
Vybrané biomarkery primárnych nádorov centrálneho nervového systému: krátky prehľad
Selected biomarkers in the primary tumors of the central nervous system: short review
Classification, grading and treatment of central nervous system tumors is currently based on morphology. Advances in molecular biology help to clarify pathogenesis, refine prognosis and detect potential targets for targeted therapy in a wide spectrum of CNS tumors. In this short review we present our view on selected diagnostic, prognostic and predictive biomarkers of primary CNS tumors, with an emphasis on application in daily praxis.
Keywords:
brain tumors – biomarkers – genetics – immunohistochemistry
:
M. Švajdler ml. 1; B. Rychlý 2; L. Fröhlichová 1; P. Grossmann 3; A. Šteňo 4; F. Pataky 5
:
Oddelenie patológie UNLP Košice, Trieda SNP1
1; Cytopathos, spol. s. r. o., Bratislava
2; Bioptická laboratoř, s. r. o., Plzeň
3; Neurochirurgická klinika, Nemocnica akad. L. Dérera, Bratislava
4; Neurochirurgická klinika UNLP, Košice
5
:
Čes.-slov. Patol., 48, 2012, No. 2, p. 65-71
:
Review Articles – Neuropathology
Klasifikácia, grading a liečba primárnych nádorov centrálneho nervového systému je v súčasnosti založená predovšetkým na morfológii. Poznatky z oblasti molekulovej biológie pomáhajú v širokom spektre nádorov CNS objasniť patogenézu, upresňujú prognózu a detekujú potencionálne ciele pre cielenú terapiu. V krátkom prehľade prinášame pohľad na vybrané diagnostické, prognostické a prediktívne biomarkery primárnych nádorov CNS, s dôrazom na aplikáciu v bežnej praxi.
Kľúčové slová:
nádory mozgu – biomarkery – genetika – imunohistochémia
Diagnostika nádorov CNS je postavená predovšetkým na histomorfológii. Vo všeobecnosti platí, že nádory konkrétneho histologického typu a stupňa malignity sa liečia v zásade identicky. Morfologicky identické nádory však často reagujú na liečbu úplne odlišne. Čoraz častejšie sa v tejto často až frustrujúco beznádejnej oblasti onkológie do popredia dostávajú poznatky z oblasti molekulovej genetiky. Tie v niektorých prípadoch znamenajú prvé pokusy o individualizovanú terapiu. Niektoré z týchto biomarkerov (detekované geneticky alebo na úrovni proteínu imunohistochemicky) sú už súčasťou bežnej praxe. Iné sú zasa prísľubom do blízkej budúcnosti. V krátkom prehľade prinášame náš pohľad na najvýznamnejšie už používané, ako aj niektoré nové diagnostické, prognostické a prediktívne biomarkery primárnych nádorov CNS.
METYLÁCIA PROMÓTERA MGMT AKO PREDIKTÍVNY A PROGNOSTICKÝ MARKER GLIOBLASTÓMU
Gén MGMT je lokalizovaný na chromozóme 10q26. Kóduje O6-metylguanín-DNA metyltransferázu, proteín ktorý zabezpečuje opravy DNA poškodenej alkyláciou guanínových zvyškov na pozícii O6. Takáto alkylácia spôsobuje nesprávne párovanie a zlomy v DNA, čo vedie k ireverzibilnému poškodeniu bunky a apoptóze. Alkylácia DNA je jedným z hlavných mechanizmov účinku alkylačných chemoterapeutík (napr. karmustín, temozolomid) používaných v liečbe glioblastómu (GBM). Vysoká hladina MGMT v nádorových bunkách efektívne eliminuje alkyláciu DNA spôsobenú chemoterapiou a zapríčiňuje tak rezistenciu na liečbu. Predpokladá sa, že metylácia promótera génu MGMT (a tým jeho inaktivácia) znižuje expresiu MGMT a tým aj reparačné schopnosti nádorovej bunky. Bolo dokázané, že pacienti s GBM, ktorí boli liečení kombinovanou rádioterapiou a alkylačnou chemoterapiou a mali metylovaný promóter MGMT, mali signifikantne lepšie prežívanie ako pacienti s nemetylovaným promóterom (1,2). Signifikantný rozdiel v prežívaní však bol zistený aj medzi skupinami pacientov s metylovaným a nemetylovaným promóterom, ktorí nedostali chemoterapiu a boli liečení iba rádioterapiou (2). Je teda otázne, či metylácia promótera MGMT je skutočne prediktívnym markerom pre alkylačnú chemoterapiu, alebo iba charakterizuje skupinu GBM s inherentne lepšou prognózou (napríklad pre možné širšie epigenetické zmeny spôsobené metyláciou DNA, asociáciu s inými prognosticky pozitívnymi genetickými zmenami alebo klinickými charakteristikami). Metylácia promótera MGMT bola napríklad identifikovaná až u 74 % (28 z 36) dlho prežívajúcich pacientov s GBM (> 3 roky). Títo pacienti však boli v lepšom výkonnostnom stave a signifikantne mladší (priemerne 51 rokov) ako ,,bežní” pacienti s GBM (> 60 rokov), čo sú dva klinicky najvýznamnejšie prognostické parametre GBM (3).
Aj keď sa zdá, že stav promótora MGMT je jedným z najvýznamnejších prognostických a pravdepodobne aj prediktívnych faktorov, jeho praktické využitie v manažmente konkrétneho pacienta s GBM je v súčasnosti limitované z viacerých príčin. Predovšetkým, neexistuje lepšia alternatíva ako je ,,zlatý štandard” resekcia + rádioterapia + temozolomid (4) a benefit tejto terapie možno pozorovať aj u malého percenta pacientov bez metylácie promótera MGMT. Výsledok vyšetrenia teda v súčasnosti výraznejšie neovplyvní rozhodovanie onkológa o iniciálnej liečbe GBM. Ďalším dôvodom je technická náročnosť (použiteľné sú viaceré techniky molekulovej genetiky, imunohistochémia je nevhodná), vysoká cena, nízka reprodukovateľnosť a chýbanie štandardov vyšetrovania a hodnotenia výsledkov. Testovanie MGMT sa však stalo nevyhnutným štandardom pri skúšaní nových liekov v klinických štúdiách (napr. CENTRIC) (5). Po odhalení ďalších mechanizmov citlivosti a rezistencie v GBM a nových možnostiach liečby (napr. cielená liečba) sa testovanie MGMT v budúcnosti pravdepodobne stane súčasťou rutínnej diagnostiky a klasifikácie GBM.
MUTÁCIA IZOCITRÁT DEHYDROGENÁZY 1 V GLIÓMOCH – PROGNOSTICKÝ A DIAGNOSTICKÝ MARKER
V roku 2008 Parsons a spol. sekvenovali 20661 proteín kódujúcich génov v 22 GBM a ako prví zachytili mutáciu izocitrát dehydrogenázy 1 (IDH1). Išlo prevažne o mladších pacientov so sekundárnym GBM (vznikajúci z gliómu nižšieho stupňa malignity). Pacienti s mutáciou IDH1 vykazovali významne dlhšie prežívanie ako pacienti bez mutácie (6). Následne sa ukázalo, že IDH1 mutácia je častá v gliómoch II. a III. stupňa malignity (astrocytóm, oligoastrocytóm a oligodendroglióm), pravdepodobne vzniká v skorom štádiu tumorigenézy a je silným priaznivým prognostickým markerom v gliómoch vysokého stupňa malignity (7).
IDH gény kódujú tri IDH izoenzýmy (IDH1, IDH2 a IDH3). Výrazne najčastejšia je mutácia IDH1 (iba 4 % mutácií postihujú IDH2 a mutácie IDH3 neboli zachytené). Normálnou funkciou IDH je katalýza oxidatívnej dekarboxylácie izocitrátu na alfa-ketoglutarát, čo vedie k redukcii NADP+ na NADPH. Mutovaná forma IDH vykazuje pokles normálnej katalytickej aktivity, zároveň ale získava schopnosť konverzie alfaketoglutarátu na 2-hydroxyglutarát (8). Mutácia IDH1 teda indukuje stratu a zároveň aj nadobudnutie funkcie, čo je paradoxné z hľadiska otázky, či je IDH1 onkogén alebo tumor supresor. Presný mechanizmus, ktorým vedie mutácia IDH1 ku vzniku gliómov, nebol zatiaľ objasnený. Fakt, že IDH mutácie boli identifikované v astrocytómoch aj v oligodendrogliómoch, poukazuje na ich možnú spoločnú prekurzorovú bunku.
Ako už bolo spomenuté, mutácia IDH1 je silným prognostickým faktorom v high grade gliómoch. Pacienti s anaplastickým astrocytómom (grade III) s nemutovaným IDH1 majú horšiu prognózu ako pacienti s IDH1 mutovaným glioblastómom (grade IV). IDH1 status je teda v high grade gliómoch silnejším prognostickým faktorom ako štandardné histologické kritériá (9). Pravdepodobne bude tiež priaznivým prognostickým faktorom aj v gliómoch II. stupňa malignity, i keď v tejto oblasti sú údaje zatiaľ kontroverzné (10).
Až na malé výnimky, ako je napr. myeloidná leukémia (11), sa IDH mutácie vyskytujú iba v grade II a III astrocytómoch, oligoastrocytómoch a oligodendrogliómoch a v sekundárnych glioblastómoch. Nevyskytujú sa v iných mozgových tumoroch, ktoré difúzne astrocytómy a oligodendrogliómy môžu napodobovať (napr. pilocytický astrocytóm, pleomorfný xantoastrocytóm, subependymálny obrovskobunkový astrocytóm, ependymóm, neurocytóm, dysembryoplastický neuroepitelový tumor, atď.) ani v nenádorových ochoreniach (reaktívna glióza, postradiačné zmeny, vírusové infekcie, infarkt, demyelinizácie) (12). To robí IDH1 aj vhodným diagnostickým markerom. K dispozícii je komerčná monoklonová protilátka voči mutantnej forme IDH1 s vynikajúcou koreláciou medzi imunohistochémiou a genetickým vyšetrením stavu IDH1 génu (13). Imunohistochemické znázornenie mutovaného IDH1 proteínu má navyše oproti genetike výhodu priamej vizualizácie nádorových gliových buniek, a to i v teréne prevažujúcej nenádorovej glie (obr. 1). To môže byť mimoriadne diagnosticky cenné v limitovaných stereotaktických biopsiách zachytávajúcich často iba infiltrujúci okraj gliómu, v ktorom môže byť denzita nádorových buniek nízka. Samozrejme, diagnózu nie je možné stanoviť len na základe pozitivity IDH1, a nález je potrebné hodnotiť v adekvátnom klinicko-patologickom kontexte. Navyše, dostupná protilátka detekuje iba najčastejšiu mutáciu IDH1 (R132H), ktorá sa vyskytuje v približne 93 %, pričom nedetekuje minoritne zastúpené mutácie IDH1 ani vzácne sa vyskytujúce mutácie IDH2 (14).
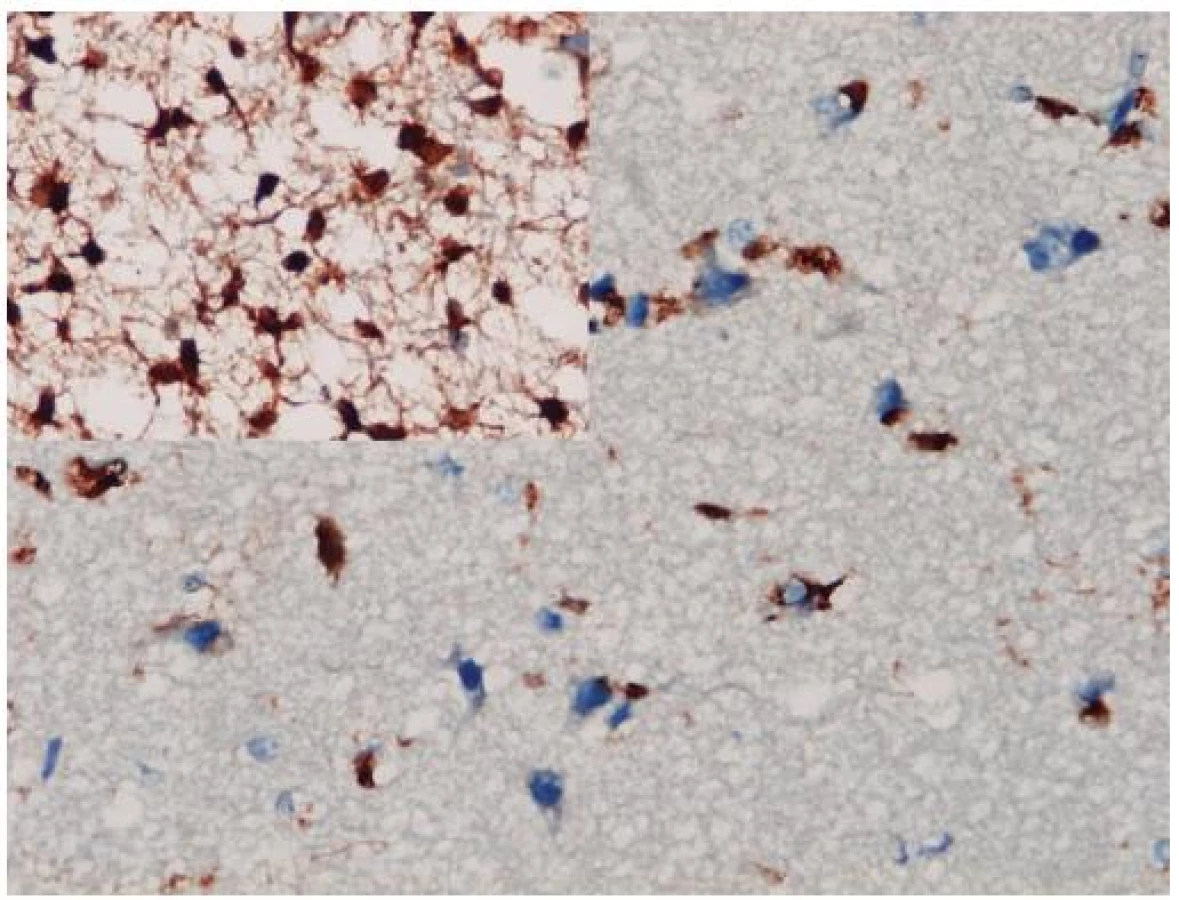
Stanovenie statusu IDH1sa v súčasnosti stáva zlatým štandardom diagnostiky gliómov a vedú sa diskusie o jeho začlenení do budúcej WHO klasifikácie mozgových nádorov. Algoritmus na pracovisku dizajnérov protilátky IDH1 (R132H) pozostáva z iniciálnej imunohistochémie a pri negativite pokračujú sekvenáciou DNA (14).
ALTERÁCIE CESTY MAPK/ERK A PI3K/AKT V LOW GRADE GLIÓMOCH
Mutácia BRAF (kináza cesty MAPK/ERK) sa na rozdiel od iných nádorov (napr. papilárny karcinóm štítnej žľazy, melanóm, kolorektálny karcinóm) v primárnych nádoroch CNS vyskytuje pomerne vzácne. Dostupnosť inhibítorov tejto cesty, ktoré by sa onedlho mohli v širšej miere dostať do klinickej praxe, však podnecuje záujem o detekciu mutovanej formy BRAF aj v primárnych nádoroch mozgu. Schindlerová a spol. (15) v analýze stavu génu BRAF v 1320 primárnych nádoroch mozgu, adenohypofýzy, meningov a periférnych nervov zistili, že až 65 % pleomorfných xantoastrocytómov (PXA, grade II) a 65 % PXA s anapláziou (a-PXA) malo mutovaný BRAF. Mutované PXA boli signifikantne asociované s detským vekom. V skupine a-PXA boli dokonca mutované všetky detské (10/10) a 38 % adultných tumorov. Druhou najpočetnejšou skupinou nádorov s BRAF mutáciou (18 %, v niektorých prácach až 50 %) je ganglioglióm (GG). To nie je až také prekvapujúce, pretože na morfologickej ako aj genetickej úrovni (častá delécia CDKN2A v PXA aj GG) sa tieto lézie často prekrývajú (15). Častá mutácia BRAF v týchto nádoroch môže byť cenným diagnostickým markerom. Detekcia mutácie v pleomorfnom glióme (najmä s neobvykle priaznivým klinickým priebehom) by mala upriamiť pozornosť na možnosť diagnózy PXA/a-PXA alebo GG. Treťou významnou BRAF mutovanou skupinou (9 %) sú pilocytické astrocytómy (PA). Dôležité je zistenie, že mutované PA sa signifikantne častejšie vyskytujú v extracerebelárnej, chirurgicky často nedostupnej lokalizácii (diencephalon), kde by možnosť cielenej liečby predstavovala lákavú alternatívu (15). Podobne ako v prípade protilátky proti mutovanej IDH1, sa nedávno tým istým autorom podarilo vyklonovať protilátku (zatiaľ komerčne nedostupnú) proti mutovanému BRAF V600E (97 % mutácií BRAF je V600E). Protilátka pri porovnaní s genetickou analýzou mala 100% senzitivitu a špecificitu (16).
Omnoho častejšie ako mutácia BRAF sa v PA vyskytuje translokácia BRAF génu, ktorá rovnako spôsobuje permanentnú aktiváciu MAPK/ERK cesty. Najčastejšie (70 %) ide o tandemovú duplikáciu na lókuse 7q34 vedúcej k fúzii génov KIAA1549 a BRAF. Spoľahlivá detekcia tejto tandemovej duplikácie je možná aj pomocou, pre patológov najprijateľnejšou, fluorescenčnou in situ hybridizáciou (FISH), na ktorú je už dostupný aj komerčný kit (17,18). V literatúre postupne pribúdajú popisy alternatívnych translokácií aktivujúcich túto cestu (napr. fúzia RAF1-SRGAP3 alebo BRAF-FAM131B) (19,20). Možno skonštatovať, že prakticky všetky PA, majú aktivovanú cestu MAPK/ERK: okrem alterácií BRAF sú takmer vždy detekované iné abnormalityv ceste a mutácia NF1 v PA asociovaných s neurofibromatózou tiež spôsobuje aktiváciu MAPK/ERK cesty. Z praktického hľadiska sú tieto poznatky významné najmä z pohľadu možnosti využitia cielenej liečby ako aj v bežnej diagnostike pri odlišovaní PA a difúzneho astrocytómu, a to najmä v kombinácii s analýzou IDH1/2 (21).
V klinicky agresívnych rekurentných PA a najmä histologicky anaplastických PA (definované ako hypercelulárne, cytologicky atypické, mitoticky aktívne PA s nekrózami alebo bez nekróz) sú v porovnaní s klinicky typickými PA signifikantne častejčie prítomné alterácie v ceste PI3K/AKT. V týchto nádoroch sa pomerne často vyskytuje delécia a zníženie expresie tumor supresora PTEN a imunohistochemicky je detekovateľná zvýšená expresia fosforylovanej formy AKT (pAKT). Dôležitým zistením je, že táto overexpresia je prítomná aj v prekurzorových tumoroch predchádzajúcich recidíve alebo zmene na histologicky anaplastický PA (22). Limitáciou tejto štúdie sú pomerne nízke počty vyšetrených prípadov. Detekcia aktivácie cesty PI3K/AKT (napr. aj imunohistochemickým dôkazom pAKT) by však v budúcnosti mohla slúžiť na odhalenie potenciálne agresívnych PA s možnosťou využitia cielenej liečby. Tu je však potrebné upozorniť, že imunohistochemická detekcia fosforylovaných proteínov je technicky náročnejšia a vyžaduje optimálnu fixáciu a spracovanie materiálu.
PNET-LIKE DIFERENCIÁCIA V GLIOBLASTÓME
Glioblastóm a CNS PNET (supratentoriálny PNET) sú na základe epidemiológie, morfológie, biologického správania sa a rozdielov v genetike považované za samostatné jednotky, s výrazne odlišným manažmentom. Sporadicky sú však publikované prípady malígnych nádorov, ktoré majú prekrývajúce sa morfologické a/alebo imunohistochemické charakteristiky. Tieto nádory sú diagnosticky náročné a sú často klasifikované ako nezvyčajné varianty GBM, CNS PNET s výraznou gliálnou diferenciáciou alebo ako malígne glioneuronálne nádory (23). Klinicko-patologické a genetické charakteristiky týchto nádorov, najmä výrazne častejšie metastázovanie cestou likvoru a benefit liečby určenej pre CNS PNET (zatiaľ iba v malom počte publikovaných prípadov) však nasvedčujú tomu, že by sa mohlo jednať o špecifickú kategóriu nádorov CNS (zatiaľ bez WHO klasifikácie), po ktorej sa v rutínnych biopsiách oplatí pátrať (24). Gliálna zložka v týchto nádoroch najčastejšie pripomína GBM alebo gliosarkóm, vzácne anaplastický oligodendroglióm. PNET-like zložka je prítomná ako ohraničené hypercelulárne noduly pripomínajúce CNS PNET alebo meduloblastóm (vrátane dezmoplastického / nodulárneho alebo anaplastického variantu) (obr. 2). Výraznou pomôckou v diagnostike je imunohistochémia a genetické (FISH) vyšetrenie. Expresia neuronálnych markerov (synaptofyzín, neurofilament protein, Neu-N) je limitovaná na PNET-like fókusy. Relatívne častá amplifikácia MYC génu (väčšinou MYCN, menej často MYC), ktorá je dosť typická pre primitívne neuronálne nádory je taktiež lokalizovaná prevažne v tejto zložke. Naproti tomu, zmeny asociované s gliómami (delécia 10q, delécia PTEN, amplifikácia EGFR, difúzna imunohistochemická pozitivita p53) sú lokalizované v oboch zložkách, čo svedčí najskôr pre sekundárny vznik PNET-like zložky dediferenciáciou v high grade glióme (24).
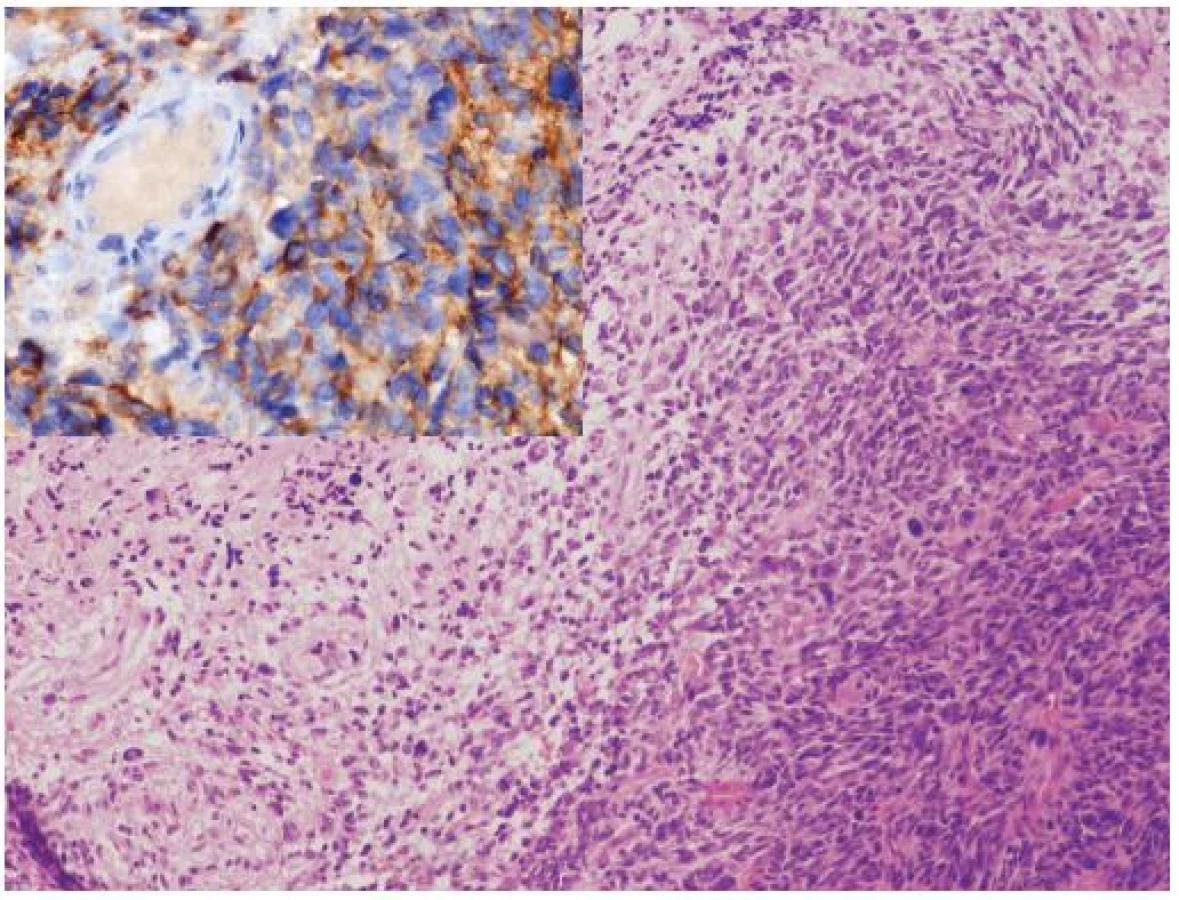
OLIGODENDROGLIÓMY A ALELICKÁ STRATA 1p/19q
Zvýšený záujem o genetiku oligodendrogliómov (ODG) bol zaznamenaný, keď sa ukázalo, že tumory s alelickou stratou na krátkom ramienku 1. chromozómu a dlhom ramienku 19. chromozómu (1p/19q LOH) vykazujú senzitivitu na chemo - a rádioterapiu (25). 1p/19q LOH je prediktívny aj prognostický marker, ktorý silno koreluje s klasickou ODG histomorfológiou (26). Nie všetky ODG s klasickou histomorfológiou však vykazujú túto genetickú zmenu. Klasická morfológia je pravdepodobne nezávislým prognostickým znakom. ODG potom môžeme rozdeliť do štyroch odlišných prognostických skupín: ODG s klasickou morfológiou a 1p/19q LOH, ODG s klasickou morfológiou bez 1p/19q LOH, ODG s neklasickou morfológiou s 1p/19q LOH a ODG s neklasickou morfológiou bez 1p/19q LOH. Posledná skupina má najhoršiu prognózu (27). Zaujímavé je, že pediatrické ODG majú zriedkavo 1p/19q LOH a tie, ktoré túto prestavbu majú, nevykazujú lepšiu prognózu (28).
Vzhľadom na to, že 1p/19q LOH je pomerne špecifická prestavba pre ODG, je možné použiť ju ako diagnostický marker a doplnok k histologickej diagnóze. Nedostatočná reproducibilita histologických diagnostických kritérií pre odlíšenie oligodendrogliových, oligoastrocytových a astrocytových tumorov viedla k snahe využiť viac reprodukovateľné a objektívnejšie genetické znaky. Takto by sa morfologicky nejasné difúzne gliómy mohli klasifikovať skôr podľa statusu 1p/19q a nie na základe subjektívneho štýlu, či diagnostických preferencií patológa. Tento prístup by mohol viesť k eliminácii nereprodukovateľnej diagnostickej kategórie oligoastrocytómu.
Dôležitou diferenciálnou diagnózou ODG je malobunkový variant anaplastického astrocytómu a glioblastómu. Malobunkový glioblastóm nemá 1p/19q LOH, ale takmer vždy vykazuje stratu 10q a amplifikáciu EGFR (29). Odlíšenie ODG od dysembryoplastického neuroepitelového tumoru (DNET) je tiež dôležité (podstatne lepšia prognóza DNET, ktorý je kurabilný resekciou, ODG je vo väčšine prípadov nakoniec fatálny a manažment sa zásadne líši). DNET môže mať časti histologicky neodlíšiteľné od ODG, čo môže byť problematické v limitovanej biopsii, DNET však nikdy nevykazuje 1p/19q LOH (30). 1p/19q LOH môže byť tiež pomôckou na odlíšenie od svetlobunkového ependymómu a centrálneho / extraventrikulového neurocytómu. Vzhľadom na (podľa niektorých údajov) častú expresiu neuronálnych markerov v ODG, pokladáme existenciu 1p/19q LOH pozitívneho neurocytómu prinajmenšom za spornú (31). Možno sa však jedná o histogeneticky príbuzné tumory, ktoré vytvárajú biologické kontinuum.
Nie je úplne jasné, či 1p/19q LOH predstavuje diagnostický znak pre ODG, alebo skôr ide o marker terapeutickej senzitivity širšej skupiny nádorov. Status 1p a 19q chromozómov je možné vyšetrovať rozličnými metodikami, pre rutínnu prax je v súčasnosti preferovaná FISH (obr. 3). I keď ešte neboli zodpovedané všetky otázky spojené s genetikou ODG, vyšetrenie statusu 1p/19q je v súčasnosti rutínnou súčasťou diagnózy gliómov ktoré sa viac alebo menej podobajú na ODG.
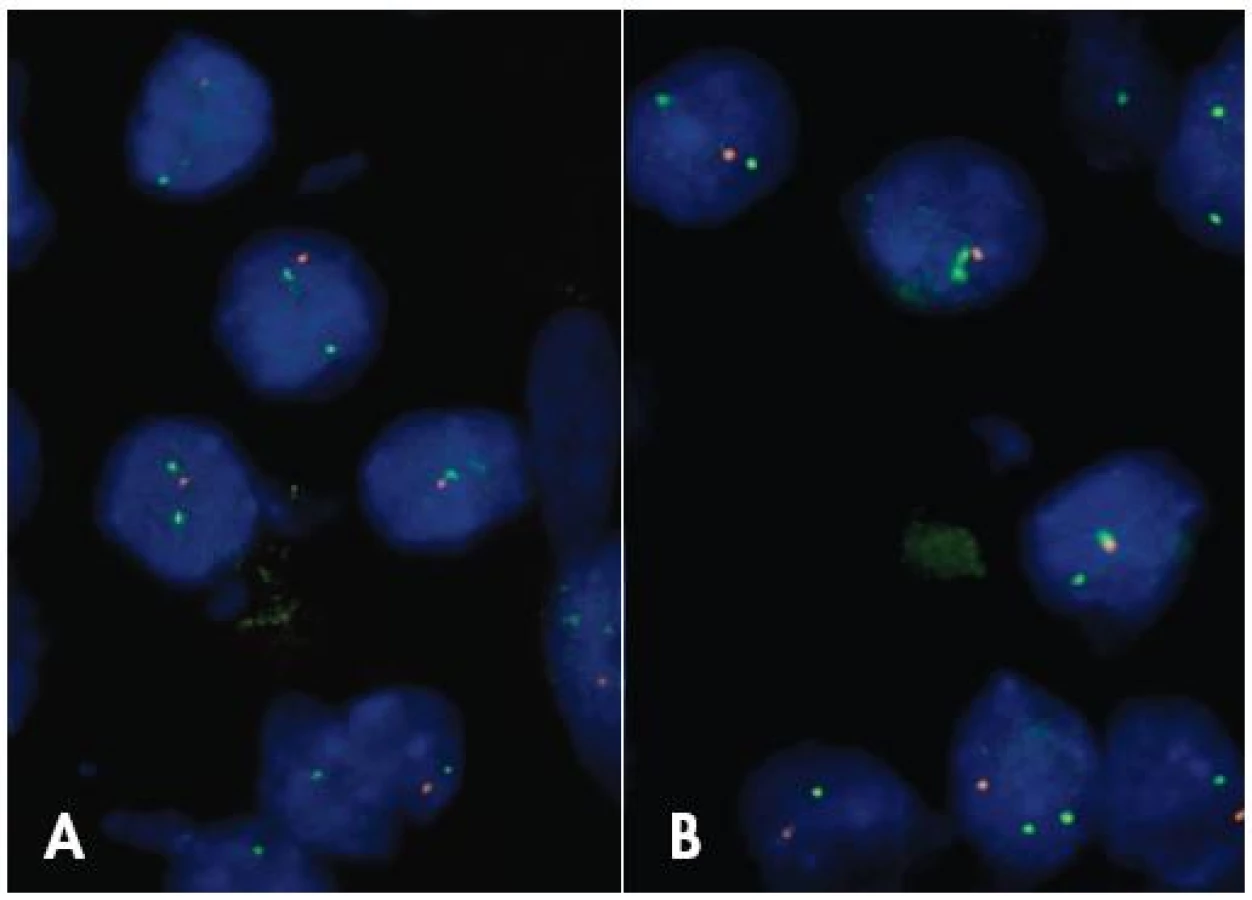
INI1 V DIAGNOSTIKE ATYPICKÉHO TERATOIDNÉHO / RABDOIDNÉHO TUMORU (AT/RT)
AT/RT je agresívny embryonálny tumor so zlou prognózou, ktorý je asociovaný so stratou funkcie INI1 génu a chýbaním expresie INI1 proteínu (32). INI1 (Integráza Interaktor 1) označovaný aj ako hSNF5, SMARCB1 alebo BAF47, je tumor supresorový gén lokalizovaný na chromozóme 22q11.2. Patrí do skupiny chromatín remodelujúceho komplexu a jeho proteínový produkt je exprimovaný v jadrách nenádorových buniek a vo väčšine ostatných malignít. Charakteristickým histologickým nálezom v AT/RT sú rabdoidné bunky s excentrickým jadrom a globulárnou inklúziou. Môže obsahovať mezenchýmové či epitelové elementy (preto názov teratoidný). Veľmi často sú tiež prítomné nediferencované malé modré bunky, pripomínajúce iné CNS PNET-y. Tieto nediferencované bunky môžu prevládať a rabdoidné bunky nemusia byť vôbec zachytené. To komplikuje diagnostiku AT/RT a tento tumor bol v minulosti poddiagnostikovaný. Odlíšenie od iných typov PNETov je ale dôležité, pretože AT/RT vyžaduje inú liečbu. Aj keď existujú iné INI1 deficientné tumory (epiteloidný sarkóm, renálny medulárny karcinóm, epiteloidný MPNST, myoepitelový karcinóm) (33), v mozgu je strata expresie INI1 relatívne špecifická pre AT/RT. Aj keď diagnóza na základe negativity expresie sa môže zdať problematickou, neistotu znižuje paušálna prítomnosť vnútornej pozitívnej kontroly, ktorou sú cievy a lymfocyty (obr. 4). Zaujímavý je vzťah AT/RT a karcinómu plexus choroideus, kde bola v niektorých prípadoch tiež pozorovaná strata expresie INI1 (34). Klinicko-patologické aspekty AT/RT a karcinómov plexu sa však natoľko prekrývajú, že mnohí autori považujú INI1 negatívne karcinómy plexus choroideus za zle diagnostikované AR/RT. Expresia proteínu Kir7.1 (považovaná doposiaľ za vysoko senzitívnu a špecifickú pre tumory plexu), bola pozorovaná aj v niektorých inak typických AT/RT (35) a straty na chromozóme 22q (nesie aj INI1 gén) boli zachytené vo väčšine karcinómov a takmer polovici papilómov plexus choroideus (36). Preto je možné, že AT/RT a karcinómy plexus chorodeus (alebo aspoň ich časť), sú histogeneticky príbuzné tumory.
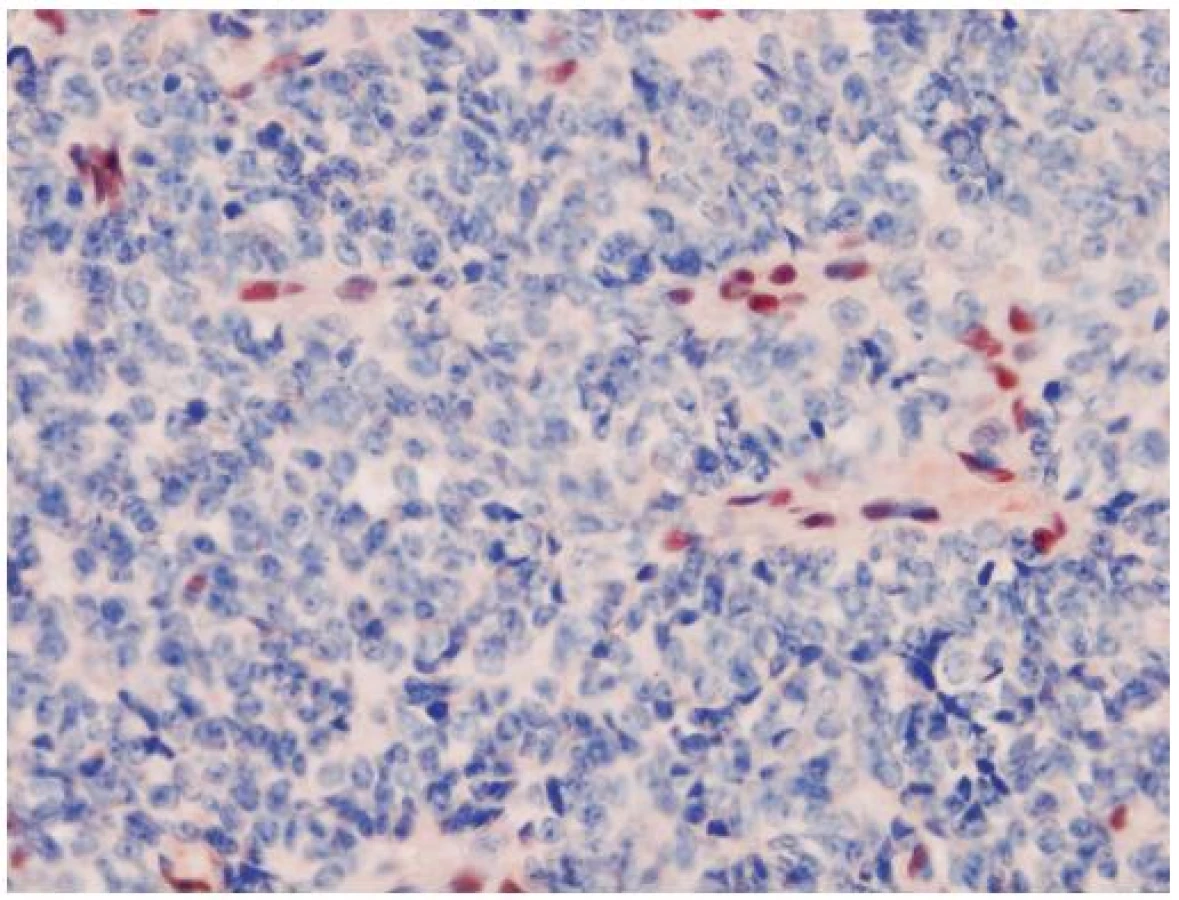
Zriedkavo sa môže vyskytnúť zárodočná mutácia INI1 génu, ktorá je podkladom syndrómu predispozície pre rabdoidné tumory s tvorbou viacpočetných mozgových, obličkových aj extrarenálnych rabdoidných tumorov (32).
V praxi by sa každý embryonálny tumor u detí (a pravdepodobne aj dospelých) mal testovať na stav INI1 génu (imunohistochémia má senzitivitu prakticky 100 %, genetickým vyšetrením sa mutácia/strata dokáže iba v približne 75–80 %). Tumory so stratou expresie INI1 by sa následne mali liečiť ako AT/RT, a to aj v prípade, že histologicky žiadne rabdoidné črty nevykazujú. Výnimkou sú snáď dezmoplastické/nodulárne meduloblastómy a meduloblastómy s extenzívnou nodularitou, kde INI1 nemá zmysel paušálne vyšetrovať, keďže strata INI1 v týchto tumoroch nebola nikdy pozorovaná. Pozor si treba ale dať v limitovaných biopsiách, pretože niektoré AT/RT môžu fokálne dezmoplastický/nodulárny meduloblastóm napodobovať.
MORFOLOGICKO–GENETICKÁ KLASIFIKÁCIA MEDULOBLASTÓMU
Liečba meduloblastómu (MB) sa v súčasnosti riadi predovšetkým klinickými charakteristikami pacienta (vek, prítomnosť metastáz pri prezentácii, resekabilita tumoru). Meduloblastóm (MB) je však heterogénna skupina nádorov s prognosticky významnými morfologickými variantami. Dezmoplastický/nodulárny MB (dnMB) a MB s extenzívnou nodularitou (MBen) majú vynikajúcu prognózu, klasický MB (cMB) stredne dobrú a veľkobunkový (lcMB) a anaplastický (aMB) zlú prognózu (37). Významným doplnkom ku klinicko-patologickým parametrom s výslednou lepšou stratifikáciou pacientov by mohli byť molekulárne markery. Kombinovaná klasifikácia by mala identifikovať pacientov s dobrou prognózou, u ktorých by bolo možné použiť menej agresívnu terapiu s menším výskytom vedľajších účinkov (najmä kognitívny deficit po rádioterapii) a na druhej strane by mala identifikovať pacientov u ktorých je súčasná liečba aj napriek svojej agresivite len málo účinná a mali by byť skôr liečení niektorou novou formou liečby. Aj keď v literatúre existuje veľké množstvo potencionálnych markerov, len veľmi málo z nich bolo (retrospektívne) overených na reprezentatívnych súboroch pacientov.
Výskyt MB v rámci Gorlinovho syndrómu (mutácia génu PTCH1) a Turcotovho syndrómu typu 2 (mutácia génu APC) upozornili na abnormality v ceste Sonic hedgehog (Shh) a ceste Wingless (Wnt). Okrem mutácií PTCH1 a APC boli v MB zatiaľ dokázané aj mutácie Shh, proteínu Smoothened a SUFU (v ceste Shh) a mutácie CTNNB1 a AXIN1/2 (v ceste Wnt). Tretia (heterogénna) skupina MB nemá up-reguláciu týchto signálnych ciest (non Shh/Wnt) a je charakterizovaná skôr chromozomálnymi abnormalitami – predovšetkým aberáciami chromozómu 17 a amplifikáciami MYC alebo MYCN (37–39).
Wnt podskupina MB (15 %) má takmer vždy klasickú morfológiu, iba vzácne lcMB/aMB a nikdy nemá dnMB morfológiu. Má výbornú prognózu a títo low risk pacienti (pri chýbaní ďalších rizikových faktorov – lc/aMB morfológia, metastáza, amplifikácia MYC – viď nižšie) by mohli byť liečení menej intenzívne. Detekcia tejto skupiny je jednoduchá, keďže cesta Wnt vedie k imunohistochemicky detekovateľnej nukleárnej akumulácii ß-catenínu. S Wnt MB a dobrou prognózou je asociovaná aj monozómia chromozómu 6 (37–39).
Do Shh podskupiny MB (25–30 %) patria prakticky všetky dnMB a MBen, ktoré majú výbornú prognózu. Približne 50 % však tvoria cMB a lcMB/aMB (37). Detetekcia aktivácie cesty Shh môže byť pre týchto pacientov kritická, keďže v súčasnosti sa už skúšajú inhibítory Shh (40). Detekcia Shh MB je tiež spoľahlivo možná pomocou imunohistochémie (cytoplazmatická expresia GAB1) (39).
Spomedzi chromozomálnych aberácií je vo väčšine štúdií uznávaným nezávislým high-risk markerom amplifikácia MYC (prevažuje MYC nad MYCN, sú vzájomne exkluzívne), ktorá je navyše často asociovaná s metastatickým ochorením a lcMB/aMB morfológiou. Amplifikácia MYC sa vyskytuje ako tzv. double minutes alebo homogenously staining region a je detekovateľná komerčne dostupnými FISH kitmi. V skupine non Shh/Wnt MB, ktoré sú MYC aplifikované sa morfologicky väčšinou jedná o cMB, v skupine Shh MB sú to dnMB aj lcMB/aMB (37–39). Prognostický význam ostatných aberácií (napr. aberácie chromozómov 17, 6, 10, 1) nie je zatiaľ celkom jednoznačný. Údaje v literatúre sú často úplne protichodné (37–39,41,42). V analýze siedmich publikovaných štúdií s dostatočnými údajmi o chromozomálnych abnormalitách sa ako jediný signifikantný faktor korelujúci so zlou prognózou ukázala izolovaná strata 17p. Izolovaný zisk 17q mal naopak tendenciu k asociácii s dobrou prognózou (42). Možná morfologicko-genetická klasifikácia MB a potencionálna stratifikácia pre nové klinické štúdie je zhrnutá v Tabuľke 1.
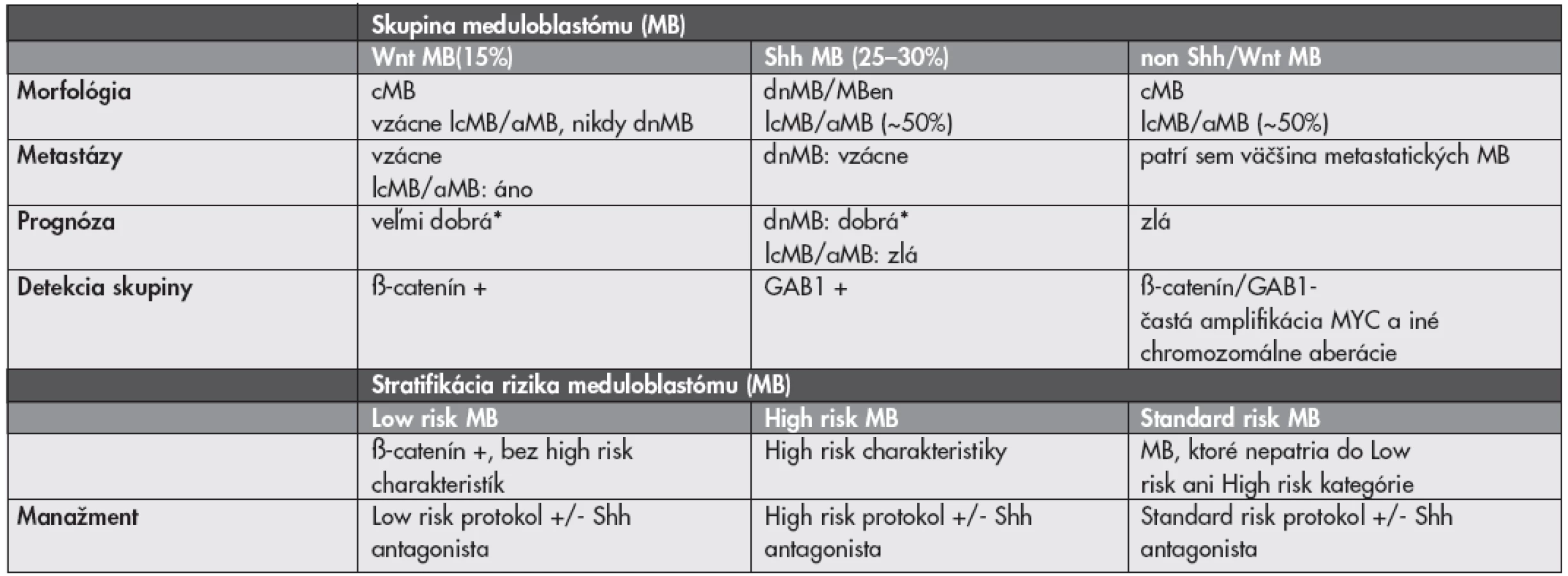
Všetky vyššie uvedené fakty platia pre detských pacientov. Zastúpenie a význam genetických aberácií v MB u dospelých sú odlišné. Napríklad, amplifikácie MYC sú pomerne vzácne a WNT aktivované MB majú signifikantne hošiu prognózu ako u detí. Klasifikácia a stratifikácia MB dospelých pacientov (aj vzhľadom na ich relatívnu vzácnosť) preto predstavuje ešte väčšiu výzvu (43,44).
BIOMARKERY NÁDOROV CNS – PERSPEKTÍVY
Pokroky v metodikách molekulovej genetiky a proteonomiky priniesli možnosť vyšetrovať v nádorovom tkanive naraz stovky až tisíce génov a proteínov. To umožňuje bioštatistikom identifikáciu molekulových tried (molekulová taxonómia) alebo predpovedať správanie nádoru a odpoveď na určitý typ liečby. Najznámejšie sú príklady z oblasti karcinómu mliečnej žľazy (klasifikácia na luminálny, bazálny, HER2+ a ďalšie typy a OncoTypeDX, MammaPrint a iné multigénne prediktory). Aj keď praktický význam týchto techník je stále prinajmenšom sporný (prebieha overovanie v prvých randomizovaných štúdiách), predstavujú nádej do budúcnosti (45). Tieto postupy možno aplikovať prakticky na všetky malignity a úspešne boli vyskúšané už aj na najčastejšom nádore CNS dospelého veku – glioblastóme. Analýzou štyroch rôznych súborov dát z microarray PCR vyšetrení GBM bol vytvorený multigénny (9 génov) prediktor odpovede na štandardnú liečbu, ktorý nezávisle od veku, výkonnostného stavu alebo stavu promótera MGMT identifikoval pacientov u ktorých je nízka pravdepodobnosť úspešnosti klasickej liečby a sú kandidátmi na experimentálnu alternatívnu liečbu (46). Tento multigénny prediktor bol navyše asociovaný s expresiou markerov tzv. gliómových kmeňových buniek (CD133, nestin), ktoré predstavujú ďalšiu sľubnú oblasť výskumu malígnych gliómov. Gliómové kmeňové bunky sú pravdepodobne nositeľmi chemo - a rádiorezistencie, spôsobujú rekurenciu a progresiu gliómov a mohli by predstavovať potenciálny terapeutický cieľ (47).
Všeobecným problémom cielenej liečby gliómov (monoklonové protilátky, inhibítory kináz) je, že napriek teoretickým predpokladom nebola (väčšinou v malých štúdiách) dokázaná ich väčšia účinnosť (viac v prehľade – (48)). Príčinou sú zatiaľ málo známe mechanizmy primárnej a/alebo sekundárnej rezistencie a nekompletné informácie o signálnych cestách v nádorových bunkách. Poučná môže byť lekcia od kolorektálneho karcinómu. Pacienti s metastatickým kolorektálnym karcinómom liečení chemoterapiou a dvojnásobnou cielenou liečbou bevacizumab (anti VEGF) + cetuximab (anti EGFR) paradoxne prežívali signifikantne kratšie (a s nižšou kvalitou života) ako pacienti liečení bez pridania cetuximabu – dvojitá blokáda teda nemusí znamenť vžy lepší účinok (49). Primárnym cieľom molekulových vyšetrovacích metód by teda okrem prognosticky významnej klasifikácie (napr. detekcia rozdielnych aberácií v primárnych a sekundárnych glioblastómoch) mala byť najmä identifikácia potencionálnych terapeutických cieľov, vrátane známych mechanizmov rezistencie. Dizajn klinických štúdií by sa mal v budúcnosti vždy opierať o molekulovú klasifikáciu. S použitím čoraz dostupnejších vysokokapacitných metód molekulovej genetiky nie sú tieto ciele nereálne.
Adresa pro korespondenci:
MUDr. Marián Švajdler ml.
Oddelenie patológie UNLP Košice
Pracovisko Trieda SNP 1, 041 66 Košice
tel.: +421 556402914
fax: +421 556402945
e-mail: svajdler@yahoo.com
Sources
1. Esteller M, Garcia-Foncillas J, Andion E, et al. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med 2000; 343(19): 1350–1354.
2. Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 2005; 352(10): 997–1003.
3. Krex D, Klink B, Hartmann C, et al. Long-term survival with glioblastoma multiforme. Brain 2007; 130(10): 2596–2606.
4. Stupp R, Hegi ME, van den Bent MJ, et al. Changing paradigms - an update on the multidisciplinary management of malignant glioma. Oncologist 2006; 11(2): 165–180.
5. Preusser M. MGMT analysis at DNA, RNA and protein levels in glioblastoma tissue. Histol Histopathol 2009; 24(4): 511–518.
6. Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008; 321(5897): 1807–1812.
7. Yan H, Parsons DW, Jin G, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med 2009; 360(8): 765–773.
8. Zhao S, Lin Y, Xu W, et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science 2009; 324(5924): 261–265.
9. Hartmann C, Hentschel B, Wick W, et al. Patients with IDH1 wild type anaplastic astrocytomas exhibit worse prognosis than IDH1-mutated glioblastomas, and IDH1 mutation status accounts for the unfavorable prognostic effect of higher age: implications for classification of gliomas. Acta Neuropathol 2010; 120(6): 707–718.
10. Kim YH, Nobusawa S, Mittelbronn M, et al. Molecular classification of low-grade diffuse gliomas. Am J Pathol 2010; 177(6): 2708–2714.
11. Mardis ER, Ding L, Dooling DJ, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med 2009; 361(11): 1058–1066.
12. Capper D, Sahm F, Hartmann C, et al. Application of mutant IDH1 antibody to differentiate diffuse glioma from nonneoplastic central nervous system lesions and therapy-induced changes. Am J Surg Pathol 2010; 34(8): 1199–1204.
13. Capper D,Weißert S, Balss J, et al. Characterization of R132H mutation specific IDH1 antibody binding in brain tumors. Brain Pathol 2010; 20(1): 245–254.
14. von Deimling A, Korshunov A, Hartman C. The next generation of glioma biomarkers: MGMT methylation, BRAF fusions and IDH1 mutations. Brain Pathology 2011; 21(1): 74–87.
15. Schindler G, Capper D, Meyer J, et al. Analysis of BRAF V600E mutation in 1.320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra-cerebellar pilocytic astrocytoma. Acta Neuropathol 2011; 121(3): 397–405.
16. Capper D, Preusser M, Habel A, et al. Assessment of BRAF V600E mutation status by immunohistochemistry with a mutation-specific monoclonal antibody. Acta Neuropathol 2011; 122(1): 11–19.
17. Pfister S, Janzarik WG, Remke M, et al. BRAF gene duplication constitutes a mechanism of MAPK pathway activation in low-grade astrocytomas. J Clin Invest 2008; 118(5): 1739–1749.
18. Jones DT, Kocialkowski S, Liu L, et al. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res 2008; 68(21): 8673–8677.
19. Jones DT, Kocialkowski S, Liu L, Pearson DM, Ichimura K, Collins VP. Oncogenic RAF1 rearrangement and a novel BRAF mutation as alternatives to KIAA1549:BRAF fusion in activating the MAPKpathway in pilocytic astrocytoma. Oncogene 2009; 28(20): 2119–2123.
20. Cin H, Meyer C, Herr R et al. Oncogenic FAM131B-BRAF fusion resulting from 7q34 deletion comprises an alternative mechanism of MAPK pathway activation in pilocytic astrocytoma. Acta Neuropathol 2011; 121(6): 763–774.
21. Korshunov A, Meyer J, Capper D, et al. Combined molecular analysis of BRAF and IDH1 distinguishes pilocytic astrocytoma from diffuse astrocytoma. Acta Neuropathol 2009; 118(3): 401–405.
22. Rodriguez EF, Scheithauer BW, Giannini C et al. PI3K/AKT pathway alterations are associated with clinically aggressive and histologically anaplastic subsets of pilocytic astrocytoma. Acta Neuropathol 2011; 121(3): 407–420.
23. Varlet P, Soni D, Miquel C, et al. New variants of malignant glioneuronal tumors: a clinicopathological study of 40 cases. Neurosurgery 2004; 55(6): 1377–1391.
24. Perry A, Miller CR, Scheithauer BW, et al. Malignant gliomas with primitive neuroectodermal tumor-like components: a clinicopathologic and genetic study of 53 cases. Brain Pathol 2009; 19(1): 81–90.
25. Bauman GS, Ino Y, Ueki K, et al. Allelic loss of chromosome 1p and radiotherapy plus chemotherapy in patients with oligodendrogliomas. Int J Radiat Oncol Biol Phys 2000; 48(3): 825–830.
26. Scheie D, Cvancarova M, MŅrk S, et al. Can morphology predict 1p/19q loss in oligodendroglial tumours? Histopathology 2008; 53(5): 578–587.
27. Giannini C, Burger PC, Berkey BA et al. Anaplastic oligodendroglial tumors: refining the correlation among histopathology, 1p 19q deletion and clinical outcome in Intergroup Radiation Therapy Oncology Group Trial 9402. Brain Pathol 2008; 18(3): 360–369.
28. Kreiger PA, Okada Y, Simon S, et al. Losses of chromosomes 1p and 19q are rare in pediatric oligodendrogliomas. Acta Neuropathol 2005; 109(4): 387–392.
29. Perry A, Aldape KD, George DH, Burger PC. Small cell astrocytoma: an aggressive variant that is clinicopathologically and genetically distinct from anaplastic oligodendroglioma. Cancer 2004; 101(10): 2318–2326.
30. Prayson RA, Castilla EA, Hartke M et al. Chromosome 1p allelic loss by fluorescence in situ hybridization is not observed in dysembryoplastic neuroepithelial tumors. Am J Clin Pathol 2002; 118(4): 512–517.
31. Mrak RE, Yasargil MG, Mohapatra G, Earel J Jr, Louis DN. Atypical extraventricular neurocytoma with oligodendroglioma like spread and an unusual pattern of chromosome 1p and 19q loss. Hum Pathol 2004; 35(9): 1156–1159.
32. Judkins AR, Eberhart GC, Wesseling P. Atypical teratoid/rhabdoid tumor. In: Louis DN, Ohgaki H, Wiestler OD, Cavenee K, eds. World health organisation classification of tumors. WHO classification of tumors of the central nervous system (4th ed). Lyon, IARC; 2007 : 147–149.
33. Hollmann TJ, Hornick JL. INI1-deficient tumors: diagnostic features and molecular genetics. Am J Surg Pathol 2011; 35(10): 47–63.
34. Gessi M, Giangaspero F, Pietsch T. Atypical teratoid / rhabdoid tumors and choroid plexus tumors: when genetics “surprise” pathology. Brain Pathol 2003; 13(3): 409–414.
35. Schittenhelm J, Nagel C, Meyermann R, Beschorner R. Atypical teratoid/rhabdoid tumors may show morphological and immunohistochemical features seen in choroid plexus tumors. Neuropathology 2011; 31(5): 461–467.
36. Rickert CH, Wiestler OD, Paulus W. Chromosomal imbalances in choroid plexus tumors. Am J Pathol 2002; 160(3): 1105–1113.
37. Ellison WE. Childhood medulloblastoma: novel approaches to the classification of a heterogeneous disease. Acta Neuropathol 2010; 120(3): 305–316.
38. Ellison DW, Kocak M, Dalton J, et al. Definition of disease-risk stratification groups in childhood medulloblastoma using combined clinical, pathologic, and molecular variables. J Clin Oncol 2011; 29(11): 1400–1407.
39. Ellison DW, Dalton J, Kocak M, et al. Medulloblastoma: clinicopathological correlates of SHH, WNT, and non-SHH/WNT molecular subgroups. Acta Neuropathol 2011; 121(3): 381–396.
40. Rudin CM, Hann CL, Laterra J, et al. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N Engl J Med 2009; 361(12): 1173–1178.
41. Pfister S, Remke M, Benner A, et al. Outcome prediction in pediatric medulloblastoma based on DNA copy-number aberrations of chromosomes 6q and 17q and the MYC and MYCN loci. J Clin Oncol 2009; 27(10): 1627–1636.
42. McCabe MG, Bäcklund LM, Leong HS, Ichimura K, Collins VP. Chromosome 17 alterations identify good-risk and poor-risk tumors independently of clinical factors in medulloblastoma. Neuro Oncol 2011; 13(4): 376–383.
43. Korshunov A, Remke M, Werft W, et al. Adult and pediatric medulloblastomas are genetically distinct and require different algorithms for molecular risk stratification. J Clin Oncol 2010; 28(18): 3054–3060.
44. Remke M, Hielscher T, Northcott PA, et al. Adult medulloblastoma comprises three major molecular variants. J Clin Oncol 2011; 29(19): 2717–2723.
45. Colombo PE, Milanezi F, Weigelt B, Reis-Filho JS. Microarrays in the 2010s: the contribution of microarray-based gene expression profiling to breast cancer classification, prognostication and prediction. Breast Cancer Res 2011; 13(3): 212.
46. Colman H, Zhang L, Sulman EP, et al. A multigene predictor of outcome in glioblastoma. Neuro Oncol 2010; 12(1): 49–57.
47. Hide T, Takezaki T, Nakamura H, Kuratsu J, Kondo T. Brain tumor stem cells as research and treatment targets. Brain Tumor Pathol 2008; 25(2): 67–72.
48. Sathornsumetee S, Reardon DA, Desjardins A, Quinn JA, Vredenburgh JJ, Rich JN. Molecularly targeted therapy for malignant glioma. Cancer 2007; 110(1): 13–24.
49. Tol J, Koopman M, Cats A, et al. Chemotherapy, bevacizumab, and cetuximab in metastatic colorectal cancer. N Engl J Med 2009; 360(6): 563–572.
Labels
Anatomical pathology Forensic medical examiner ToxicologyArticle was published in
Czecho-Slovak Pathology

2012 Issue 2
Most read in this issue
- Neurodegenerative Disorders: Review of Current Classification and Diagnostic Neuropathological Criteria
- Neuropathology of refractory epilepsy: the structural basis and mechanisms of epileptogenesis
- Selected biomarkers in the primary tumors of the central nervous system: short review
- Peripheral neuropathy in Whipple’s disease: A case report