
Neuropatologie farmakorezistentní epilepsie - strukturální podklad a mechanismy epileptogeneze
Neuropathology of refractory epilepsy: the structural basis and mechanisms of epileptogenesis
In recent years, the expansion of surgical treatment of patients with refractory epilepsy brought unique opportunity to analyse resected epileptic brain tissue and to define the morphological and molecular basis of this heterogeneous disease. The most common clinicopathological entities identified in epilepsy surgical brain specimens are hippocampal sclerosis, malformations of cortical development, glioneuronal tumors, vascular malformations, glial scarring or inflammation. In addition to the diagnostics and classification of the lesions, the text provides a summary of current knowledge about the pathogenesis and mechanisms, by which they contribute to the genesis and spread of epilepsy.
Keywords:
epilepsy - hippocampal sclerosis - focal cortical dysplasia - epileptogenesis
:
J. Zámečník
:
Ústav patologie a molekulární medicíny, 2. LF UK a FN Motol, Praha
:
Čes.-slov. Patol., 48, 2012, No. 2, p. 76-82
:
Review Articles – Neuropathology
V posledních letech přinesl rozvoj epileptochirurgické léčby pacientů s farmakorezistentní epilepsií unikátní možnost vyšetřovat resekovanou epileptickou mozkovou tkáň a definovat tak morfologický a molekulární podklad tohoto heterogenního onemocnění. Nejčastěji diagnostikovanými klinicko-patologickými jednotkami jsou hipokampální skleróza, malformace kortikálního vývoje (zejména fokální kortikální dysplazie), glioneuronální nádory, cévní malformace, gliové jizvení nebo záněty. V textu je kromě histopatologické diagnostiky a klasifikace těchto lézí podán souhrn současných poznatků o jejich patogenezi a o mechanismech, kterými se podílejí na vzniku a šíření epilepsie.
Klíčová slova:
epilepsie - hipokampální skleróza - fokální kortikální dysplazie - epileptogeneze
Rozvoj epileptochirurgie jako důležitého terapeutického postupu u pacientů s farmakorezistentní epilepsií (1) přinesl v posledních letech unikátní možnost vyšetřovat resekovanou epileptickou mozkovou tkáň a definovat tak morfologický podklad tohoto heterogenního onemocnění. Dostupnost čerstvé a také dobře elektrofyziologicky i neuroradiologicky charakterizované lidské epileptické tkáně otevřela bránu výzkumu, který do té doby probíhal jen na experimentálních zvířecích modelech nebo na úrovni elektrofyziologie. V posledních letech tak mohlo dojít k alespoň částečnému rozkrytí mechanismů, kterými různé léze CNS farmakorezistentní epilepsii vyvolávají, což přineslo také důležité informace pro pochopení jejich patogeneze (včetně molekulárních mechanismů). Tyto poznatky by mohly v budoucnu ukázat nové směry v terapii pacientů s tímto dnes konzervativně neřešitelným onemocněním.
Podle zkušeností jak v naší sestavě čítající nyní přes 400 vyšetřených pacientů, tak i ve větších souborech (2), je některá z definovaných epileptopatologických jednotek diagnostikována až v 95 % resekátů. Nejčastěji jsou to hipokampální skleróza, malformace kortikálního vývoje (zejména fokální kortikální dysplazie, ale také tuberózní skleróza nebo hemimegalencefalie), low-grade nádory mozku (většinou smíšené glioneuronální tumory), vzácněji pak cévní malformace mozku, gliové jizvy (posttraumatické, postischemické) nebo změny po proběhlé (meningo)encefalitidě. Kazuisticky lze pozorovat i zvláštní unihemisferální autoimunitní zánět - Rasmussenovu encefalitidu. Po celou dobu rozvoje epileptopatologie se ale vedou vzrušené debaty o tom, jakou skutečnou roli v rozvoji epilepsie tyto strukturální změny hrají a do jaké míry se na rozvoji epilepsie podílejí spíše okolní struktury (2). Nejevidentnější je to na příkladech astrocytárních nádorů a kavernomů, které schopnost generovat akční potenciály nemají.
Asi v 5 % případů se však dnes ani extenzivním neuropatologickým vyšetřením žádnou konkrétní morfologickou poruchu odhalit nepodaří. A to i přesto, že resekovaná tkáň byla z hlediska klinického i elektrofyziologického jednoznačným zdrojem záchvatovité aktivity.
HIPOKAMPÁLNÍ SKLERÓZA
U pacientů s farmakorezistentní epilepsií temporálního laloku (TLE, temporal lobe epilepsy) je nečastějším strukturálním podkladem onemocnění hipokampální skleróza (HS; synonymum: mesiotemporální skleróza) (3-5). HS nemusí být v resekovaném temporálním laloku přítomna izolovaně, ale může se kombinovat s jinou další poruchou ve zbylé části temporálního laloku (většinou se kromě mesiotemporální oblasti s hipokampem resekuje i pól temporálního laloku - tzv. anteromediální temporální reskce). Pro tuto situaci se v epileptologii vžilo nepříliš zdařilé označení “duální patologie”. Nejčastěji to bývají fokální kortikální dysplazie (4,6), případně nádory nebo gliové jizvení.
Ačkoli zatím není příčina HS zcela objasněna, anamnéza velké části těchto pacientů bývá poměrně charakteristická. Asi u 50 % pacientů s HS se podaří odhalit některý z tzv. iniciálních inzultů (IPI, incial precipitating injury) většinou před 4. rokem života. Nejčastěji to bývají febrilní křeče, méně často je to perinatální poškození CNS, kraniocerebrální úraz nebo meningoencefalitida (3,4,6).
Zajímavé jsou klinické charakteristiky pacientů s HS (2,6). HS se poprvé manifestuje průměrně kolem 11. roku života ve formě komplexních parciálních záchvatů. Doba trvání epileptických záchvatů rezistentních k antiepileptické farmakoterapii před epileptochirurgickým výkonem je ve velkých sestavách téměř 24 let a průměrný věk pacientů s HS v neuropatologických sestavách pak dosahuje dokonce 35 let. Obě pohlaví bývají postižena stejně často a v naprosté většině případů nebývá ohledně epilepsie pozitivní rodinná anamnéza. Hereditární vlivy tedy v rozvoji TLE na podkladě HS roli nejspíše nehrají.
Histopatologie
Na histopatologické úrovni je HS charakterizovaná segmentálním úbytkem pyramidových neuronů v hipokampálních sektorech CA1 (Sommerův sektor) a CA4 (endfolium/hilus), méně často v CA3; neurony v gyrus dentatus a sektoru CA2 (tzv. rezistentní zóna) naopak bývají ušetřeny (obr. 1). Postiženy ale nejsou jen velké pyramidové neurony hipokampu - zaznamenán byl i úbytek některých interneuronů (např. pozitivních v imunohistochemickém průkazu somatostatinu a neuropeptidu Y) stejně jako mechových buněk v sektoru CA4 (7,8). Úbytek neuronů v postižených hipokampálních sektorech vždy doprovází astroglióza, která vede k tužší konzistenci tkáně (z toho tradiční název skleróza hipokampu). Kromě výše popsaných změn lze někdy mikroskopicky nalézt i poruchu uspořádání gyrus dentatus a to ve formě tzv. disperze granulárních buněk (9,10).
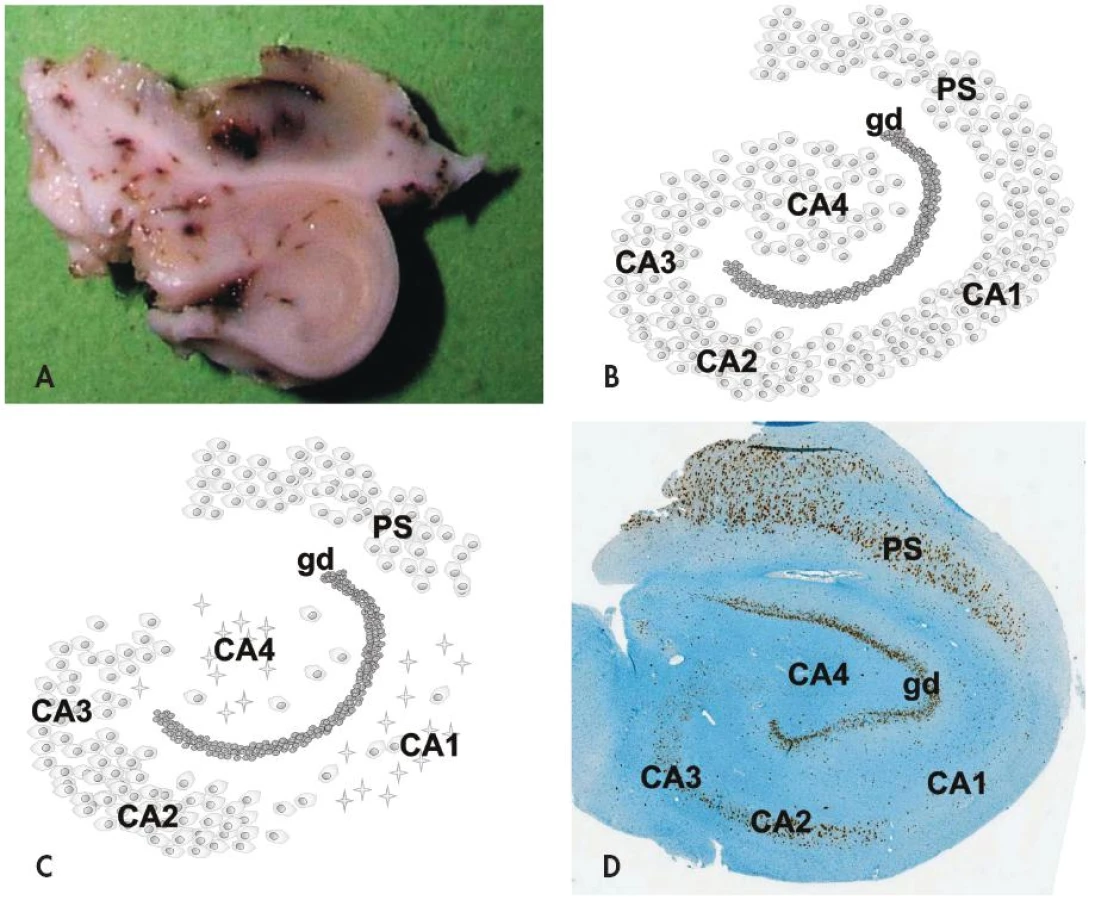
Mechanismus selektivní vulnerability morfologicky podobných neuronů v různých sektorech hipokampu je předmětem intenzivního výzkumu, závěr však zatím není jednoznačný. V patogenezi HS hraje roli jednak vznik abnormálních neuronálních okruhů, které indukuje iniciální inzult (mechanismem pučení mechových vláken - tzv. “mossy fiber sprouting”) (11), dále pak molekulární přestavba různých iontových kanálů a změněná exprese řady receptorů pro neurotransmitery (12).
Klasifikace HS
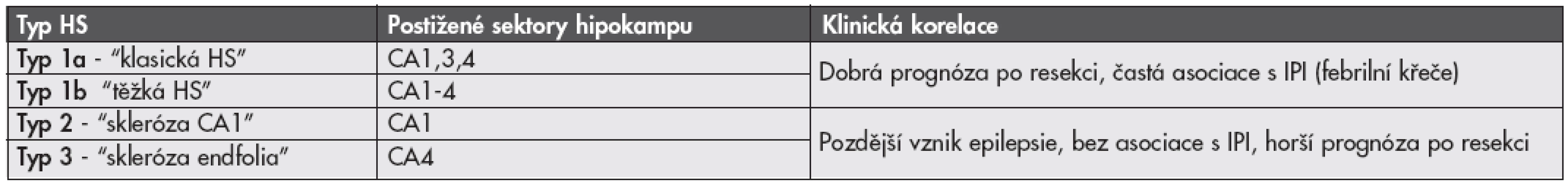
Vzhledem k tomu, že rozsah i distribuce postižení jednotlivých oblastí hipokampu se u jednotlivých případů liší (3,4), je zde dlouhodobá snaha nalézt vhodnou histopatologickou klasifikaci tohoto onemocnění. Byla navržena celá řada různých klasifikačních systémů, žádný z nich však nebyl neurology akceptován pro svou nízkou výpovědní hodnou ohledně prognózy pacienta po chirurgické resekci nebo následných poruch paměti. Poměrně slibným se ukazuje být návrh klasifikace HS německých autorů (13), kteří na velké sestavě pacientů prokázali její korelaci s důležitými klinickými aspekty onemocnění včetně psychofyziologické prognózy a pooperačního postižení procesu učení. Návrh této klasifikace je uveden v tabulce 1.
FOKÁLNÍ KORTIKÁLNÍ DYSPLAZIE A DALŠÍ MALFORMACE VÝVOJE MOZKOVÉ KŮRY
Malformace kortikálního vývoje mozku (MCD, malformations of cortical development) jsou také poměrně častým podkladem farmakorezistentní epilepsie pacientů, kteří dospějí k neurochirurgickému výkonu (14). Spektrum těchto malformací je velmi široké - od rozsáhlých strukturálních poruch zřetelných už makroskopicky (resp. při vyšetření MRI - hemimegalencefalie, polymikrogyrie), až po poměrně diskrétní poruchy. Nodulární heterotopie neuronů nebo fokální kortikální dysplazie (FCD) mohou být tak drobné, že při vyšetření MRI unikají a jsou detekovány až mikroskopicky v resekátu. Existují však i FCD rozsáhlé - lobární až multilobární.
Fokální kortikální dysplazie (FCD) - klasifikace
FCD jsou heterogenní skupinou poruch, charakterizovanou porušenou cytoarchitektonikou mozkové kůry, u některých případů navíc s přítomností cytologicky aberantních neuronů. Hledání vhodné klasifikace těchto poruch, která by měla nejen morfologický, ale i klinický a prognostický význam, bylo v posledních letech věnováno mnoho úsilí.
Palminiho klasifikace FCD z roku 2004 (15). FCD se podle této stále ještě nejpoužívanější klasifikace rozdělují do dvou velkých skupin, které mají své morfologické charakteristiky, ale také rozdílný klinický průběh před operací i prognózu (16). FCD mohou být lokalizovány kdekoli; FCD I. typu ale bývají nejčastěji v temporálním laloku, FCD II. typu zase spíše extratemporálně (tj. ve frontální, okcipitální nebo parietální kůře).
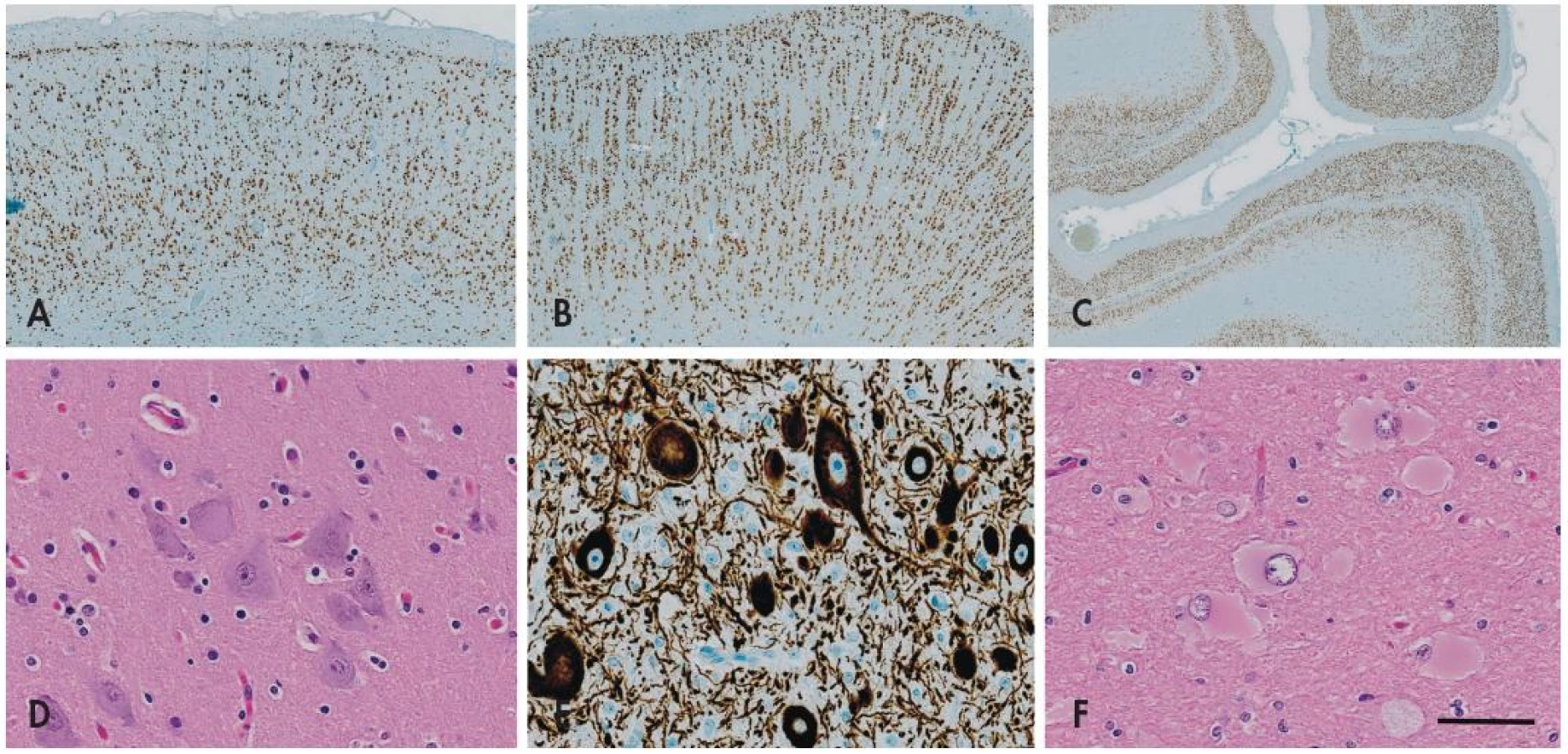
FCD I. typu je charakterizovaná poruchou korové cytoarchitektoniky - mizí pravidelné hexalaminární uspořádání mozkové kůry, vznikají vertikální neuronální sloupce a clustery neuronů s nepravidelnou orientací (obr. 2). Pokud se jedná jen o takovou poruchu architektoniky, označuje se léze jako FCD typu IA, pokud jsou navíc přítomny skupinky nezralých neuronů nebo obrovských neuronů (velikosti až Betzových neuronů) v atypické lokalizaci jako jsou např. svrchní vrstvy kůry, hovoříme o FCD typu IB.
FCD I. typu často nejsou při MRI vyšetření zřetelné, proto je obtížné před chirurgickým výkonem i peroperačně identifikovat lézi v celém rozsahu - v tom je neurochirurg při resekci závislý na invazivní peroperační elektrokortikografii a předpokládané lokalizaci odvozené se symptomatologie záchvatů. Pacienti s FCD I. typu tak mají kvůli riziku nekompletní resekce celkově horší prognózu ohledně vymizení záchvatů po operaci a vyšší riziko recidivy onemocnění (16).
FCD II. typu je charakterizovaná kromě kompletní disrupce laminární stavby mozkové kůry také přítomností charakteristických cytologicky aberantních buněk - tzv. dysmorfních neuronů (FCD typu IIA), případně navíc tzv. balónovitých buněk (FCD typu IIB) (obr. 2). Tyto poruchy bývají většinou rozeznávány jako ložiskové signálové změny už během předoperačního MRI vyšetření, bývají poměrně dobře ohraničené, a jsou-li lokalizované v odstranitelné oblasti mozku, mívají po odstranění výrazně lepší prognózu než FCD typu I (až 75% pacientů je po operaci trvale bez záchvatů) (16-18).
V uvedené klasifikaci z roku 2004 je kromě FCD I. a II. typu rozlišována ještě kontroverzní jednotka - tzv. mírná malformace kortikálního vývoje (“mild MCD”), která je charakterizovaná přítomností heterotopních neuronů v 1. korové vrstvě nebo v přilehlé bílé hmotě. Tato jednotka nebyla široce akceptovaná zejména z důvodu, že heterotopní neurony v daných lokalizacích (zejména v temporálním laloku) mohou být přítomny i fyziologicky - nalézáme je prakticky ve všech resekovaných temporálních pólech, a rozlišení by nejspíše spočívalo v přesné stereologické kvantifikaci. K tomu však chybějí komparativní data ze zdravého nemalformovaného mozku, ale i vůle neuropatologů stereologické kvantifikace pro extrémní časovou i technickou náročnost v resekátech rutinně provádět.
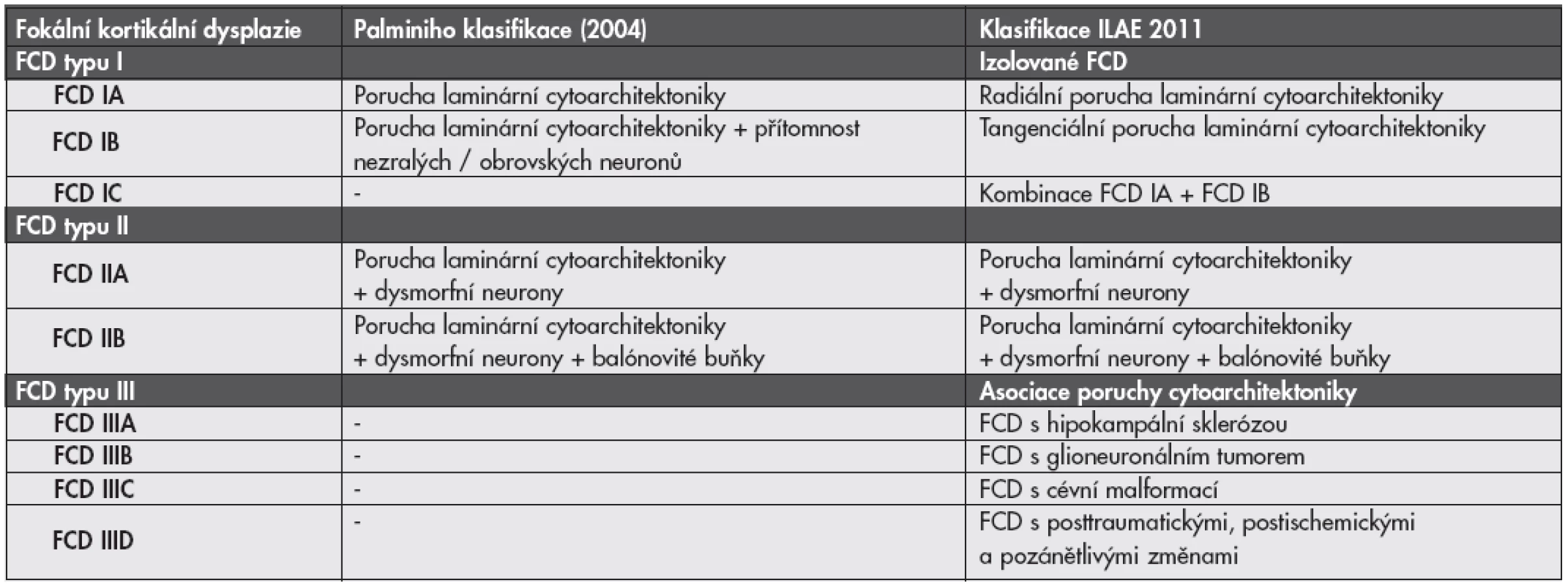
Nová klasifikace FCD - ILAE 2011. Po konsenzuálním zasedání skupiny expertů ILAE (International League Against Epilepsy) byl v minulém roce podán návrh na změnu klasifikace FCD (19,20). Podrobnosti a porovnání s Palminiho klasifikací jsou uvedeny v tabulce č. 2.
Důvody k zavedení nového klasifikačního systému vycházejí podle expertů ILAE z několika skutečností, které se ukázaly během používání předešlého klasifikačního systému:
- nízká interpersonální (i intrapersonální) reproducibilita diagnózy FCD I. typu (21)
- FCD I. typu je heterogenní skupinou onemocnění s rozdílnou prognózou i klinickým průběhem (17,18)
- FCD I. typu je často přítomna současně s jinou závažnou poruchou CNS (zejména s HS) a u těchto “asociovaných FCD” byly v porovnání s izolovanými lézemi prokázány signifikantní rozdíly v začátku záchvatů i jejich frekvenci (22). Na druhou stranu ale byly mezi těmito dvěma podskupinami FCD I. typu ukázány jen minimální rozdíly v elektrofyziologických nálezech a prognóze (6)
- izolovaná FCD (tj. bez další morfologické odchylky) je častěji MRI-negativní (16).
Panel expertů ILAE proto rozhodl, že kategorie FCD II. typu, která je dobře patologicky, klinicky i prognosticky charakterizovanou jednotkou, zůstane nezměněna. FCD I. typu ale bude rozdělena do dvou nových skupin:
- a. izolovaná FCD - nově FCD I. typu; definice podtypů A-C jsou ale změněny: ze hry vypadávají nezralé a obrovské neurony a nově se zřetel klade na přítomnost vertikální (radiální) nebo horizontální (tangenciální) poruchy laminárního uspořádání (obr. 2C)
- b. FCD asociovaná s další ložiskovou lézí CNS (nově FCD III. typu), která se dělí na podtypy A - D podle typu asociované léze.
Zda tento nový návrh kategorizace FCD přinese pouze pozitiva, ukáže až čas. Už krátce po představení nové klasifikace je ale jasné, že např. ponechání původního alfanumerického systému (avšak s jinou definicí stejně vyhlížejících kategorií) by mohlo být v budoucnu zdrojem řady odborných nedorozumění. Stejně tak není zřejmý důvod pro zavedení zvláštní kategorie pro FCD s horizontální poruchou laminace, neboť se jedná o velmi vzácný jev (zejména v izolované formě - nově FCD typu IB). Navíc, kouzlo nové kategorie “FCD asociovaných s jinou lézí”, tj. FCD typu III, kazí např. v případě asociace s HS skutečnost, že pokud pacientovi z různého důvodu zůstane hipokampus zachován (tj. HS není histologicky ověřena), zařadí se taková FCD zase do kategorie izolovaných FCD (tj. FCD I. typu) a nikoli do FCD typu IIIA, kam by měla patřit.
Patogeneze FCD
Ačkoli jsou dnes známy geny, jejichž mutace vedou ke vzniku rozsáhlých malformací korového vývoje mozku (např. lissencefalie/pachygyrie - geny LIS1, DCX, RELN; subkortikální a periventrikulární heterotopie - geny DCX a FLN1A; syndrom tuberózní sklerózy - TSC1, TSC2) (23), u nejvýznamnější malformace vedoucí k farmakorezistentní epilepsii - FCD - nebyl zatím molekulární podklad onemocnění odhalen. Pro podobnost s morfologií kortikálních tuberů u tuberózní sklerózy se ale předpokládá podobná patogeneze (24). Podle současného názoru vzniká FCD abnormální migrací, maturací a buněčnou smrtí neuronů během ontogeneze (25,26). Na druhé straně existují i teorie o rozvoji FCD v důsledku ložiskového perinatálního nebo časného postnatálního poškození mozku (23). Na jednom experimentálním modelu bylo ukázáno, že by balónovité buňky i dysmorfní neurony mohly vznikat z progenitorových buněk radiální glie ve ventrikulární zóně telencefala (27). FCD tak asi bude patogeneticky nejspíše heterogenním poruchou (23) a přesný mechanismus rozvoje těchto poruch teprve čeká na své rozkrytí.
Mechanismy epileptogeneze u FCD
Existuje několik studií, které se snažily analýzou resekované epileptické tkáně přinést odpověď na otázku, jak se FCD podílejí na vzniku a šíření epilepsie. Žádný konečný závěr však zatím nepadl (26). Většina hypotéz je založena na poruše rovnováhy mezi excitačními a inhibičními neuronálními okruhy v postižené kůře. K tomu přispívají poruchy perikaryálních synapsí (28) i změněná exprese glutamátových receptorů a membránových transportérů v oblastech FCD (26,29). Inhibiční okruhy v mozkové kůře představují zejména GABA-ergní interneurony, kterých je několik podskupin a dělí se podle toho, jaké exprimují kalcium-vážící proteiny. Našim výzkumem jsme prokázali, že v FCD je významně snížena hustota a změněná distribuce parvalbumin-pozitivních (30) i calretinin-pozitivních inhibičních interneuronů (31).
Nově se nám ale podařilo ukázat i nový směr, kterým by se FCD mohly podílet na vzniku epilepsie - prokázali jsme, že v FCD dochází k výrazným změnám geometrie mezibuněčného prostoru (32), což se odráží na změněných difúzních parametrech takto změněné kůry. Neurony mezi sebou nekomunikují jenom pomocí synapsí, ale také extrasynapticky difúzí uvolněných neuroaktivních látek v mezibuněčném prostoru. Tento typ přenosu informací v mozku se nazývá tzv. extrasynaptická neurotransmise, a je to proces, jehož význam byl prokázán u celé řady plastických procesů v CNS, včetně tvorby paměti, vzniku depresí a dalších poruch (33). Alterace difúze v mezibuněčném prostoru FCD by tedy mohla tuto cestu komunikace nervových buněk pozměnit a přispívat tak k jejich epileptogenicitě. Příčinou změn v objemu a geometrii mezibuněčného prostoru kůry postižené FCD se ukázala být depozita patologicky akumulované extracelulární matrix, jako třeba tenascinů R a C nebo versicanu, a zmnožení astrocytárních výběžků (34).
TUBERÓZNÍ SKLERÓZA
Tuberózní skleróza (TSC) je multisystémové onemocnění způsobené mutací jednoho ze dvou tumorsupresorových genů - TSC1 (9q34) nebo TSC2 (16p13.3), s variabilní fenotypovou expresí (35). Mozek bývá postižen nejčastěji, ale hamartogenní léze lze nalézt i v jiných orgánech (renální angiomyolipom, srdeční rhabdomyom atd.). Až u dvou třetin pacientů je onemocnění důsledkem spontánní mutace, jen u jedné třetiny lze vystopovat autozomálně dominantní přenos.
U pacientů s TSC lze v CNS nalézt tři typy lézí (obr. 3) - první dvě jsou statické (kortikální tubery a subkortikální heterotopní noduly). Třetí (subependymální noduly) mohou být pomalu progresivní a dávají pak vzniku subependymálnímu obrovskobuněčnému astrocytomu (SEGA).
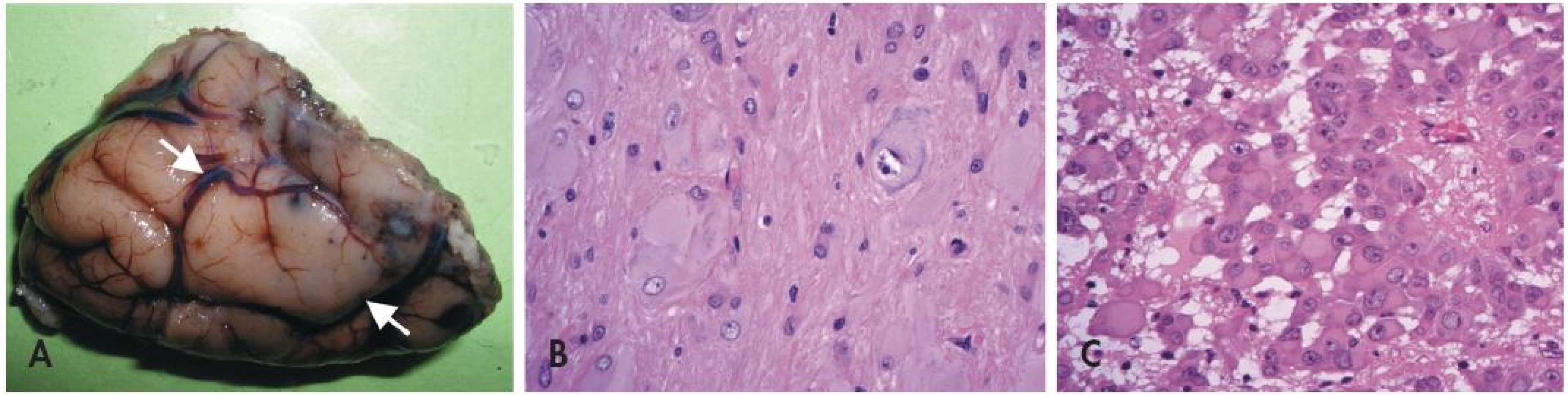
Neuropatologická diagnóza kortikálního tuberu není obtížná (36) - místo normální laminární struktury je v oblasti tuberů přítomno nakupení špatně orientovaných aberantních obrovských neuronů. Navíc bývají v kůře i přilehlé bílé hmotě přítomny obrovské bizarní buňky s obšírnou eozinofilní cytoplazmou. Typická je navíc výrazná subpiální piloidní glióza, které pomůže rozlišit tubery např. od konvenčních FCD nebo od změn při hemimegalencefalii.
FOKÁLNÍ EPILEPSIE ASOCIOVANÁ S NÁDORY
Nádory mozku asociované s farmakorezistentní epilepsií mají celou řady společných aspektů a v literatuře jsou řazeny společně do skupiny tzv. LEATs (“long-term epilepsy-associated tumors”). Jedná se o nádory s převážně temporální lokalizací (80 %) (37), obtížně vysvětlitelnou bifazickou glioneuronální diferencí, pomalým růstem a častou přítomností poruch kortikálního vývoje (FCD) v okolní kůře.
Do skupiny LEATs patří zejména gangliogliom a potom dysembryoplastický neuropiteliální tumor (DNET). Jen vzácně bývá farmakorezistentní epilepsie spojena s pilocytárními astrocytomy, pleomorfním xantoastrocytomem, SEGA nebo jinými typy mozkových nádorů. Naopak do této skupiny nepatří maligní mozkové nádory, jejichž první klinickou prezentací také může být epileptický paroxysmus.
WHO klasifikace přičítá nádorům ze skupiny LEATs grade I a existuje několik indicií, které dokonce zpochybňují jejich skutečně nádorový charakter. Recentní molekulárně genetické studie například ukázaly, že v této skupině nádorů jsou postiženy spíše vývojové signalizační dráhy (38,39) než geny, o kterých je známo, že se podílejí na onkogenezi gliomů (např. TP53, PTEN, EGFR).
Přestože se jedná o nádory povětšinou zcela indolentní, mohou být hypercelulární a jejich bifazičnost může být v mikroskopu (zejména ve zmraženém řezu) chybně interpretována jako atypie. To může vést k chybnému zařazení takových nádorů do skupiny high grade tumorů CNS.
(Nádorům ze skupiny LEATs je věnována rubrika “Jaká je vaše diagnóza” na straně 91 tohoto čísla, pozn. red.)
Mechanismy epileptogeneze u LEATs
V mechanismu rozvoje epilepsie u nádorů ze skupiny LEATs není zdaleka jasno. Recentními nálezy jsou podpořeny dva směry v uvažování.
Jednak by se na vzniku epilepsie mohla podílet neuronální komponenta vlastního nádoru - v imunohistochemických studiích byla v dysplastických neuronech gangliogliomů ukázána aberantní exprese různých molekul, zejména glutamatergních receptorů (40). Intracerebrální EEG navíc nedávno ukázalo iniciální epileptické výboje z vlastní nádorové tkáně gangliogliomu (41). Hyperexcitabilní nádorové neurony tohoto tumoru jsou tedy pravděpodobně zapojeny do excitačních neuronálních okruhů mozkové kůry.
Druhou možností je podíl okolní mozkové tkáně na rozvoji záchvatů - v peritumorózní mozkové tkáni byla prokázána patologická exprese neuroaktivních molekul (42). Řada klinických pozorování navíc tvrdí, že epileptogenní oblast je podstatně větší než vlastní nádorová masa. Většině pacientů totiž nepřinese ústup záchvatů prosté odstranění nádoru, ale je třeba resekovat i okolní nenádorovou mozkovou kůru.
ZÁVĚR
I přes dramatický rozvoj v neuroradiologii zůstává role patologa pro stanovení strukturálního podkladu epilepsie v resekované mozkové tkáni nezastupitelná. Přestože jsou dnes pacienti s farmakorezistentní epilepsií před operací vyšetřování kromě MRI i MR spektroskopií, podstupují vyšetření PET a SPECT, stále je určité procento pacientů, kteří k operaci dospějí jako případy tzv. “MRI-negativní” a až histopatologická analýza resekované tkáně prokáže strukturální podklad onemocnění. Přesná klasifikace a subtypizace onemocnění má navíc význam pro stanovení prognózy záchvatovitého onemocnění a s tím související následnou dispenzarizaci a volbu optimálního léčebného režimu po operaci. Navíc, někdy teprve histopatologické vyšetření prokáže, zda ložisko pozorované při předoperačním zobrazení je korovou malformací a nikoli nádorem.
Epileptopatologie je poměrně mladou oblastí bioptické diagnostiky, proto bude ještě nějaký čas trvat, než budou nalezena optimální klasifikační schémata jednotlivých poruch s dostatečným klinickým i prognostickým korelátem. Dostupnost lidské epileptické tkáně ale otevřela široký prostor pro výzkum těchto dříve poněkud přehlížených chorob. Poznání jejich patogeneze ale i mechanismu, kterým se tyto poruchy na vzniku a šíření epilepsie podílejí, by do budoucna mohlo nabídnout nové směry v jejich léčbě.
PODĚKOVÁNÍ
Podpora: IGA MZČR NS9915-4.
Adresa pro korespondenci:
Doc. MUDr. Josef Zámečník, Ph.D.
Ústav patologie a molekulární medicíny
UK 2. LF a FN v Motole
V Úvalu 84, 150 06 Praha 5
Tel: 224 435 635, Fax: 224 435 620
Email: josef.zamecnik@lfmotol.cuni.cz
Sources
1. Elger CE. Epilepsy: disease and model to study human brain function. Brain Pathol 2002; 12(2): 193-198.
2. Blümcke I. Neuropathology of focal epilepsies: a critical review. Epilepsy Behav 2009; 15(1): 34-39.
3. Blümcke I, Thom M, Wiestler OD. Ammon’s horn sclerosis: a maldevelopmental disorder associated with temporal lobe epilepsy. Brain Pathol 2002; 12(2): 199-211.
4. Zámečník J, Kršek P, Marusič P et al. Mikroskopické poruchy kortikálního vývoje mozku a etiopatogenetický význam jejich detekce u pacientů s temporální epilepsií při skleróze hipokampu. Cesk Patol 2003; 39(4): 178-184.
5. Thom M, Zhou J, Martinian L, Sisodiya S. Quantitative post-mortem study of the hippocampus in chronic epilepsy: seizures do not inevitably cause neuronal loss. Brain 2005; 128(Pt 6): 1344-1357.
6. Marusič P, Tomášek M, Kršek P et al. Clinical characteristics in patients with hippocampal sclerosis with or without cortical dysplasia. Epileptic Disord 2007; 9(Suppl 1): S75-S82.
7. Blümcke I, Suter B, Behle K et al. Loss of hilar mossy cells in Ammon’s horn sclerosis. Epilepsia 2000; 41 Suppl 6(S174-S180.
8. de Lanerolle NC, Kim JH, Williamson A et al. A retrospective analysis of hippocampal pathology in human temporal lobe epilepsy: evidence for distinctive patient subcategories. Epilepsia 2003; 44(5): 677-687.
9. Houser CR. Granule cell dispersion in the dentate gyrus of humans with temporal lobe epilepsy. Brain Res 1990; 535(2): 195-204.
10. Blümcke I, Kistner I, Clusmann H et al. Towards a clinico-pathological classification of granule cell dispersion in human mesial temporal lobe epilepsies. Acta Neuropathol 2009; 117(5): 535-544.
11. Sutula T, Cascino G, Cavazos J, Parada I, Ramirez L. Mossy fiber synaptic reorganization in the epileptic human temporal lobe. Ann Neurol 1989; 26(3): 321-330.
12. Becker AJ, Chen J, Zien A et al. Correlated stage - and subfield-associated hippocampal gene expression patterns in experimental and human temporal lobe epilepsy. Eur J Neurosci 2003; 18(10): 2792-2802.
13. Blümcke I, Pauli E, Clusmann H et al. A new clinico-pathological classification system for mesial temporal sclerosis. Acta Neuropathol 2007; 113(3): 235-244.
14. Barkovich AJ, Kuzniecky RI, Jackson GD, Guerrini R, Dobyns WB. A developmental and genetic classification for malformations of cortical development. Neurology 2005; 65(12): 1873-1887.
15. Palmini A, Najm I, Avanzini G et al. Terminology and classification of the cortical dysplasias. Neurology 2004; 62(Suppl 3): S2-S8.
16. Kršek P, Pieper T, Karlmeier A et al. Different presurgical characteristics and seizure outcomes in children with focal cortical dysplasia type I or II. Epilepsia 2009; 50(1): 125-137.
17. Blümcke I, Vinters HV, Armstrong D et al. Malformations of cortical development and epilepsies: neuropathological findings with emphasis on focal cortical dysplasia. Epileptic Disord 2009; 11(3): 181-193.
18. Lerner JT, Salamon N, Hauptman JS et al. Assessment and surgical outcomes for mild type I and severe type II cortical dysplasia: a critical review and the UCLA experience. Epilepsia 2009; 50(6): 1310-1335.
19. Blümcke I, Spreafico R. An international consensus classification for focal cortical dysplasias. Lancet Neurol 2011; 10(1): 26-27.
20. Blümcke I, Thom M, Aronica E et al. The clinicopathologic spectrum of focal cortical dysplasias: a consensus classification proposed by an ad hoc Task Force of the ILAE Diagnostic Methods Commission. Epilepsia 2011; 52(1): 158-174.
21. Chamberlain WA, Cohen ML, Gyure KA et al. Interobserver and intraobserver reproducibility in focal cortical dysplasia (malformations of cortical development). Epilepsia 2009; 50(12): 2593-2598.
22. Tassi L, Garbelli R, Colombo N et al. Type I focal cortical dysplasia: surgical outcome is related to histopathology. Epileptic Disord 2010; 12(3): 181-191.
23. Guerrini R, Parrini E. Neuronal migration disorders. Neurobiol Dis 2010; 38(2): 154-166.
24. Becker AJ, Urbach H, Scheffler B et al. Focal cortical dysplasia of Taylor’s balloon cell type: mutational analysis of the TSC1 gene indicates a pathogenic relationship to tuberous sclerosis. Ann Neurol 2002; 52(1): 29-37.
25. Ying Z, Gonzalez-Martinez J, Tilelli C, Bingaman W, Najm I. Expression of neural stem cell surface marker CD133 in balloon cells of human focal cortical dysplasia. Epilepsia 2005; 46(11): 1716-1723.
26. Najm IM, Tilelli CQ, Oghlakian R. Pathophysiological mechanisms of focal cortical dysplasia: a critical review of human tissue studies and animal models. Epilepsia 2007; 48 Suppl 2(21-32.
27. Lamparello P, Baybis M, Pollard J et al. Developmental lineage of cell types in cortical dysplasia with balloon cells. Brain 2007; 130(Pt 9): 2267-2276.
28. Aronica E, Gorter JA, Jansen GH et al. Expression and cell distribution of group I and group II metabotropic glutamate receptor subtypes in taylor-type focal cortical dysplasia. Epilepsia 2003; 44(6): 785-795.
29. Wong M. Mechanisms of epileptogenesis in tuberous sclerosis complex and related malformations of cortical development with abnormal glioneuronal proliferation. Epilepsia 2008; 49(1): 8-21.
30. Zámečník J, Kršek P, Druga R et al. Densities of parvalbumin-immunoreactive neurons in non-malformed hippocampal sclerosis-temporal neocortex and in cortical dysplasias. Brain Res Bull 2006; 68(6): 474-481.
31. Barinka F, Druga R, Marusič P, Kršek P, Zámečník J. Calretinin immunoreactivity in focal cortical dysplasias and in non-malformed epileptic cortex. Epilepsy Res 2010; 88(1): 76-86.
32. Vargová L, Homola A, Cicanic M et al. The diffusion parameters of the extracellular space are altered in focal cortical dysplasias. Neurosci Lett 2011; 499(1): 19-23.
33. Vizi ES, Kiss JP, Lendvai B. Nonsynaptic communication in the central nervous system. Neurochem Int 2004; 45(4): 443-451.
34. Zámečník J, Homola A, Cicanic M et al. Extracellular matrix and diffusion barriers in focal cortical dysplasias. Eur J Neurosci 2012, in press.
35. Jozwiak J, Jozwiak S, Wlodarski P. Possible mechanisms of disease development in tuberous sclerosis. Lancet Oncol 2008; 9(1): 73-79.
36. Mizuguchi M, Takashima S. Neuropathology of tuberous sclerosis. Brain Dev 2001; 23(7): 508-515.
37. Blümcke I, Wiestler OD. Gangliogliomas: an intriguing tumor entity associated with focal epilepsies. J Neuropathol Exp Neurol 2002; 61(7): 575-584.
38. Hoischen A, Ehrler M, Fassunke J et al. Comprehensive characterization of genomic aberrations in gangliogliomas by CGH, array-based CGH and interphase FISH. Brain Pathol 2008; 18(3): 326-337.
39. Fassunke J, Majores M, Tresch A et al. Array analysis of epilepsy-associated gangliogliomas reveals expression patterns related to aberrant development of neuronal precursors. Brain 2008; 131(Pt 11): 3034-3050.
40. Aronica E, Yankaya B, Jansen GH et al. Ionotropic and metabotropic glutamate receptor protein expression in glioneuronal tumours from patients with intractable epilepsy. Neuropathol Appl Neurobiol 2001; 27(3): 223-237.
41. Ferrier CH, Aronica E, Leijten FS et al. Electrocorticographic discharge patterns in glioneuronal tumors and focal cortical dysplasia. Epilepsia 2006; 47(9): 1477-1486.
42. Aronica E, Redeker S, Boer K et al. Inhibitory networks in epilepsy-associated gangliogliomas and in the perilesional epileptic cortex. Epilepsy Res 2007; 74(1): 33-44.
Labels
Anatomical pathology Forensic medical examiner ToxicologyArticle was published in
Czecho-Slovak Pathology

2012 Issue 2
Most read in this issue
- Neurodegenerative Disorders: Review of Current Classification and Diagnostic Neuropathological Criteria
- Neuropathology of refractory epilepsy: the structural basis and mechanisms of epileptogenesis
- Selected biomarkers in the primary tumors of the central nervous system: short review
- Peripheral neuropathy in Whipple’s disease: A case report