestinální trakt
Cca v 50 % se u pacientů s MTS vyskytuje kolorektální karcinom, který většinou vzniká na rozdíl od normální populace v proximální části tlustého střeva. Vzácně se také mohou vyskytovat karcinomy rekta. V kontrastu se sporadickými případy, kde karcinom střeva vzniká často po 60. roce života, kolorektální karcinomy asociované s MTS či HNPCC/Lynchovým syndromem vznikají u pacientů mladšího věku (průměr 40–45 let). Ploché adenomy představují často prekurzorové léze, které bývají klinicky těžko detekovatelné a doba jejich maligní transformace bývá krátká. Většina pacientů však nemá zvýšený počet polypů, ačkoliv přítomnost dvou i více kolorektálních karcinomů není výjimkou.
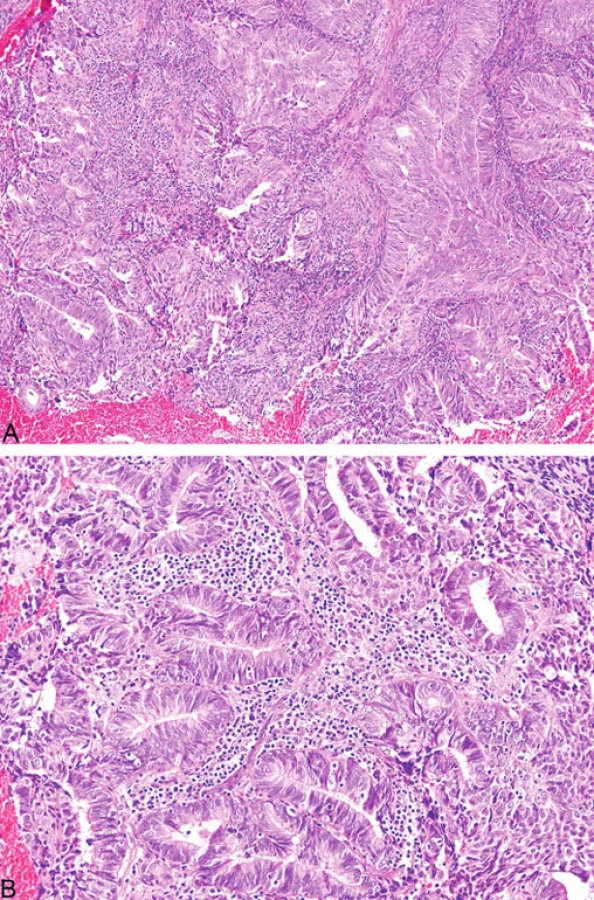
Histopatologicky vykazují nádory tlustého střeva spojené s MTS stejné morfologické rysy jako u HNPCC/Lynchova syndromu, a to mucinózní charakter, prstenčité buňky, peri - a intratumorální infiltraci lymfocyty a medulární růst (obr. 8) (18). Někteří autoři zahrnují mezi tyto znaky i nepřítomnost tzv. špinavé nekrózy v luminech žlázek, která je naopak typickým nálezem u sporadických případů (19). Mucinózní karcinomy jsou obyčejně dobře diferencované a epitel vykazuje podobnost s epitelem vilózních adenomů (19).
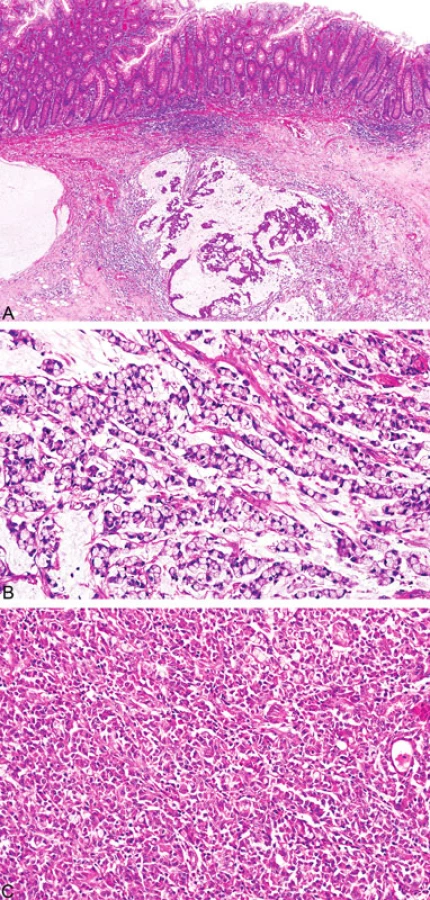
Ženský pohlavní systém
Zatímco HNPCC/Lynchův syndrom zahrnuje 2,3 % endometriálních karcinomů, u pacientů s MTS se zdají být gynekologické malignity vzácnější, což může být vysvětleno asociací endometriálních karcinomů s mutací v MSH6 genu. Endometriální karcinomy se ve srovnání s běžnou populací vyskytují v mladším věku, avšak vzácně mohou vznikat i u starších žen nad 60 let (20). Nejčastějším histologickým typem je endometriální endometrioidní karcinom.
Mikroskopicky typickým rysem podobně jako u kolorektálních karcinomů je intra - a peritumorální přítomnost lymfocytů (obr. 9). Rovněž charakteristickým nálezem je heterogenita tumoru definovaná přítomností dvou morfologicky odlišných nádorových populací tvořící minimálně 10 % tumorózní tkáně (21). Ostatní morfologické nálezy u endometriálních karcinomů publikované v kontextu s MSI zahrnují kribriformní uspořádání, mucinózní oblasti a nekrózu (20).
Urogenitální trakt
Nejčastějším typem nádoru vylučovacího traktu u pacientů s HNPCC/Lynchovým syndromem je uroteliální karcinom, jehož incidence je cca 14–22krát vyšší než u běžné populace. Typicky vzniká v horním močovém traktu (ureter a ledvinná pánvička). Močový měchýř bývá naopak postižen méně často (22). U pacientů s MTS vznikají tyto tumory jak v močovodech a v ledvinné pánvičce, tak i v močovém měchýři (23). Urologické malignity se v rámci obou syndromů objevují o 10–15 let dříve než jejich sporadické protějšky vznikající u běžné populace (22).
Jiné orgány
U pacientů s MTS a HNPCC/Lynchovým syndromem mohou nádory zřídka postihovat i jiné orgány včetně prsu, plic, mediastina, CNS, slinivky břišní, hlavy a krku, biliárního traktu, varlat, ledvin a prostaty (23–25). Tumory ve výše zmíněných lokalizacích bývají vzácným nálezem stejně tak jako hematologické malignity a melanomy.
Molekulární biologie MTS syndromu
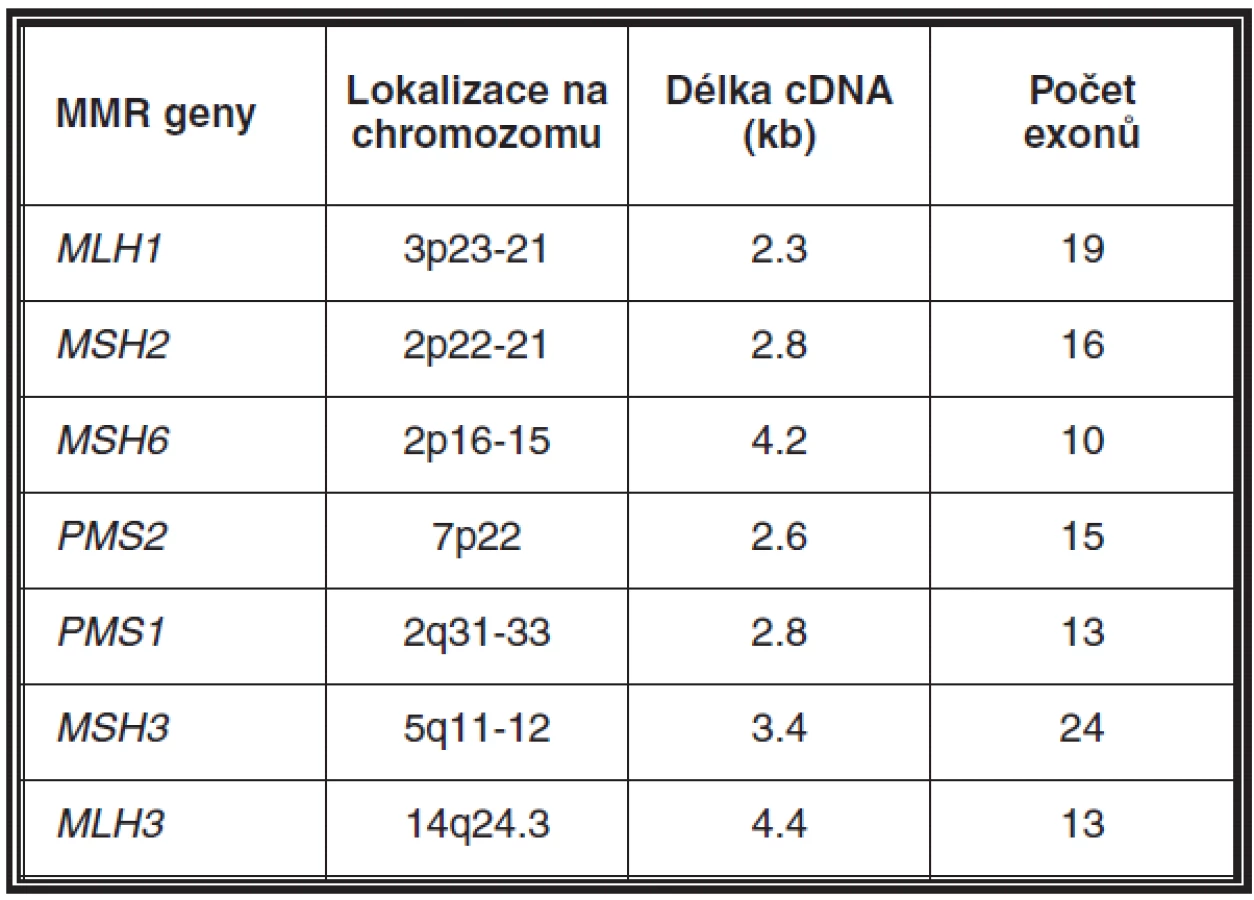
Příčinou vzniku MTS je ve většině případů stejně jako u HNPCC/Lynchova syndromu autozomálně dominantně dědičná zárodečná mutace v některém z genů zodpovědných za opravy replikačních chyb v DNA, tzv. mismatch repair (MMR) genů (viz tabulka 1) (26). Jelikož MMR geny jsou typické tumor supresorové geny, k nástupu onemocnění dochází až somatickou inaktivací druhé alely téhož genu (27).
Nejsou-li MMR geny poškozeny, exprimované MMR proteiny se spojují do funkčních komplexů, a to v heterodimery MSH2/MSH6 (příp. MSH2/MSH3) a MLH1/PMS2 (příp. MLH1/MLH3). První z těchto dimerů rozpoznává chyby v DNA (nespárované nebo špatně spárované nukleotidy) a signalizuje poškození, druhý chyby opravuje. Zároveň se podílí na zastavení buněčného cyklu a indukci apoptózy v reakci na poškození DNA. Pokud některý z proteinů není funkční následkem inaktivace obou alel jeho genu, dochází ke vzniku nádorů charakteristických vysokým stupněm nestability v krátkých tandemových repeticích molekuly DNA (v tzv. mikrosatelitech) (28, 29). Tumory jevící mikrosatelitní nestabilitu (MSI) v 30 a více procentech sledovaných mikrosatelitních sekvencí jsou označovány jako léze s vysokým stupněm mikrosatelitní nestability (microsatellite instability-high; MSI-H) (30). Právě vysokou mikrosatelitní nestabilitu vykazuje přibližně 70 % nádorů u pacientů s MTS (31, 32).
Ačkoliv do skupiny MMR genů podílejících se na vzniku HNPCC/Lynchova syndromu řadíme v současné době 7 genů (MLH1, MSH2, MSH6, PMS2, PMS1, MSH3 a MLH3), na vzniku MTS se podílí především zárodečné mutace v genu MSH2 (cca 90 % případů) (34). Mutace v MLH1 genu zahrnují zhruba 10 % případů. V poslední době byly detekovány také mutace v MSH6 genu (35, 36). U MTS byla zachycena celá škála mutací, a to krátké delece a inzerce, bodové missense a nonsense mutace a rozsáhlé delece či duplikace celých exonů (37).
Fenotypové projevy MTS mohou být vyvolány i jinými mechanismy než přímým poškozením MMR genů. Příčinou může být i zárodečná delece několika posledních exonů genu TACSTD1 kódujícího Ep-CAM. Tento gen se nachází na druhém chromozomu v těsné blízkosti před genem MSH2. Delecí exonů tak dochází k zasažení jeho transkripce do oblasti MSH2 genu. Promotor MSH2 genu ležící na stejném chromozomu jako terminálně mutovaný TACSTD1 je proto epigeneticky metylován a nedochází k transkripci této alely MSH2 genu (38). Popsány byly také případy vzniku sebaceózních lézí u pacientů s bi-alelickými mutacemi v MYH (MutYH) genu, jehož proteinový produkt se podílí na excizní reparaci DNA v reakci na oxidativní poškození DNA (39). V takových případech léze mohou být stabilní v mikrosatelitních sekvencích (40).
Diagnostika MMR deficience, MSI a MMR zárodečné mutace
V případě klinicko-patologického podezření na MTS jsou nezbytná další laboratorní vyšetření k potvrzení nebo vyloučení MMR deficience a MSI. Senzitivita vyšetření nestability mikrosatelitů založená na PCR metodě versus imunohistochemické vyšetření je shodná. Prvním krokem skríningového testu je imunohistochemická studie exprese MMR proteinů, která představuje rychlou, efektivní a spolehlivou metodu s vysokou prediktivní hodnotou diagnózy MMR deficience (33). Charakter exprese MMR proteinů v kožních tumorech koreluje s molekulárně-genetickými výsledky. Ztráta exprese těchto proteinů je indikátorem pravděpodobného zárodečného defektu MMR genů a s tím spojené MSI (obr. 10) (33). V naší rutinní praxi používáme standardně panel čtyř protilátek (MLH1, PMS2, MSH2, MSH6), ačkoliv nedávné studie poukázaly na možnost použití pouze dvou-protilátkového panelu (MSH6 a PMS2), jehož prediktivní hodnota je údajně shodná jako u vyšetření s použitím 4 protilátek (41).

MLH1 a MSH2 jsou tzv. základní proteiny, zatímco PMS2 a MSH6 představují tzv. sekundární proteiny. Abnormality v základních proteinech (MLH1, MSH2) vedou k proteolytické degradaci heterodimerů a následně ke ztrátě jak základních, tak sekundárních proteinů. Jestliže se mutace vyskytne v genech sekundárních proteinů (tj. geny PMS2, MSH6), heterodimery zůstávají stabilní a nedochází k současné ztrátě jejich partnerů, tj. základních proteinů. Proto v případě použití dvou-protilátkového panelu je třeba mít vždy na mysli, že většina případů se ztrátou MSH6 proteinu může mít defekt v MSH2 genu, zatímco u případů se ztrátou PMS2 proteinu může být naopak genetický defekt v MLH1 genu. Jestliže je využito čtyřprotilátkového panelu, často je ztráta MLH1 nebo MSH2 spojena ztrátou PMS2 a MSH6 proteinu.
Několik postřehů k imunohistochemickému vyšetření exprese MMR proteinů:
- Sporadické sebaceózní léze mohou příležitostně vykazovat ztrátu MMR proteinů (42, 43).
- Někteří pacienti mohou mít zárodečnou mutaci, která se neprojevuje ztrátou MMR proteinů v tumoru. Taková situace může nastat u některých zárodečných missense mutací vedoucích k expresi nefunkčních, ale antigenně intaktních proteinů (33, 44).
- U různých sebaceózních tumorů jednoho pacienta je očekáván identický barvící obraz. Z tohoto důvodu postačí k testování osob s mnohočetnými projevy na kůži jen jedna léze (9, 32). Je však třeba zdůraznit, že sebaceózní hyperplazie obyčejně nevykazuje ztrátu MMR proteinů, ačkoliv ostatní typy sebaceózních tumorů u téhož jedince (např. sebaceózní adenom, sebaceózní karcinom) simultánně vykazují MMR deficienci (9). Předpokládaným nálezem je taktéž imunohistochemická shoda mezi všemi tumory od jednoho pacienta včetně kožních a viscerálních tumorů, avšak vzácně může být u kožních a viscerálních tumorů detekován rozdílný charakter imunohistochemického vyšetření MMR proteinů (45).
- Pomocí imunohistochemického vyšetření nelze rozlišit mezi ztrátou MLH1 exprese způsobené zárodečnou mutací versus somatickou hypermetylací promotoru (44).
- Známým a často diskutovaným faktem je existence několika kvalit barvení včetně tzv. slabého barvení neoplastických elementů a nedostatku přesvědčivé pozitivní interní kontroly. Někteří autoři zastávají názor, že jenom kompletní ztráta exprese MMR proteinu v tumorózní tkáni s validní pozitivní kontrolou může být považována za interpretativní. Podle našich zkušeností je možné zaznamenat částečnou ztrátu exprese MMR proteinu, někdy s nápadným gradientním profilem (tj. ztráta barvení v centru léze s naopak pozitivním, i když jen slabě, barvením na periferii tumoru) (33). V problematických případech je doporučováno vyhodnocení vzorku nejméně dvěma patology, kteří mají zkušenosti s interpretací imunohistochemických nálezů MMR proteinů. I přesto se vzácně vyskytnou případy s nevyhodnotitelným výsledkem imunohistochemického vyšetření (tzv. neinterpretovatelné barvení) (46).
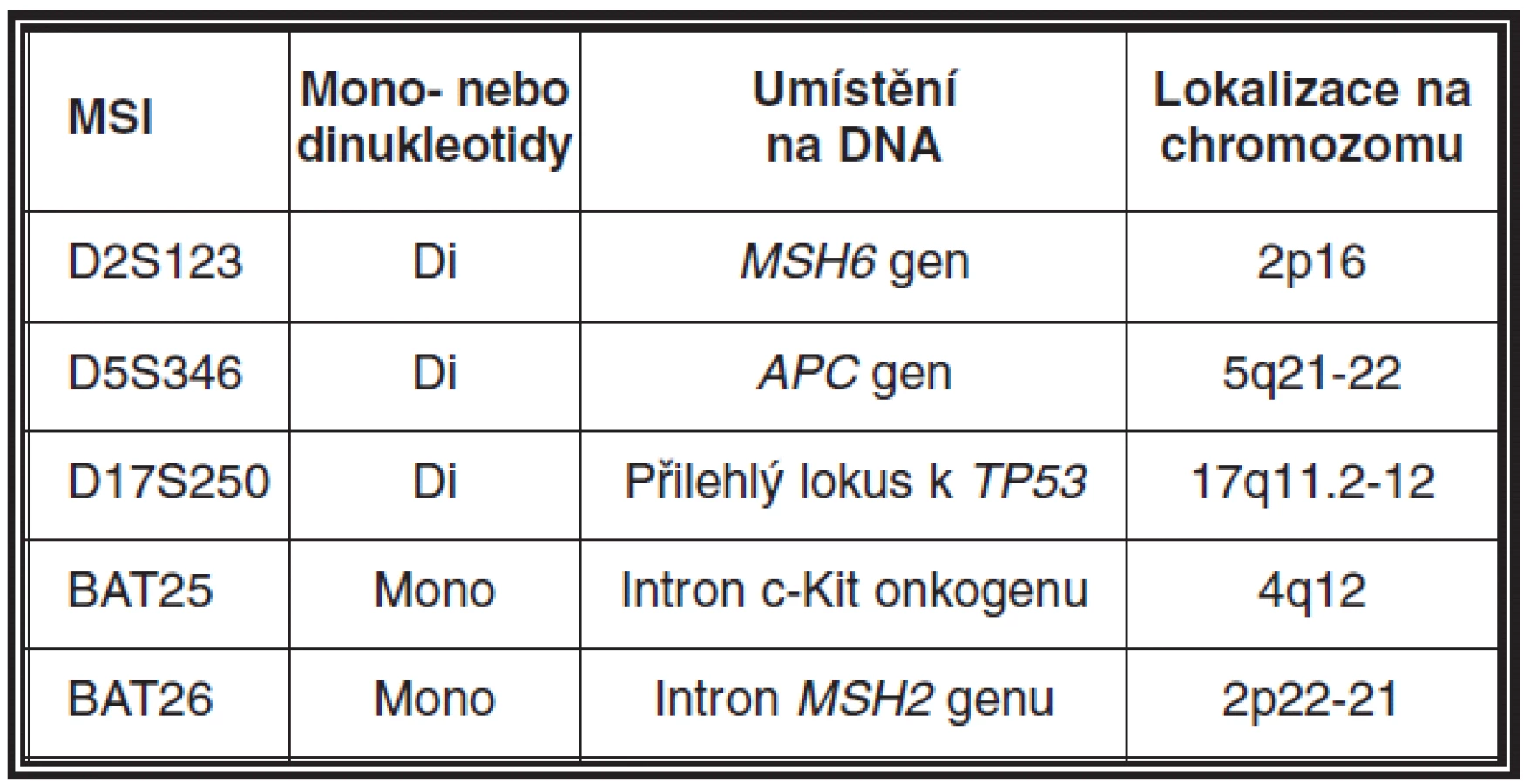
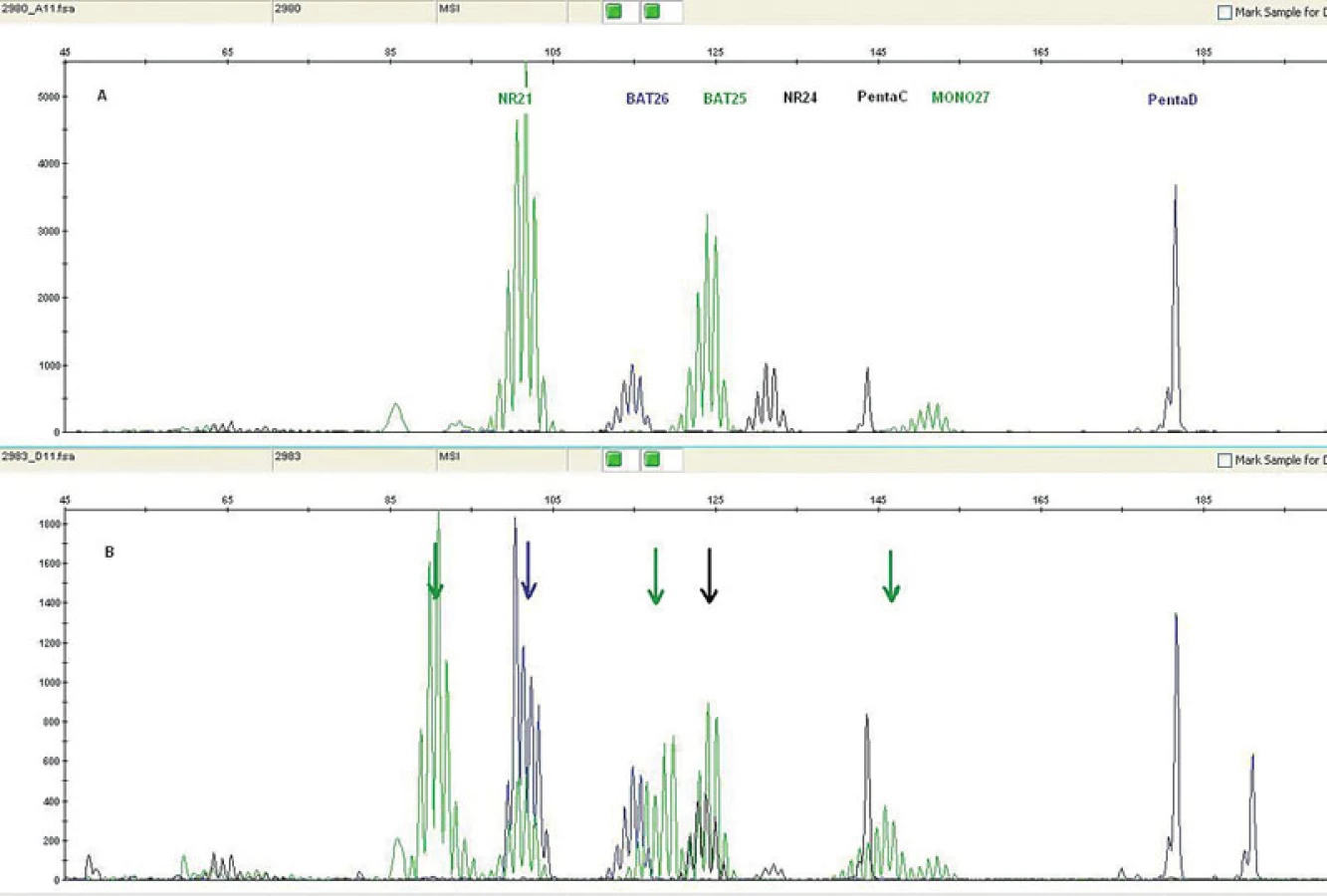
MSI může být detekována pomocí PCR technik a fragmentační analýzy zjišťující délku určitého segmentu DNA v lezionální tkáni a srovnávající ji se stejnými segmenty v normální tkáni přilehlé k tumoru. Pro tyto účely je nutná ve formolu fixovaná tkáň a dále pro dermatopatologickou praxi toto vyšetření předpokládá využití mikrodisektoru k obdržení optimálního výsledku. Snadnější a z praktického hlediska výhodnější alternativou získání kontrolního vzorku je odběr periferní krve pacienta, která může být také použita pro detekci zárodečné mutace. Současně nejvíce používaný panel mikrosatelitních markerů zahrnuje D2S123, D5S346, D17S250, BAT25, BAT26 (viz tabulka 2). V naší praxi navíc používáme i NR21, NR24 a MONO27. Výsledkem analýzy mohou být dva fenotypy MSI, a to mikrosatelitní nestabilita vysokého (high degree of MSI; MSI-H) (obr. 11) a nízkého stupně (low degree of MSI; MSI-L) (46). Zatímco MSI-H u sporadických sebaceózních kožních lézí a u sebaceózní hyperplazie je detekována pouze vzácně, u ostatních kožních lézí pacientů s MTS je častým nálezem (26, 31, 45, 47).
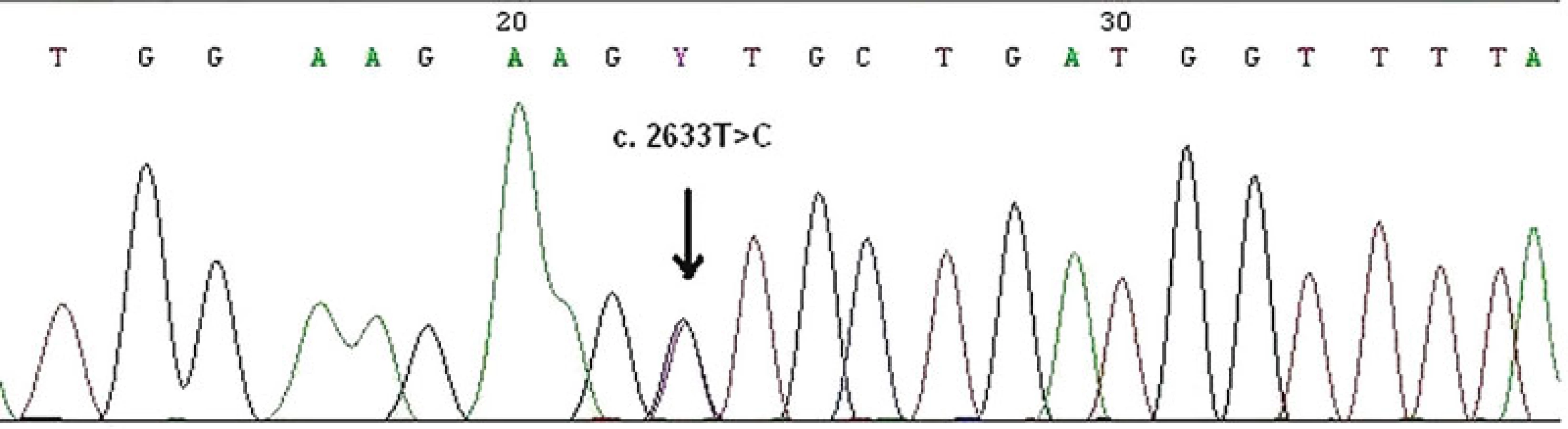
Zárodečná mutace v MMR genech může být detekována u pacienta a jeho pokrevních příbuzných mnoha metodami, např. testem na přítomnost zkrácených proteinů (protein truncation test), analýzou konformačního polymorfismu jednořetězcových segmentů (single strand conformation polymorphism analysis, SSCP), analýzou heteroduplexů. U pacientů s MTS je zárodečná mutace v MMR genech detekována v 60–70 % (26, 34). Na našem pracovišti je k detekci malých mutací používána PCR metoda amplifikující všechny exony a exon-intronové spoje daných genů s následnou přímou sekvenací korespondujících PCR fragmentů (obr. 12). K detekci rozsáhlých delecí a duplikací je využívána MLPA (multiplex ligation-dependent probe amplification) (obr. 13). Součástí genetického vyšetření je i test na onkogenní V600E (dříve známý jako V599E) hotspot mutaci v genu BRAF. Tato metoda spolu s vyšetřením MSI byla nedávno přidána k prospektivnímu skríningu pacientů s kolorektálním karcinomem, u kterých bylo vysloveno podezření na HNPCC/Lynchův syndrom. BRAF mutace je identifikovaná ve významné části kolorektálních karcinomů s MSI-H vyvolaných hypermetylací MLH1 promotoru (tzv. sporadické karcinomy), ale není přítomna u tumorů s MSI vznikajících v rámci HNPCC/Lynchova syndromu a MTS. Zjednodušeně lze shrnout, že identifikace V600E BRAF mutace prakticky vylučuje možnost HNPCC/Lynchova syndromu a MTS, a lze ji proto použít před samotným testováním zárodečné mutace v MLH1 genu u kolorektálních karcinomů s MSI-H (48).

Genotyp – fenotyp korelace u MTS
Mezi případy deficience MMR je naprosto unikátní významná korelace mezi genotypem a fenotypem, a to konkrétně mezi mutací MSH2 genu a klinickým nálezem typickým pro MTS. Rodiny s HNPCC/Lynchovým syndromem, které jsou nositeli mutace c.942+3A>T v MSH2 genu mají vyšší frekvenci vzniku MTS nežli rodiny, které jsou nositeli jiné mutace v témže genu (48).
MTS pacienti s postiženým MSH2 genem mají ve srovnání s nosiči MLH1 mutace signifikantně vyšší celoživotní riziko vzniku karcinomů na jiných místech než ve střevě. Jak již bylo zmíněno, jen menšina pacientů s MTS má mutován MSH6 gen (35). Je třeba ale mít na paměti, že takovéto případy jsou obyčejně velmi těžko identifikovatelné, neboť u HNPCC/Lynchova syndromu mutace v MSH6 genu vykazují méně závažný klinický fenotyp, a to i s ohledem na kolorektální a endometriální karcinom (např. pozdní vznik nádoru, histologické rysy jsou více podobné tumorům u běžné populace) (50).
Nápadná je i korelace mezi anatomickou lokalizací tumoru, jeho typem, architekturou a deficiencí MMR. Bylo zjištěno, že deficience MMR je signifikantně spojena s anatomickou lokalizací nádoru (MMR deficientní tumory se převážně vyskytují na trupu a končetinách a jen velice vzácně na hlavě a krku), s typem tumoru (pokud léze vniká na hlavě a krku, mnohem častěji se jedná o sebaceózní adenom než karcinom) a s architekturou (architektura sebaceózních lézí připomíná keratoakantom) (9, 51). Tato korelace vykazuje analogii s kolorektálními tumory, kde rovněž vpravo lokalizované karcinomy tlustého střeva jsou častěji spojeny s deficiencí MMR ve srovnání s levostrannými.
Závěr
MTS, fenotypická varianta HNPCC/Lynchova syndromu, bývá nejčastěji diagnostikovaný dermatology, dermatopatology/patology, gastroenterology a gynekology. Identifikace těchto pacientů a jejich pokrevních příbuzných vyžaduje další vyšetření, které lze provádět jen na specializovaných patologických pracovištích disponujících imunohistochemickými a molekulárně biologickými metodami. Tento přístup pak dovoluje včasné odhalení postižených a jejich pokrevních příbuzných, kteří jsou nositeli zárodečné mutace v MMR genech. V zemích, kde existuje registr těchto osob včetně striktního systému vyšetřování, je doba přežití pacientů srovnatelná s normální populací (52).
Práce byla zčásti podpořena Specifickým vysokoškolským výzkumným záměrem Univerzity Karlovy v Praze (č. projektu 260 809).
Korespondenční adresa:
prof. MUDr. Michal Michal
Šiklův patologicko-anatomický ústav
Alej Svobody 80
304 60 Plzeň
tel.: +420-377104631
e-mail: michal@medima.cz
Sources
1. Cohen, P.R., Kohn, S.R., Kurzrock, R.: Association of sebaceous gland tumors and internal malignancy: the Muir-Torre syndrome. Am J Med., 90, 1991, s. 606–613.
2. Lynch, H.T., Fusaro, R.M., Roberts, L., Voorhees, G.J., Lynch, J.F.: Muir-Torre syndrome in several members of a family with a variant of the Cancer Family Syndrome. Br J Dermatol., 113, 1985, s. 295–301.
3. Esche, C., Kruse, R., Lamberti, C., et al.: Muir-Torre syndrome: clinical features and molecular genetic analysis. Br J Dermatol., 136, 1997, s. 913–917.
4. Graham, R., McKee, P., McGibbon, D., Heyderman, E.: Torre-Muir syndrome. An association with isolated sebaceous carcinoma. Cancer, 55, 1985, s. 2868–2873.
5. Schwartz, R.A., Torre, D.P.: The Muir-Torre syndrome: a 25-year retrospect. J Am Acad Dermatol., 33, 1995, s. 90–104.
6. Rutten, A., Burgdorf, W., Hugel, H., et al.: Cystic sebaceous tumors as marker lesions for the Muir-Torre syndrome: a histopathologic and molecular genetic study. Am J Dermatopathol., 21, 1999, s. 405–413.
7. Burgdorf, W.H., Pitha, J., Fahmy, A.: Muir-Torre syndrome. Histologic spectrum of sebaceous proliferations. Am J Dermatopathol., 8, 1986, s. 202–8.
8. Greenson, J.K., Bonner, J.D., Ben-Yzhak, O., et al.: Phenotype of microsatellite unstable colorectal carcinomas: Well-differentiated and focally mucinous tumors and the absence of dirty necrosis correlate with microsatellite instability. Am J Surg Pathol., 27, 2003, s. 563–570.
9. Orta, L., Klimstra, D.S., Qin, J., et al.: Towards identification of hereditary DNA mismatch repair deficiency: sebaceous neoplasm warrants routine immunohistochemical screening regardless of patient‘s age or other clinical characteristics. Am J Surg Pathol., 33, 2009, s. 934–944.
10. Fahmy, A., Burgdorf, W.H., Schosser, R.H., Pitha, J.: Muir-Torre syndrome: report of a case and reevaluation of the dermatopathologic features. Cancer 1982;49 : 1898–903.
11. Misago N, Narisawa Y.: Sebaceous neoplasms in Muir-Torre syndrome. Am J Dermatopathol., 22, 2000, s. 155–161.
12. Kazakov, D.V., Kutzner, H., Rutten, A., Mukensnabl, P., Michal, M.: Carcinoid-Like Pattern in Sebaceous Neoplasms: Another Distinctive, Previously Unrecognized Pattern in Extraocular Sebaceous Carcinoma and Sebaceoma. Am J Dermatopathol. 27, 2005, s.195–203.
13. Fukai, K., Sowa, J., Ishii, M.: Reticulated acanthoma with sebaceous differentiation. Am J Dermatopathol., 28, 2006, s.158–161.
14. Haake, D.L., Minni, J.P., Nowak, M., Abenoza, P., Nousari, C.H.: Reticulated acanthoma with sebaceous differentiation. Lack of association with Muir-Torre syndrome. Am J Dermatopathol., 31, 2009, s. 391–392.
15. Rishi, K., Font, R.L.: Sebaceous gland tumors of the eyelids and conjunctiva in the Muir-Torre syndrome: a clinicopathologic study of five cases and literature review. Ophthal Plast Reconstr Surg., 20, 2004, s. 31-36.
16. Stockl, F.A., Dolmetsch, A.M., Codere, F., Burnier, M.N., Jr.: Sebaceous carcinoma of the eyelid in an immunocompromised patient with Muir-Torre syndrome. Can J Ophthalmol., 30, 1995, s. 324–326.
17. Ackerman, A.B., Lee, S.N.: Neoplasms in all organs of Muir-Torre syndrome are carcinomas: sebaceous carcinomas and squamous-cell carcinomas (keratoacanthomas) in skin and adenocarcinomas, squamous-cell carcinomas, and transitional-cell carcinomas in internal organs. Dermatopathology: Practical & Conceptual, 5, 1999, s. 312–318.
18. Bellizzi, A.M., Frankel, W.L.: Colorectal cancer due to deficiency in DNA mismatch repair function: a review. Adv Anat Pathol., 16, 2009, s. 405–417.
19. Jass, J.R.: Role of the pathologist in the diagnosis of hereditary non-polyposis colorectal cancer. Dis Markers, 20, 2004, s. 215–224.
20. Karamurzin, Y., Rutgers, J.K.: DNA mismatch repair deficiency in endometrial carcinoma. Int J Gynecol Pathol., 28, 2009, s. 239–255.
21. Garg, K., Soslow, R.A.: Lynch syndrome (hereditary non-polyposis colorectal cancer) and endometrial carcinoma. J Clin Pathol., 62, 2009, s. 679–684.
22. Roupret, M., Yates, D.R., Comperat, E., Cussenot, O.: Upper urinary tract urothelial cell carcinomas and other urological malignancies involved in the hereditary nonpolyposis colorectal cancer (lynch syndrome) tumor spectrum. Eur Urol., 54, 2008, s. 1226–1236.
23. Akhtar, S., Oza, K.K., Khan, S.A., Wright, J.: Muir-Torre syndrome: case report of a patient with concurrent jejunal and ureteral cancer and a review of the literature. J Am Acad Dermatol., 41, 1999, s. 681–686.
24. Sparr, J.A., Bandipalliam, P., Redston, M.S., Syngal, S.: Intraductal papillary mucinous neoplasm of the pancreas with loss of mismatch repair in a patient with Lynch syndrome. Am J Surg Pathol., 33, 2009, s. 309–312.
25. Oman, S.A., Ballinger, L., Cerilli, L.A.: Small cell carcinoma: arising in Lynch syndrome: a previously undocumented occurrence. Int J Surg Pathol., 17, 2009, s. 46–50.
26. Kruse, R., Rutten, A., Lamberti, C., et al.: Muir-Torre phenotype has a frequency of DNA mismatch-repair-gene mutations similar to that in hereditary nonpolyposis colorectal cancer families defined by the Amsterdam criteria. Am J Hum Genet., 63, 1998, s. 63–70.
27. Yuen, S.T., Chan, T.L., Ho, J.W., et al.: Germline, somatic and epigenetic events underlying mismatch repair deficiency in colorectal and HNPCC-related cancers. Oncogene, 21, 2002, s. 7585–7592.
28. Peltomaki, P., Lothe, R.A., Aaltonen, L.A., et al: Microsatellite instability is associated with tumors that characterize the hereditary non-polyposis colorectal carcinoma syndrome. Cancer Res., 53, 1993, s. 5853–855.
29. Thibodeau, S.N., Bren, G., Schaid, D.: Microsatellite instability in cancer of the proximal colon. Science, 260, 1993, s. 816–819.
30. Boland, C.R., Thibodeau, S.N., Hamilton, S.R., et al.: A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res., 58, 1998, s. 5248–5257.
31. Entius, M.M., Keller, J.J., Drillenburg, P., Kuypers, K.C., Giardiello, F.M., Offerhaus, G.J.: Microsatellite instability and expression of hMLH-1 and hMSH-2 in sebaceous gland carcinomas as markers for Muir-Torre syndrome. Clin Cancer Res., 6, 2000, s. 1784–9.
32. Ponti, G., Losi, L., Di Gregorio, C., et al.: Identification of Muir-Torre syndrome among patients with sebaceous tumors and keratoacanthomas: role of clinical features, microsatellite instability, and immunohistochemistry. Cancer, 103, 2005, s. 1018–1025.
33. Mathiak, M., Rutten, A., Mangold, E., et al.: Loss of DNA mismatch repair proteins in skin tumors from patients with Muir-Torre syndrome and MSH2 or MLH1 germline mutations: establishment of immunohistochemical analysis as a screening test. Am J Surg Pathol., 26, 2002, s. 338–343
34. Mangold, E., Pagenstecher, C., Leister, M., et al.: A genotype-phenotype correlation in HNPCC: strong predominance of msh2 mutations in 41 patients with Muir-Torre syndrome. J Med Genet., 41, 2004, s. 567–572.
35. Mangold, E., Rahner, N., Friedrichs, N., et al.: MSH6 mutation in Muir-Torre syndrome: could this be a rare finding? Br J Dermatol., 156, 2007, s. 158–162.
36. Arnold, A., Payne, S., Fisher, S., et al.: An individual with Muir-Torre syndrome found to have a pathogenic MSH6 gene mutation. Fam Cancer, 6, 2007, s. 317–321.
37. Ponti, G., Losi, L., Pedroni, M., et al.: Value of MLH1 and MSH2 mutations in the appearance of Muir-Torre syndrome phenotype in HNPCC patients presenting sebaceous gland tumors or keratoacanthomas. J Invest Dermatol., 126, 2006, s. 2302–2307.
38. Ligtenberg, M.J., Kuiper, R.P., Chan, T.L., et al.: Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3’exons of TACSTD1. Nat Genet., 41, 2009, s. 112–117.
39. Barnetson, R.A., Devlin, L., Miller, J., et al.: Germline mutation prevalence in the base excision repair gene, MYH, in patients with endometrial cancer. Clin Genet., 72, 2007, s. 551–555.
40. Ponti, G., Ponz de Leon, M., Maffei, S., et al.: Attenuated familial adenomatous polyposis and Muir-Torre syndrome linked to compound biallelic constitutional MYH gene mutations. Clin Genet., 68, 2005, s. 442–447.
41. Shia, J., Tang, L.H., Vakiani, E., et al.: Immunohistochemistry as first-line screening for detecting colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome: a 2-antibody panel may be as predictive as a 4-antibody panel. Am J Surg Pathol., 33, 2009, s. 1639–1645.
42. Cesinaro, A.M., Ubiali, A., Sighinolfi, P., Trentini, G.P., Gentili, F., Facchetti, F.: Mismatch repair proteins expression and microsatellite instability in skin lesions with sebaceous differentiation: a study in different clinical subgroups with and without extracutaneous cancer. Am J Dermatopathol., 29, 2007, s. 351–358.
43. Morales-Burgos, A,, Sanchez, J.L., Figueroa, L.D., et al.: MSH-2 and MLH-1 protein expression in Muir Torre syndrome-related and sporadic sebaceous neoplasms. P R Health Sci J., 27, 2008, s. 322–327.
44. Doxey, B.W., Kuwada, S.K., Burt, R.W.: Inherited polyposis syndromes: molecular mechanisms, clinicopathology, and genetic testing. Clin Gastroenterol Hepatol., 3, 2005, s. 633–641.
45. Machin, P., Catasus, L., Pons, C., et al.: Microsatellite instability and immunostaining for MSH-2 and MLH-1 in cutaneous and internal tumors from patients with the Muir-Torre syndrome. J Cutan Pathol,., 29, 2002, s. 415–420.
46. Overbeek, L.I., Ligtenberg, M.J., Willems, R.W., et al.: Interpretation of immunohistochemistry for mismatch repair proteins is only reliable in a specialized setting. Am J Surg Pathol., 32, 2008, s. 1246–1251.
47. Chung, D.C., Rustgi, A.K.: The hereditary nonpolyposis colorectal cancer syndrome: genetics and clinical implications. Ann Intern Med., 138, 2003, s. 560–570.
48. Domingo, E., Laiho, P., Ollikainen, M., et al.: BRAF screening as a low-cost effective strategy for simplifying HNPCC genetic testing. J Med Genet., 41, 2004, s. 664–668.
49. South, C.D., Hampel, H., Comeras, I., Westman, J.A., Frankel, W.L., de la Chapelle, A.: The frequency of Muir-Torre syndrome among Lynch syndrome families. J Natl Cancer Inst., 100, 2008, s. 277–281.
50. Plaschke, J., Engel, C., Kruger, S., et al.: Lower incidence of colorectal cancer and later age of disease onset in 27 families with pathogenic MSH6 germline mutations compared with families with MLH1 or MSH2 mutations: the German Hereditary Nonpolyposis Colorectal Cancer Consortium. J Clin Oncol., 22, 2004, s. 4486–4494.
51. Singh, R.S., Grayson, W., Redston, M., et al.: Site and tumor type predicts DNA mismatch repair status in cutaneous sebaceous neoplasia. Am J Surg Pathol., 32, 2008, s. 936–942.
52. Järvinen, H.J., Renkonen-Sinisalo, L., Aktán-Collán, K., Peltomäki, P., Aaltonen, L.A., Mecklin, J.P.: Ten years after mutation testing for Lynch syndrome: cancer incidence and outcomes in mutation-positive and mutation-negative family members. J Clin Oncol., 27, 2009, s. 4793–4797.
Labels
Anatomical pathology Forensic medical examiner ToxicologyArticle was published in
Czecho-Slovak Pathology

2010 Issue 4
Most read in this issue
- Muir-Torre syndrom – fenotypická varianta Lynchova syndromu
- Lymfatický systém: novinky v morfologii a patologii
- Systémová sklerozující choroba spojená s imunoglobuliny IgG4 – současné poznatky
- Dlhodobá dialyzačná liečba chronickej obličkovej nedostatočnosti sprevádzaná získanou cystickou chorobou obličiek a súčasnými prednádorovými zmenami a mnohopočetnými nádormi ľavej obličky