
Primární neuroendokrinní karcinom jater
Primary hepatic neuroendocrine carcinoma
Primary neuroendocrine carcinoma of the liver is a rare tumour, probably arising from scattered neuroendocrine cells of the bile duct. We present the case of a 72-year-old male who experienced gradual weight loss and diarrhoea. Given the fact that he had stayed in the Dominican Republic, a parasitic disease was initially suspected. However, this was not confirmed. Further examination showed tumour infiltration of the liver. Fine needle aspiration cytology of the tumour site was performed.
The diagnostic procedure revealed neuroendocrine carcinoma. The tumour cells expressed the following neuroendocrine markers (chromogranin, synaptophysin, CD56 and NSE) as well as the epithelial marker AE1-AE3. The tumour was considered metastasis of the primary tumour located in the gastrointestinal tract. A thorough clinical examination was performed including gastroscopy, colonoscopy, In-111 Octreoscan scintigraphy, computed tomography and magnetic resonance imaging. These methods revealed metastases in the vertebrae, pelvis, long bones and skull. No other tumour sites were found in the lungs, gastrointestinal tract or pancreas. The patient became increasingly cachexic and later died.
An autopsy showed massive multicentric tumour infiltration of the liver. Histological examination revealed well differentiated neuroendocrine carcinoma which transformed into intermediate and small cells. The autopsy found no tumour sites in the gastrointestinal tract, lungs or pancreas. The results were suggestive of primary neuroendocrine carcinoma of the liver.
Keywords:
neuroendocrine carcinoma – liver – primary tumour
Authors:
J. Dvořáčková 1; J. Mačák 1; F. Fakhouri 1; J. Horáček 1; J. Plášek 2
Authors‘ workplace:
Ústav patologie Lékařské fakulty Ostravské univerzity a Fakultní nemocnice v Ostravě
1; Interní klinika Lékařské fakulty Ostravské univerzity a Fakultní nemocnice v Ostravě
2
Published in:
Čes.-slov. Patol., 48, 2012, No. 1, p. 49-52
Category:
Original Article
Overview
Primární neuroendokrinní karcinom jater patří ke vzácně se vyskytujícím nádorům, který pravděpodobně vychází z roztroušených neuroendokrinních buněk žlučových cest. V našem případě šlo o 72-letého muže, u kterého docházelo postupně k úbytku váhy a k průjmům. Vzhledem k tomu, že pacient pobýval v Dominikánské republice, pomýšlelo se na parazitární onemocnění. To se však neprokázalo. Dalším vyšetřením byla zjištěna nádorová infiltrace jater. Byl proveden cytologický odběr tenkou jehlou z místa nádoru.
Vyšetření ukázalo, že se jedná o neuroendokrinní karcinom. Nádorové buňky exprimovaly neuroendokrinní markery (chromogranin, synaptofyzin, CD56 a NSE) stejně tak i epitelový marker AE1-AE3. Nádor byl považován za metastázu primárního nádoru, který se pravděpodobně nachází v gastrointestinálním traktu. Pacient se podrobil zevrubnému klinickému vyšetření včetně gastroskopie, kolonoskopie, 111In-oktreoskenové scintigrafii, počítačové tomografii a vyšetření magnetickou rezonancí. Metody ukázaly metastázy v obratlech, pánvi, dlouhých kostech a lebce. Žádné další nádorové ložisko v plicích, v gastrointestinálním traktu nebo v pankreatu nebylo zjištěno. Pacient postupně kachektizoval a zemřel.
Pitva ukázala masivní multicentrickou nádorovou infiltraci jater. Histologicky šlo o dobře diferencovaný neuroendokrinní karcinom, který přecházel do intermediálních až malých buněk. V gastrointestinálním traktu, plicích a ani v pankreatu se při pitvě žádné nádorové ložisko nenašlo. Domníváme se, že šlo o primární neuroendokrinní karcinom jater.
Klíčová slova:
neuroendokrinní karcinom – játra – primární nádor
Většina dobře diferencovaných neuroendokrinních nádorů (WDNET) a dobře diferencovaných neuroendokrinních karcinomů (WDNEC) vzniká v gastrointestinálním traktu (GIT), méně často v plicích a v pankreatu. V játrech je jejich výskyt vzácný. Ojediněle jsou v játrech popsány i špatně diferencované neuroendokrinní karcinomy (malobuněčné karcinomy) (1–3). WDNET popsal jako první v játrech Edmondson v r. 1958 (4,5). Množství publikovaných případů je několik desítek, Modlin a spol. v r. 2005 (6) uvádějí 95 případů. Soga (2) v japonském registru zjistili 126 takových nádorů. Většina patří k WDNET. Nádory se odvozují pravděpodobně z neuroendokrinních buněk, které jsou roztroušeny mezi buňkami sliznice žlučových cest. Obdobně je tomu tak i u primárních neuroendokrinních nádorů žlučníku. Dřívější koncepce odvozovaly neuroendokrinní buňky z neurální lišty. V současné době se jejich embryologický původ odvozuje z endodermu. Podle některých autorů (4) mohou tyto nádory vznikat i z aberantních tkání pankreatu v jaterním parenchymu a případně i pluripotentních jaterních buněk (5,6).
MATERIÁL A METODIKA
72-letý muž byl přijatý do nemocnice s průjmy, které trvaly 2 měsíce, a s úbytkem váhy. Zavádějící informací byl pacientem uváděný pobyt v Dominikánské republice. V klinických úvahách bylo parazitární onemocnění. Vyšetření ultrazvukem ukázalo mnohočetná ložiska v játrech. To bylo potvrzeno počítačovou tomografií (CT) a magnetickou rezonancí (MR). Kromě jater se objevil ložiskový proces v obratlech. Zjistilo se, že v moči bylo výrazně zmnožené množství 5-hydroxyindoloctové kyseliny a chromograninu. Vyšetření 111In-octreoskenovou scintigrafií ukázalo zvýšenou denzitu somatostatinových receptorů v jaterním parenchymu bez pozitivních ložisek v GIT, pankreatu a plících. Pro morfologické vyšetření jsme měli k dispozici tenkojehlovou cytologii a nekroptický materiál. Ten byl zpracován parafinovou technikou a řezy byly standardně barveny hematoxylinem a eozinem.
Imunohistologické vyšetření. Imunohistologické vyšetření bylo provedeno metodou avidin-biotin komplex (ABC metoda). Přitom byly provedeny pozitivní a negativní kontroly. Pro vyšetření byly použity následující protilátky (v závorce je pracovní ředění): pancytokeratin AE1-AE3, klon AE1-AE3 (1 : 50); CK20, klon Ks 20.8 (předředěná); CK7, klon OV-TL 12/13 (1 : 50); NSE, klon 2F111 (1 : 50); anti-human hepatocyte, klon OCH1E5 (1 : 50); polyclonal rabbit anti-human gastrin (1 : 2000); polyclonal rabbit anti-human somatostatin code A0566 (1 : 1000); monoclonal mouse anti-human serotonin, klon 5HT-H209 (1 : 100); polyclonal rabbit anti-human glucagon, code A0565 (1 : 1000). Tyto protilátky jsou produkty fy DAKO Glostrup, DK; synaptofyzin, klon 27G12 (1 : 100); chromogranin, klon 5H7 (1 : 100), protilátky pocházejí od fy NOVOCASTRA, Newcastle-upon-Tyne, UK; CA 19-9, klon 1116-NS-19-9 (1 : 100) LSBio, Seattle; rabbit anti-Pancreatic Polypeptide, klon 18-0043 (1 : 100), Invitrogen GmbH, Lofer, Rakousko; VIP (vasoactive Intestinal Peptide), (1 : 500), Immunostar, USA.
POPIS PŘÍPADU
Cytologické vyšetření tenkou jehlou ukázalo, že jde o nádor tvořený středně velkými a malými hyperchromními buňkami. Nádorové buňky vykazovaly pozitivitu s následujícími markery: AE1-AE3, chromograninen, synpatofyzinem, CD56 a NSE. Závěr vyšetření konstatoval, že se jedná nejspíše o metastázu špatně diferencovaného neuroendokrinního karcinomu do jater.
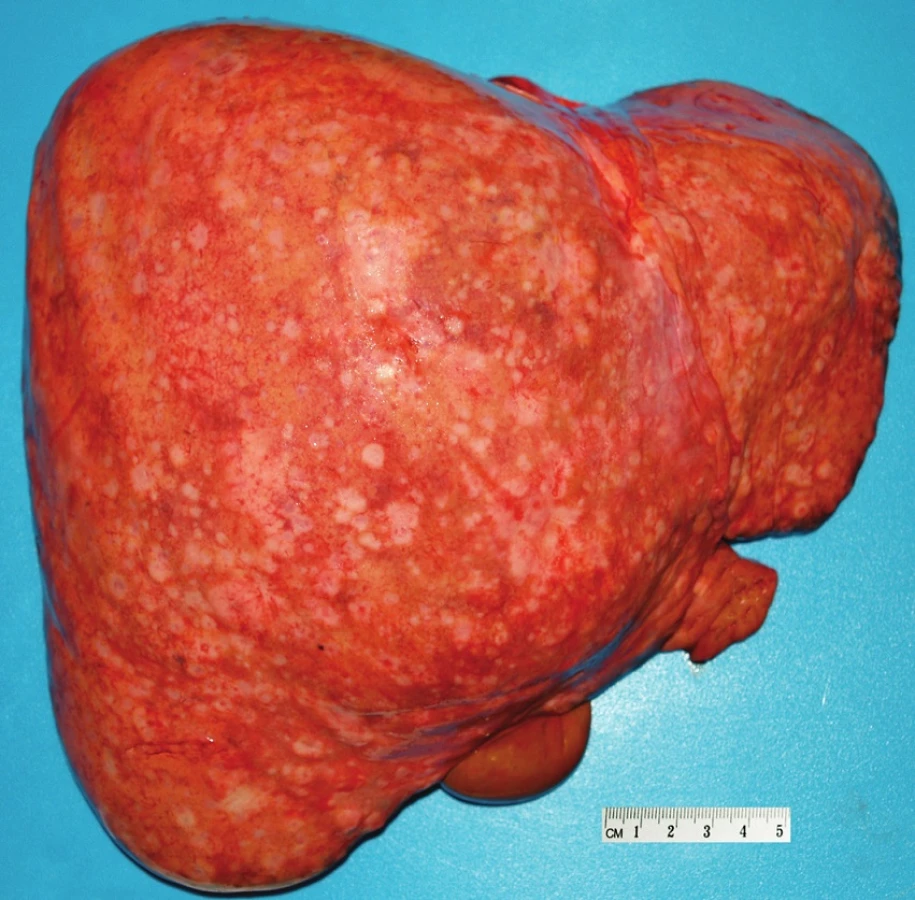
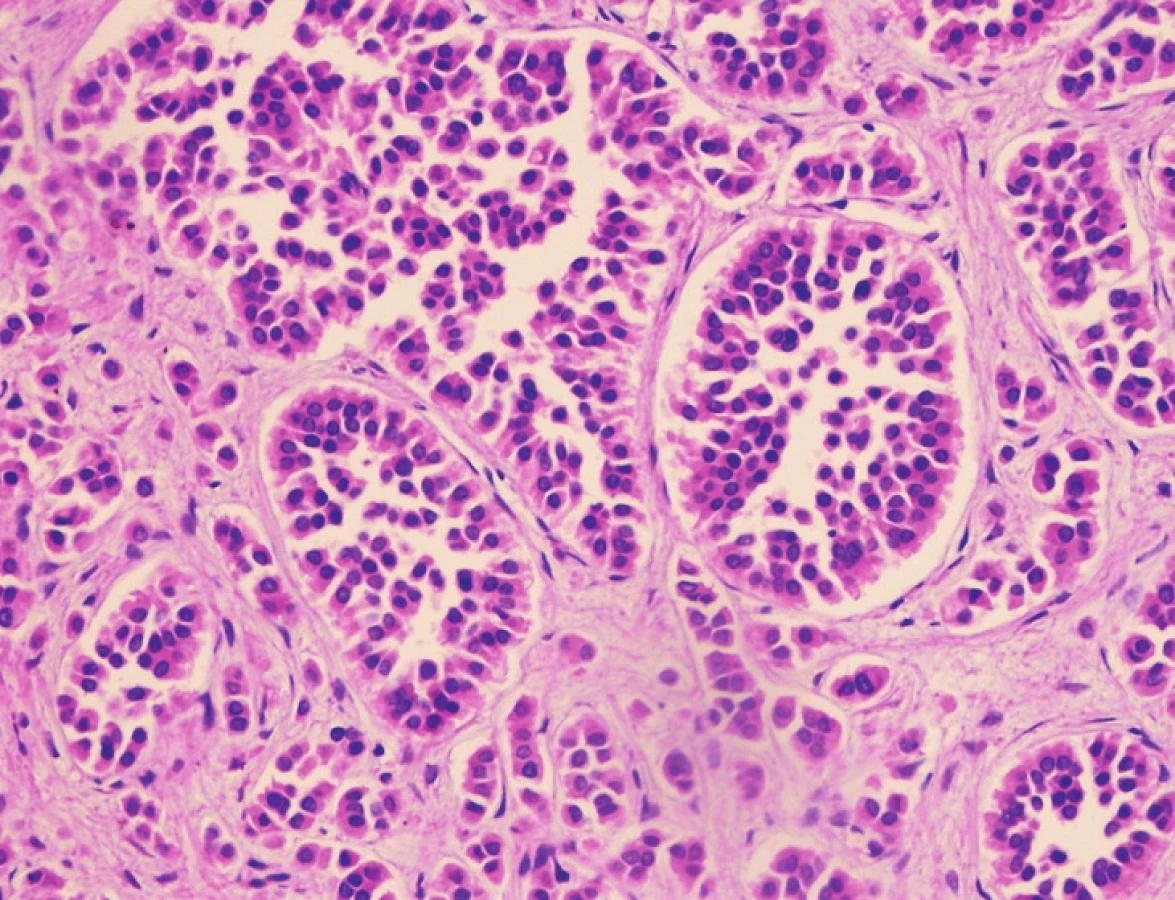
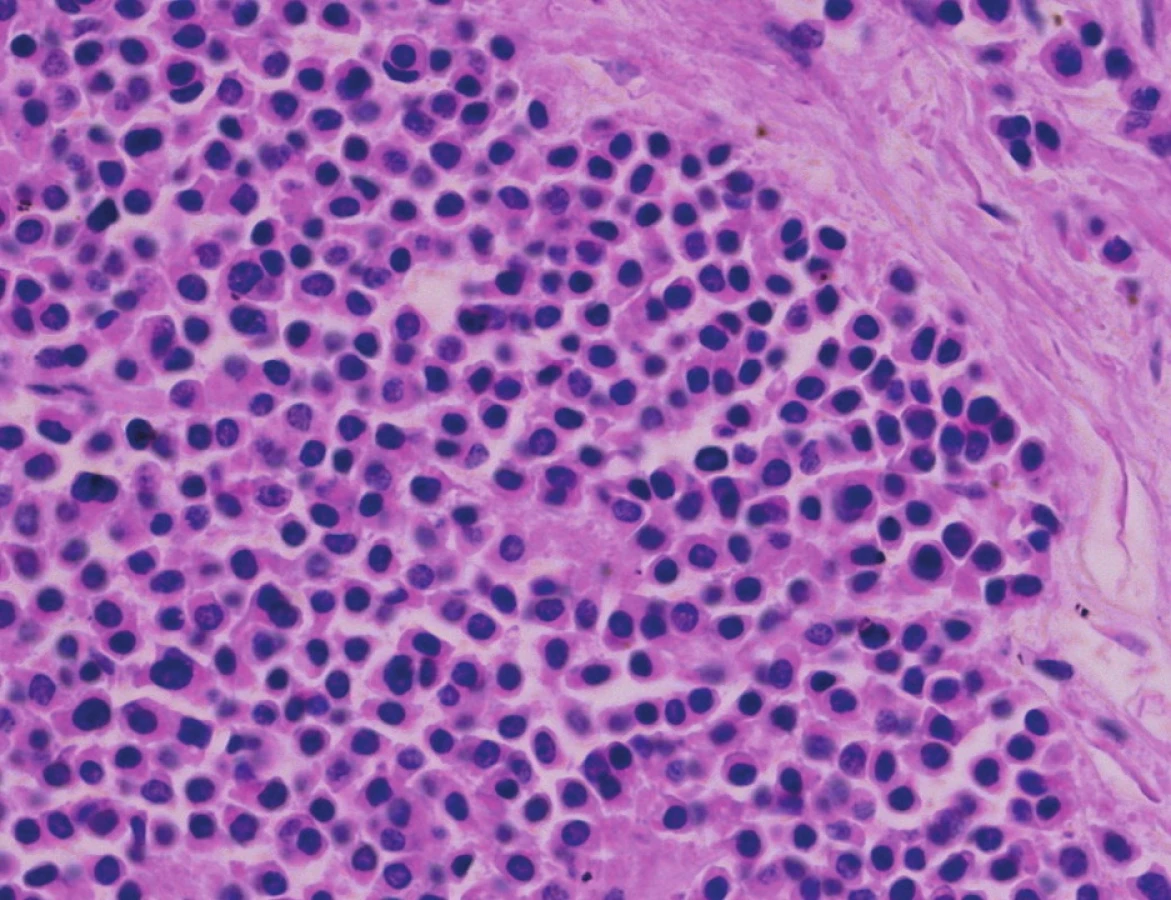
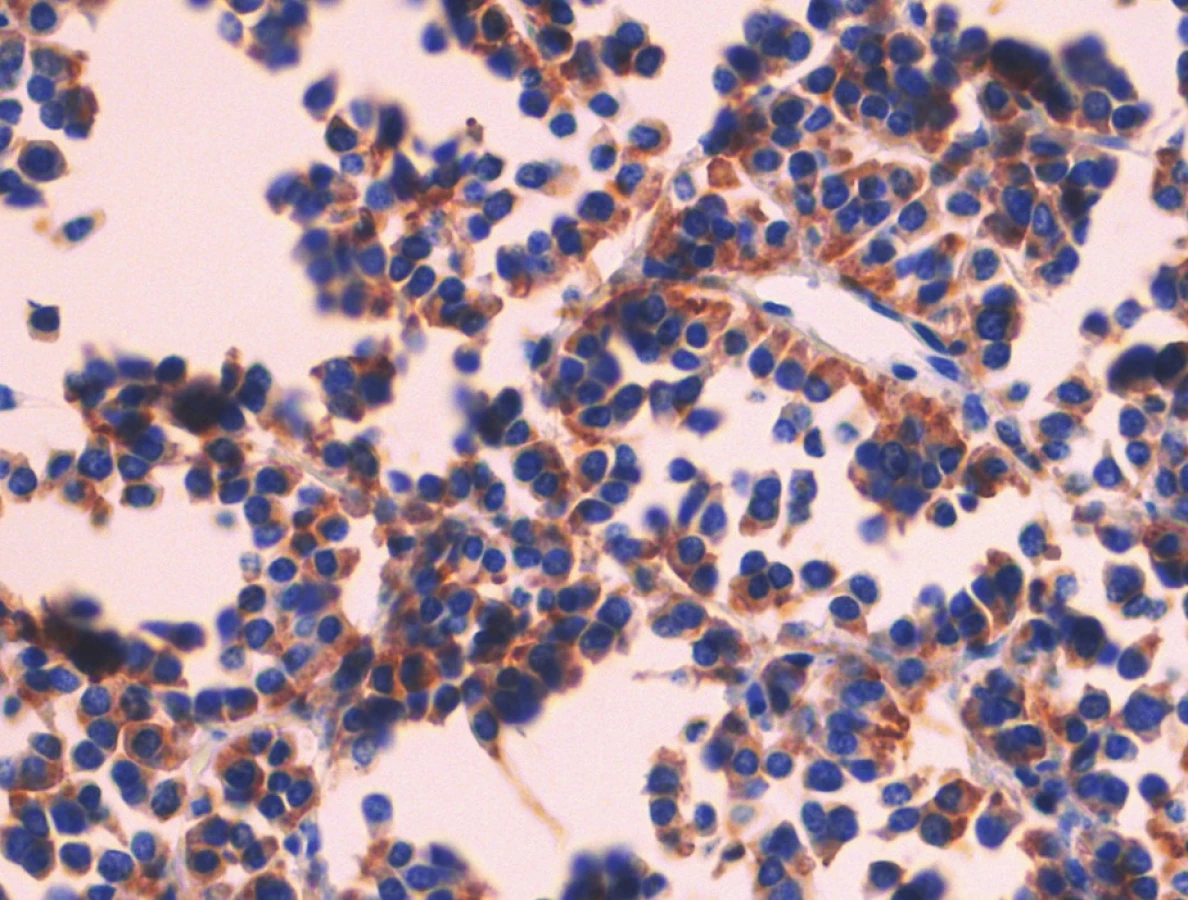
Pacient stále trpěl kolikovitými bolestmi břicha, průjmy a úbytkem váhy. Vzhledem k tomuto závěru byla provedena série klinických vyšetření (včetně gastroskopie a kolonoskopie) s cílem zjistit primární nádorové ložisko. To se však nepodařilo. Vyšetření močového měchýře ani ledvin neukázalo nádorové změny. I přes zevrubné klinické vyšetření pomocí CT, MR a oktreoskenové scintigrafie nebylo, kromě jater, odhaleno žádné jiné nádorové ložisko v GIT nebo v plicích. V kostech se prokázala metastatická ložiska (obratle, pánev, dlouhé kosti, lebka). Pacient postupně kachektizoval a za 8 měsíců od stanovení diagnózy zemřel. Při pitvě bylo zjištěno, že játra jsou zvětšená a infiltrovaná velmi četnými ložisky a uzly velikosti až 3,5 cm (Obr. 1,2). Při pitvě GIT, pankreatu a plic se nádorové změny nenašly. Nádorová ložiska byla přítomna v obratlech Th6–L5 a dále podle oktreoskenové scintigrafie ve sternu, dlouhých kostech, pánvi a lebce. Histologicky byla v játrech přítomna nádorová ložiska tvořená středně velkými a malými nádorovými buňkami s kulatými nebo lehce oválnými hyperchromními jádry. Buňky byly uspořádány do acinů na periferii s palisádovitým uspořádáním (Obr. 3). Tato ložiska plynule přecházela v pruhy, snopce a ložiska malých hyperchromních buněk (Obr. 4). Mitotická byla < 2 mitózy/10 HPF, proliferační marker Ki-67 byl v úsecích nejvyšší aktivity asi 5 %/10 HPF. Imunohistologicky nádor exprimoval obdobné markery jako bylo uvedeno výše u punkčního cytologického vyšetření (Obr. 5,6,7). Vzhledem k tomu, že jak opakovaná klinická vyšetření tak pitva neobjevily žádné jiné primární nádorové ložisko, případ byl uzavřen jako WDENC jater. Histologicky nádor přecházel do středně velkých a malých buněk. Protilátky proti serotoninu, somatostatinu, gastrinu, VIP, glukagonu, pankreatickému polypeptidu, proti lidským hepatocytům se v nádorových buňkách neexprimovaly.



DISKUZE
Většina (asi 85 %) WDNET a WDNEC se objevuje v GIT, pouze asi 10 % vzniká v plicích. Ve zbývajících případech se tyto nádory objevují v larnygu, tymu, ledvinách ováriích a v kůži. Nejčastější lokalizace neuroendokrinních nádorů v GIT jsou: apendix, ileum, jejunum, rektum a tlusté střevo. V plicích se objevují převážně WDNET a jen vzácně jde o WDNEC (7). V játrech je přítomnost všech primárních neuroendokrinních nádorů neobvyklá. Stejně je tomu tak i ve žlučníku. Zde se nádory pravděpodobně také odvozují z roztroušených neuroendokrinních buněk sliznice a mohou sekundárně prorůstat do jater (8–11). Množství neuroendokrinních buněk v epitelu žlučovodů může výrazně kolísat. Roskams a spol. (12) ukázali, že některé buňky drobných žlučovodů při regeneraci jater získávají neuroendokrinní charakteristiky jak po stránce imunohistologické, tak i elektronově mikroskopické. Slouží jako fakultativní kmenové buňky, které se aktivují, když je regenerace hepatocytů nedostatečná nebo potlačená. Obdobně je tomu tak i u některých hepatocytů v periportální oblasti. V pozdějších fázích regenerace, buňky transformují v jiný buněčný typ, který již nemá neuroendokrinní charakteristiky. Pravděpodobně látky ze sekretorických granulí vedou k apokrinnímu či parakrinnímu ovlivnění a transformaci do jiných buněčných linií.
Játra jsou orgánem, kam velmi často metastazují nádory GIT, včetně neuroendokrinních karcinomů. Proto jsou zprávy o výskytu primárních neuroendokrinních nádorů jater přijímány s určitou nedůvěrou. Dala a spol. (13) naopak uvádějí, že přehnaná nedůvěra nedovolí některé nádory diagnostikovat jako primární jaterní nádory. Ty pak unikají ze statistických přehledů. V současné době je v klinické praxi řada sofistikovaných a dostatečně citlivých metod, které mohou primární ložisko v GIT odhalit. Tyto metody byly užity i v našem případě (oktreoskenová scintigrafie, CT, MR, gastroskopie, kolonoskopie). Samotné imunohistochemické vyšetření nádoru nemůže v současné době rozlišit mezi metastatickým rozsevem a primárním neuroendokrinním nádorem jater. U nádorových buněk jsme se snažili pomoci protilátek zjistit tvorbu některých hormonů (somatostatinu, glukagonu, serotoninu, gastrinu, VIP, PP), ale výsledek vyšetření byl zřejmě negativně ovlivněný autolýzou nekroptického materiálu. Nádorové buňky rovněž nereagovaly s protilátkou proti lidským hepatocytům a také výsledek reakce nádorových buněk s proliferačním markerem Ki-67 byl zřejmě ovlivněný autolytickým procesem.
Na molekulárně genetické úrovni Zhao a spol. (7) hledali rozdíly na mezi WDNET v GIT a v plicích. Prokazovali alterace genomu metodami komparativní genové hybridizace a pomocí interfázové fluorescenční in situ hybridizace (FISH). Domnívají se, že nádory v obou lokalizacích se vyvíjejí rozdílnou molekulárně genetickou cestou. U nádorů GIT prokázali inaktivaci jednoho nebo několika supresorových genů na chromozomu 18, zatím co u plicních WDNEC jde spíše o inaktivaci genů na chromozomu 11q.
Spolehlivou metodou, kterou můžeme primární mimojaterní lokalizaci zjistit je pitva a následné histologické vyšetření. Ta byla v našem případě provedena a ani podrobné vyšetření GIT včetně pankreatu neukázalo nádorovou infiltraci. Do širšího rámce neuroendokrinních nádorů jater je možné zahrnout i případy dříve publikovaných nádorů: hepatocelulárního karcinomu či cholangiocelulárního karcinomu s neuroedokrinním syndromem (5,6,14). Hepatocelulární karcinomy se většinou objevují u nemocných s jaterní cirhózou zatímco primární neuroendokrinní nádory se nacházejí v necirhotických játrech. Histologicky často buňky hepatocelulárního karcinomu připomínají hepatocyty. Mají poměrně obsáhlou eozinofilní cytoplazmu. Imunohistologicky jsou pozitivní s protilátkou proti lidským hepatocytům a také s protilátkou proti karcinoembryonálnímu antigenu (CEA) v intercelulárních kanalikulech. Tyto markery se v našem případě neexprimovaly. Avšak podle některých sdělení (15,16) mohou neuroendokrinní nádory rovněž exprimovat markery adenokarcinomů jako jsou CEA a CA19-9. Cholangiocelulární karcinom zpravidla exprimuje cytokeratin 7 (17,18).
Rovněž radiologický obraz může přispět k diagnóze. Hepatocelulární karcinomy jsou často hypervaskulární, s prominujícími novotvořenými cévami, arterio-venózními shunty a vaskulárními jezírky.
Primární neuroendokrinní nádory jater se objevují nejčastěji u pacientů v průměrném věku kolem 50 let. Byly však popsány i případy u dětí ve věku 8–11 let (19). Zpravidla se nádor projevuje jako jedno nádorové ložisko velikosti kolem 10 cm, místy v okolí s menšími satelitními nádorovými uzly. V zobrazovacích metodách má nádorové ložisko často cystickou úpravu připomínající až hemangiom. Metastazují do kostí a do lymfatických uzlin (2,20). V našem případě nádor rostl multicentricky. Dispersní nádorový rozsev v jaterním parenchymu značně připomínal metastázy. Podobné případy byly již publikovány v několika předcházejících sděleních (4,21–24,26). Ojediněle jsou v literatuře zaznamenány i případy primárního špatně diferencovaného neuroendokrinního karcinomu (malobuněčného karcinomu) jater, který je histologicky neodlišitelný od obdobného nádoru plic (27–29).
Primární neuroendokrinní nádory jater se objevují vzácně. Vždy je nutné vyloučit metastatický rozsev do jater. Mnohdy se to v průběhu života pacientů nepodaří a pak teprve pitva může vést k jednoznačnému závěru.
PODĚKOVÁNÍ
Autoři děkují Dr. O. Koperek z Klinisches Institute für Pathologie, AKH Vídeň, za pomoc při imunohistologickém vyšetření speciálními protilátkami.
Adresa pro korespondenci:
Prof. MUDr. J. Mačák, CSc.
Ústav patologie LF OU a FN Ostrava-Poruba
Tř. 17. listopadu 1790, 708 52 Ostrava-Poruba
tel: 737054581
e-mail: macak.jirka@seznam.cz
Sources
1. Edmondson HA. Carcinoid tumor. In: Edmondson HA. Tumors of the Liver and Intrahepatic Bile Ducts. Atlas of tumor Pathology, fascicle 25. Washington: AFIP; 1958 : 105–111.
2. Soga J. Primary hepatic endocrinomas (carcinoids and variant neoplasms). A statistical evaluation of 126 reported cases. J Exp Clin Cancer Res 2002; 21 : 457–468.
3. Modlin IM, Kidd M, Latich I, et al. Current status of gastrointestinal carcinoids. Cancer 1997; 79 : 813–829.
4. Yp X, JY Y. Primary neuroendocrine carcinoma of the liver. Ultrastruct Pathol 1986; 10 : 331–336.
5. Alpert LI, Zak FG, Werthamer S, Bochetto JF. Cholangiocarcinoma: a clinicopathologic study of five cases with ultrastructural observations. Hum Pathol 1974; 5 : 709–728.
6. Primack A, Wilson J, OęConnor GT, et al. Hepatocellular carcinoma with the carcinoid syndrome. Cancer 1971; 27 : 1182–1189.
7. Zhao J, de Krijger RR, Meier D, et al. Genomic alterations in well-differentiated gastrointestinal neuroendocrine tumors (carcinoids). Marked differences indicating diversity in molecular pathogenesis. Am J Pathol 2000; 157 : 1431–1438.
8. Deehan DJ, Heys SD, Kernohan N, Eremin O. Carcinoid tumour of the gallbladder: two case reports and a review of published works. GUT 1993; 34 : 1274–1276.
9. Khetan N, Bose NCH, Arya SV. Carcinoid tumor of the gallbladder: report of a case. Surg Today 1995; 25 : 1047–1049.
10. Ozawa K, Kinoshita M. A case of double carcinoid tumors of the gallbladder. Dig Dis Sci 2003; 48 : 1760–1761.
11. Zou YP, Li WM, Liu HR, Li N. Primary carcinoid tumor of the gallbladder: a case report and brief review of the literature. World J Surg Oncol 2010; 8 : 12.
12. Roskams T, de Vos R, van der Oord JJ, Desmet V. Cells with neuroendocrine features in regenerating human liver. APMIS Suppl. 1991; 23 : 32–39.
13. Dala R, Shoosmith J, Lilenbaum R, Cabello-Inchausti B. Primary hepatic neuroendocrine carcinoma: an underdiagnosed entity. Ann Diagn Pathol 2006; 10 : 28–31.
14. Nwokediuko SCH, Uchenna I, Esther O, Ocheukwu O, Augustine O, Charity A. Relating long survival in hepatocellular carcinoma presentig with carcinoid syndrome. Gastroenterol Res 2010; 3 : 46–49.
15. Blobel GA, Gould VE, Moll R, et al. Coexpression of neuroendocrine markers and epithelial cytoskeletal proteins in bronchopulmonary neuroendocrine neoplasms. Lab Invest 1985; 52 : 39–51.
16. Kimura N, Sasano N, Namiki T, Nakazato Y. Coexpression of cytokeratin, neurofilament and vimentin in carcinoid tumors. Virchows Arch A Pathol Anat Histopathol 1989; 415 : 69–77.
17. Ishak KG, Goodman ZD, Stocker JT. Tumors of the liver and intrahepatic bile ducts, third edition, Atlas of tumor Pathology, fascicle 31. Washington: AFIP; 2001 : 274–275.
18. Lau PPL, Tint KA, Tse GMK, et al. Primary hepatic carcinoids: report of two cases. Pathology 2006; 38 : 458–461.
19. Broaddus RR, Herzog CE, Hicks MJ. Neuroendocrine tumors (carcinoids and neuroendocrine carcinoma) presentig at extra-appendiceal sites in childhood and adolescence. Arch Pathol Lab Med 2003; 127 : 1200–1203.
20. Nikfarjam M, Muralidharan V, Christophi C. Primary hepatic carcinoid tumours. HPB 2004; 6 : 13–17.
21. Ferrero A, Gallino C, DęAloisio G, Gandini G, Garavoglia M. Primary neuroendocrine carcinoma of the liver: difficult diagnosis of a rare neoplasm. Acta Chir Belg 1999; 99 : 299–302.
22. Ishizu A, Yokoyama K, Yoshiki T. Primary neuroendocrine carcinoma of the liver diagnosed at autopsy. J Gastroenterol Hepatol 2003; 18 : 1002–1004.
23. Iwao M, Nakamuta M, Enjoji M, et al. Primary hepatic carcinoid tumor: case report and review of 53 cases. Med Sci Monit 2001; 7 : 746–750.
24. Kaya G, Pasche Ch, Osterheld MC, Chaubert P, Fontolliet Ch. Primary neuroendocrine carcinoma of the liver: an autopsy case. Pathol Int 2001; 51 : 874–878.
25. Pilichowska M, Kimura N, Ouchi A, et al. Primary hepatic carcinoid and neuroendocrine carcinoma: clinicopathological and immunohistochemical study of five cases. Pathol Int 1999; 49 : 318–324.
26. Yosuda E, Takeshita A, Murata S, et al. Neuroendocrine carcinoma of the liver associated with dermatomyositis: autopsy case and review of the literature. Pathol Int 2003; 56 : 749–754.
27. Kim YH, Kwon R, Jung GJ, et al. Extrapulmonary small-cell carcionoma of the liver. J Hepatobiliary Pancreat Surg 2004; 11 : 333–337.
28. Sengoz M, Abacioglu U, Salepci T, et al. Extrapulmonary small cell carcinoma: multimodality treatment results. Tumori 2003; 89 : 274–277.
29. Zanconati F, Falconieri G, Lamovec J, Zidar A. Small cell carcinoma of the liver: a hitherto unreported variant of hepatocellular carcinoma. Histopathology 1996; 29 : 449–453.
Labels
Anatomical pathology Forensic medical examiner ToxicologyArticle was published in
Czecho-Slovak Pathology

2012 Issue 1
Most read in this issue
- Gynekologické prekancerózy z pohledu klinika dnes a zítra
- Prekancerózní léze vulvy
- Co je nového v cytodiagnostice cervikálních prekanceróz?
- Prekancerózy endometria, děložní tuby a ovaria: přehled současné problematiky