
Gliosarcoma with alveolar rhabdomyosarcoma-like component: Report of a case with a hitherto undescribed sarcomatous component
Gliosarkóm s komponentou pripomínajúcou alveolárny rabdomyosarkóm: popis prípadu s doposiaľ nepopísanou sarkómovou zložkou
Gliosarkóm (GS) je relatívne vzácny variant glioblastómu, charakterizovaný bifázickou gliálnou a mezenchymálnou diferenciáciou. Sarkomatózna časť najčastejšie pripomína fibrosarkóm alebo tzv. malígny fibrózny histiocytóm. Vzácne je v GS prítomná heterológna diferenciácia vo forme osteosarkómu, chondrosarkómu, liposarkómu, leiomyosarkómu, skvamóznej alebo žľazovej malígnej epiteliálnej diferenciácie, alebo diferenciácie pripomínajúcej primitívny neuroektodermálny tumor (PNET). Keď je v GS prítomná rabdomyosarkómová diferenciácia, je vo forme malígnych vretenovitých buniek s priečne pruhovanými bunkami alebo okrúhlymi rabdomyoblastami, pripomínajúca embryonálny rabdomyosarkóm. V kazuistike popisujeme GS s komponentou pripomínajúcou alveolárny rabdomyosarkóm. Nádor rástol v solídnych a alveolárnych formáciách a bol zložený z nediferencovaných primitívnych malých okrúhlych buniek s minimálnou cytoplazmou, nápadne zvýšenou mitotickou aktivitou a početnými apoptózami. Rabdomyosarkomatózna diferenciácia bola potvrdená pozitívnou imunohistochemickou reakciou na dezmín a myogenín. Podľa naších vedomostí, takýto histologický vzor nebol v GS doposiaľ popísaný. V krátkosti je prebraná diferenciálna diagnóza prípadu.
Kľúčové slová:
gliosarkóm – alveolárny rabomyosarkóm – myogenín - desmín
Authors:
M. Švajdler jr. 1; B. Rychlý 2; M. Gajdoš 3; F. Pataky 3; L. Fröhlichová 1; A. Perry 4
Authors‘ workplace:
Department of pathology UNLP Košice, Slovakia
1; Cytopathos, spol. s. r. o., Bratislava, Slovakia
2; Department of neurosurgery UNLP and P. J. Safarik University, Faculty of Medicine Košice, Slovakia
3; Department of Pathology, Division of Neuropathology, University of California, San Francisco, USA
4
Published in:
Čes.-slov. Patol., 48, 2012, No. 4, p. 210-214
Category:
Original Articles
Overview
Gliosarcoma (GS) is a relatively rare glioblastoma variant characterized by biphasic glial and mesenchymal differentiation patterns. The sarcomatous part most commonly resembles fibrosarcoma or so-called malignant fibrous histiocytoma. Rarely, GS shows heterologous lines of differentiation in the form of osteosarcoma, chondrosarcoma, liposarcoma, leiomyosarcoma, squamous or glandular malignant epithelial differentiation, or primitive neuroectodermal tumor (PNET)-like foci. When rhabdomyoblastic differentiation occurs, it is in the form of malignant spindle cells, with cross-striated strap cells or rounded rhabdomyoblasts reminiscent of the embryonal type of rhabdomyosarcoma. We are reporting a case of GS with an alveolar rhabdomyosarcoma-like component. The tumor consisted of poorly differentiated primitive small round cells growing in a solid and alveolar pattern, with minimal cytoplasm, markedly elevated mitotic activity and numerous apoptotic nuclei. Rhabdomyosarcomatous differentiation was confirmed by desmin and myogenin immunopositivity. To the best of our knowledge, this histologic pattern has not been previously reported in GS. Differential diagnostic considerations are discussed.
Keywords:
gliosarcoma – alveolar rhabdomyosarcoma – myogenin – desmin
According to the 2007 WHO classification of tumors of the central nervous system (,,blue book“), gliosarcoma (GS) is ,, a glioblastoma variant characterized by a biphasic tissue pattern with alternating areas displaying glial and mesenchymal differentiation“. These tumors are rare, and represent 2 to 8 % of all glioblastomas (GBM) (1). Gliosarcomas typically occur in older population, but exceptional cases can be found in young adults, and even in children and infants (2). The glial part is a high grade astrocytic neoplasm, mostly resembling classical GBM, whereas the sarcomatous part most commonly resembles fibrosarcoma or so-called malignant fibrous histiocytoma (1). Rarely, GS may show heterologous lines of differentiation in the form of osteosarcoma (3), chondrosarcoma (4), liposarcoma (5), leiomyosarcoma (6), squamous or glandular malignant epithelial differentiation (7), or primitive neuroectodermal tumor (PNET)-like foci (8).
Rhabdomyoblastic differentiation in GS was first described in the English literature by Goldman in 1969 (9), and after that by a few other authors (10–13). Rhabdomyosarcomatous differentiation in these cases was in the form of malignant spindle cells, with typical elongate strap cells or rounded rhabdomyoblastic elements with cross striations and eosinophilic fibrillar cytoplasm, the latter reminiscent of the embryonal type of rhabdomyosarcoma (RMS).
Herein, we report a case of GS with an alveolar RMS-like component and discuss its differential diagnosis. To the best of our knowledge, this histologic pattern has not been previously reported in the English literature.
CASE REPORT
A 78-year-old woman was admitted to the Department of Neurology, complaining of headaches lasting for three days, along with progressive weakness of her left extremities and gait / balance disturbances. Her past medical history included essential hypertonic disease, ischemic heart disease with paroxysmal atrial fibrillation, diaphragmatic myocardial infarction two years previously and type 2 diabetes mellitus.
On neurological examination left sided central facial nerve palsy and severe left hemiparesis was found. A routine head CT revealed an intracranial mass lesion in the right occipito-temporo-parietal region with a significant pseudocystic portion and perifocal edema. A subsequent MRI confirmed the presence of a large (6.0 x 4.2 x 5.5 cm) mass with ring enhancement, suggestive of GBM (Fig. 1).

A right sided occipito-temporo-parietal craniotomy was performed. After opening the dura mater, it was found to be infiltrated by the tumor. The solid component of the lesion appeared to be heterogeneous in consistency, with alternating soft and hard areas and dense pathological vascularisation. After evacuation of the cystic fluid, the solid part of the tumor was nearly totally removed by ultrasound aspirator under surgical microscope.
Post-operatively, partial neurological recovery was seen and the patient underwent a course of fractionated whole brain radiotherapy (30 Gray in 15 doses) with a boost to the tumor bed (30 Gray in 15 doses). Because of poor performance, chemotherapy was not given. However, after several weeks, the patient died of progressive disease.
The specimen submitted for histopathology consisted of several pieces of the dura and bloody soft tissue fragments measuring 3.5 x 2.5 x 0.5 cm in aggregate. Microscopically, sections from routinely processed paraffin embedded tissue showed dural based tumor with two morphologically distinct malignant components. The first was characterized by pleomorphic and mitotically active fibrillary astrocytic cells (Fig. 2) with focal microvascular proliferation. The second component consisted of poorly differentiated primitive small round cells, with minimal cytoplasm, markedly elevated mitotic activity, numerous apoptotic cells, and geographic infarct-like necrosis (Fig. 3). The nuclei of the small cell component were round to irregular, hyperchromatic, and without prominent nucleoli. Increased pleomorphism was present focally. The small cell component grew in solid sheets with focally nested and discohesive growth patterns, reminiscent of the solid and alveolar growth pattern of alveolar RMS (ARMS).


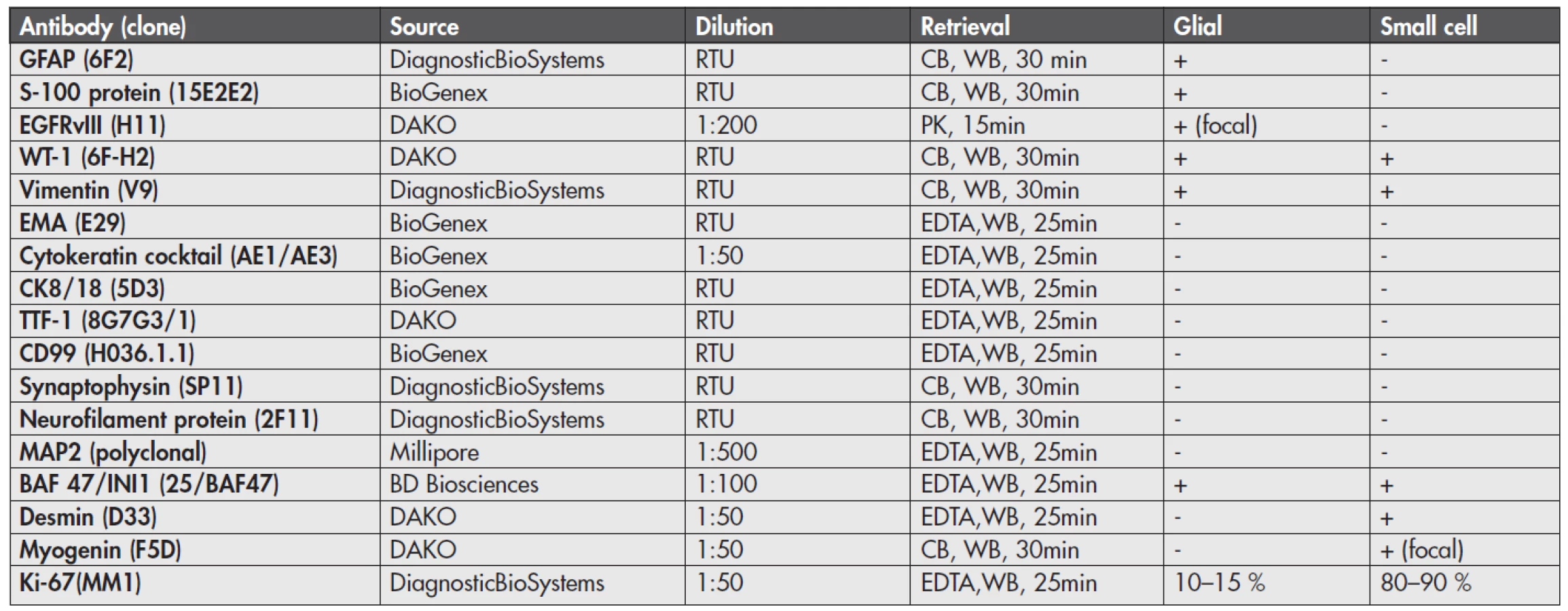
By immunohistochemistry (Fig. 4), the glial component showed strong and diffuse cytoplasmic GFAP and S-100 protein positivity and focal membranous positivity of the epidermal growth factor receptor variant type III (EGFRvIII). In contrast, the small round cell part of the tumor was GFAP, S-100 protein and EGFRvIII negative. Both components showed cytoplasmic WT-1 and vimentin positivity and were negative for epithelial membrane antigen (EMA), cytokeratin cocktail, CK8/18, TTF-1, CD99, synaptophysin, neurofilament protein and MAP2. Nuclear INI-1 expression was retained in both parts. Importantly, the small cell component was diffusely desmin and focally myogenin positive, consistent with rhabdomyosarcomatous differentiation. The proliferative labelling index as assessed by Ki-67 immunostaining was 10–15 % in the glial part and 80–90 % in the sarcomatous part. These results are illustrated in Figure 4 and sumarized in Table 1.

By fluorescence in situ hybridization (FISH, results not shown) of EGFR gene (Poseidon EGFR, HER-1 (7p11) & SE7 control probe, Kreatech, Nl) there was an evidence of polysomy of chromosome 7, but no evidence of EGFR gene amplification. FISH of PTEN gene (Poseidon PTEN (10q23) & SE10 control probe, Kreatech, Nl) did not reveal any abnormalities (deletion). Break apart FISH of PAX7 and PAX3 genes (PAX7 Breakapart probe, and PAX3 Breakapart probe, Cytocell, UK), did not reveal gene rearrangement (PAX3/PAX7-FKHR translocation is characteristic of AMRS).
DISCUSSION
Rhabdomyosarcomatous and other heterologous lines of differentiation are exceedingly rare in GS. When rhabdomyosarcomatous differentiation occurs, it most often resembles the embryonal type of RMS. We believe that this report is the first description of GS with alveolar RMS-like differentiation, with unique differential diagnostic considerations.
Firstly, PNET-like foci have been known to occur in rare high grade gliomas and such cases have recently been referred to as malignant gliomas with primitive neuroectodermal tumor-like component (MG-PNET) (8,14). These tumors are difficult to classify and pose therapeutic challenges as well. The glioma component in these tumors most often resembles GBM, GS, lower-grade forms of diffuse glioma, or rarely, anaplastic oligodendroglioma. The PNET-like component may resemble other ,,small round blue cell tumors”, including alveolar RMS. Unlike our case, the PNET-like component typically resembles medulloblastomas or CNS PNETs in general. Neuroblastic (Homer Wright) rosettes can be found in a subset, as well as desmoplastic / nodular or linear streaming patterns. Nuclear anaplasia and large cell changes are frequent findings. Immunohistochemically, the PNET-like component expresses one or several ,,neuronal markers“, including synaptophysin, Neu-N, MAP2 or neuron specific enolase. Importantly, significant number of cases (43 %) from the largest series of MG-PNET showed MYC gene amplifications (mostly N-myc), which was restricted to the PNET-like component. Leptomeningeal spread occurred in eight of 39 patients with follow-up, which is an unusually high number in comparison with classical GBM, and is very close to the incidence of cerebrospinal fluid dissemination in medulloblastoma or CNS PNET. At least three patients benefited from platinum-based PNET therapy after switching from routine GBM therapy (14).
WHO defines central nervous system primitive neuroectodermal tumor (CNS PNET) as ,,an embryonal tumor composed of undifferentiated or poorly differentiated neuroepithelial cells which have the capacity for, or display, divergent differentiation along neuronal, astrocytic, muscular or melanotic lines“ (15). Although CNS PNET occurs predominantly in children and adolescents, rare cases have been described in adults (16,17). At the molecular level, it seems that adult cases of CNS PNET are distinct from pediatric cases (17). CNS PNET with a glial component would clearly enter the differential diagnosis for our case. However, as in the case of MG-PNET, immunohistochemical expression of several neuronal markers is typical, even in undifferentiated parts of CNS PNET. In one study, one or more muscle markers (desmin, h-caldesmon, a-smooth muscle actin) were positive in eight of 18 CNS PNETs. However, none of these tumors showed myogenin expression (18).
Another differential diagnosis is a small cell glioblastoma. In some series PNET-like tumors are erroneously included in this category. However, typical small cell glioblastoma is composed of more monomorphic and cytologically bland tumor cells with minimal cytoplasm, oval minimally hyperchromatic nuclei, and an unexpectedly high mitotic rate and proliferation index (Ki-67 immunohistochemistry). This variant often behaves as a typical glioblastoma even in the absence of necrosis or microvascular proliferation and has a very high frequency of EGFR gene amplifications, analogous to the primary or de novo type of GBM (19).
Lastly, collision of two clonally distinct tumors, GBM and ARMS needs to be ruled out. Although several cases of primary intracranial RMS exist, it would be highly improbable to be present in such a close spatial relationship with a glioma purely by chance. In our case, a systemic origin of ARMS (tumor to tumor metastasis) was additionally ruled out by complete examination of the patient.
Regarding the management and prognosis of GS, some studies suggest that the outcome of GS is not very different or is slightly worse than that of conventional GBM (20, 21). Tumor excision, as opposed to biopsy only and adjuvant radiotherapy seem to better the prognosis (21). Of interest, when gliosarcomas are divided into two groups based on macroscopic description by the surgeon as meningioma-like or GBM-like, the median survival time differs significantly (16.0 months versus 9.5 months). This survival difference is most likely related to the extent of resection performed. Importantly, adjuvant radiotherapy and temozolomide administered to a limited number of patients did not improve survival in this study (22). Interestingly, in an another study of 30 patients with secondary GS (GS as the recurrent tumor following the treatment of GBM), patients with GS who had received concurrent and adjuvant temozolomide for GBM had significantly worse outcomes than those who had not. This may, perhaps, reflect a unique molecular profile of GBM that eventually recurs as GS. In the future, molecular subclassification could potentially change the therapy plan for these patients. In that same study, secondary GS had significantly worse median survival as compared to literature data for primary GS (4.6 month versus 6.25–11.25 months). However, these data should be read with caution, as patients with recurrent GS may not be eligible for a number of therapeutic modalities (23). Interestingly, approximately 11 % of gliosarcomas have extracranial metastases and a few cases with intraspinal metastases are also described (24,25).
In conclusion, in this case report we have described a novel ARMS-like pattern of differentiation in a GS. This should be added to the list of the more common patterns of heterologous sarcomatous differentiation in GS. This pattern of differentiation has unique and therapy relevant differential diagnostic and prognostic considerations.
Correspondence address:
Marian Švajdler jr., MD
Department of Pathology, L. Pasteur University Hospital
Trieda SNP 1, 041 66 Košice, Slovakia
tel.: +421 556402914, fax: +421 556402945
E-mail: svajdler@yahoo.com
Sources
1. Kleihues P, Burger PC, Adalpe KD, et al. Glioblastoma. In: Louis DN, Ohgaki H, Wiestler OD, Cavenee K, eds. World health organisation classification of tumors. WHO classification of tumors of the central nervous system (4th ed). Lyon, IARC; 2007 : 33–49.
2. Okami N, Kawamata T, Kubo O, Yamane F, Kawamura H, Hori T. Infantile gliosarcoma: a case and a review of the literature. Childs Nerv Syst 2002; 18(6–7): 351–355.
3. Charfi S, Ayadi L, Khabir A, et al. Gliosarcoma with osteosarcomatous features: a short illustrated review. Acta Neurochir 2009; 151(7): 809–813.
4. Banerjee AK, Sharma BS, Kak VK, Ghatak NR. Gliosarcoma with cartilage formation. Cancer 1989; 63(3): 518–523.
5. Vlodavsky E, Konstantinesku M, Soustiel JF. Gliosarcoma with liposarcomatous differentiation: the new member of the lipid-containing brain tumors family. Arch Pathol Lab Med 2006; 130(3): 381–384.
6. Khanna M, Siraj F, Chopra P, Bhalla S, Roy S. Gliosarcoma with prominent smooth muscle component (gliomyosarcoma): A report of 10 cases. Indian J Pathol Microbiol 2011; 54(1): 51–54.
7. Ozolek JA, Finkelstein SD, Couce ME. Gliosarcoma with epithelial differentiation: immunohistochemical and molecular characterization. A case report and review of the literature. Mod Pathol 2004; 17(6): 739–745.
8. Kaplan KJ, Perry A. Gliosarcoma with primitive neuroectodermal differentiation: case report and review of the literature. J Neurooncol 2007; 83(3): 313–318.
9. Goldman RL. Gliomyosarcoma of the cerebrum. Report of a unique case. Am J Clin Pathol 1969; 52(6): 741–744.
10. Barnard RO, Bradford R, Scott T, Thomas DG. Gliomyosarcoma. Report of a case of rhabdomyosarcoma arising in a malignant glioma. Acta Neuropathol 1986; 69(1–2): 23–27.
11. Stapleton SR, Harkness W, Wilkin PR, Uttley D. Gliomyosarcoma: An Immunohistochemical analysis. J Neurol Neurosurg Psychiatry 1992; 55(8): 728–730.
12. Sarkar C, Sharma MC, Sudha K, Gaikwad S, Varma A. A clinico-pathological study of 29 cases of gliosarcoma with special reference to two unique variants. Indian J Med Res 1997; 106 : 229–235.
13. Sethi S, Siraj F, Roy S. Gliosarcoma with rhabdomyomatous differentiation: A case report. Indian J Cancer 2011; 48(1): 129–131.
14. Perry A, Miller CR, Scheithauer BW, et al. Malignant gliomas with primitive neuroectodermal tumor-like components: a clinicopathologic and genetic study of 53 cases. Brain Pathol 2009; 19(1): 81–90.
15. McLendon RE, Judkins AR, Eberhart CG, Fuller GN, Sarkar C, Ng HK. Central nervous system primitive neuroectodermal tumors. In: Louis DN, Ohgaki H, Wiestler OD, Cavenee K, eds. World health organisation classification of tumors. WHO classification of tumors of the central nervous system (4th ed). Lyon, IARC; 2007 : 141–146.
16. Shingu T, Kagawa T, Kimura Y, Takada D, Moritake K, Hoshii Y. Supratentorial primitive neuroectodermal tumor in an aged patient. Case report. Neurol Med Chir 2005; 45(10): 530–535.
17. Gessi M, Setty P, Bisceglia M, zur Muehlen A, et al. Supratentorial primitive neuroectodermal tumors of the central nervous system in adults: molecular and histopathologic analysis of 12 cases. Am J Surg Pathol 2011; 35(4): 573–582.
18. Visée S, Soltner C, Rialland X, et al. Supratentorial primitive neuroectodermal tumours of the brain: multidirectional differentiation does not influence prognosis. A clinicopathological report of 18 patients. Histopathology 2005; 46(4): 403–412.
19. Perry A, Aldape KD, George DH, Burger PC. Small cell astrocytoma: an aggressive variant that is clinicopathologically and genetically distinct from anaplastic oligodendroglioma. Cancer 2004; 101(10): 2318–2326.
20. Galanis E, Buckner JC, Dinapoli RP, et al. Clinical outcome of gliosarcoma compared with glioblastoma multiforme: North Central Cancer Treatment Group results. J Neurosurg 1998; 89(3): 425–430.
21. Kozak KR, Mahadevan A, Moody JS. Adult gliosarcoma: epidemiology, natural history, and factors associated with outcome. Neuro Oncol 2009; 11(2): 183–191.
22. Han SJ, Yang I, Ahn BJ, et al. Clinical characteristics and outcomes for a modern series of primary gliosarcoma patients. Cancer 2010; 116(5): 1358–1366.
23. Han SJ, Yang I, Otero JJ, et al. Secondary gliosarcoma after diagnosis of glioblastoma: clinical experience with 30 consecutive patients. J Neurosurg 2010; 112(5): 990–996.
24. Beaumont TL, Kupsky WJ, Barger GR, Sloan AE. Gliosarcoma with multiple extracranial metastases: case report and review of the literature. J Neurooncol 2007; 83(1): 39–46.
25. Demirci S, Akalin T, Islekel S, Ertan Y, Anacak Y. Multiple spinal metastases of cranial gliosarcoma: a case report and review of the literature. J Neurooncol 2008; 88(2): 199–204.
Labels
Anatomical pathology Forensic medical examiner ToxicologyArticle was published in
Czecho-Slovak Pathology

2012 Issue 4
Most read in this issue
- Pleomorphic adenoma of salivary glands: diagnostic pitfalls and mimickers of malignancy
- Histiocytic necrotizing lymphadenitis / Kikuchi-Fujimoto disease (HNL/K-F) and its differential diagnosis: analysis of 19 patients
- Pseudotumors of the central nervous system
- Expression of p57 marker in differential diagnosis of complete and partial mole – correlation with DNA analysis