
Průkaz chromozomálních změn u nádorových onemocnění pomocí CGH, array-CGH a SNP array
Detection of chromosome changes by CGH, array-CGH and SNP array techniques in tumours
New molecular biology methods have specified the evidence of chromosomal changes in the tumor tissue. These alterations can be proven to exist in the majority of malignant tumors. The fast progress of whole genome molecular biological methods has helped to improve the knowledge of tumor genetics. The evidence of genetic changes is a component of currently used diagnostic and prognostic schemes in particular cancer diseases. Karyotyping was the first method used in the clinical practice but its importance has decreased with the arrival of new molecular biological methods. The most common methods used for the detection of chromosomal deletions or amplifications are CGH, array-CGH and SNP array. The first two methods are based on the principle of comparison between tumor DNA and control DNA. The principle of SNP array uses the presence of single nucleotide polymorphisms that are located in the whole genome in each individual. SNP array can prove not only deletions or amplifications of the chromosomes but unlike CGH techniques it can also detect a loss of heterozygosity or uniparental disomy. The screening of chromosomal changes has nowadays become routine. These techniques are used for diagnosis, prognosis and treatment of cancer disease in certain cases.
Keywords:
cytogenetic – CGH – array-CGH – SNP array – genetic aberration
Authors:
Tatiana Vosecká 1; Zdeněk Musil 1,2; Aleš Vícha 1
Authors‘ workplace:
Klinika dětské hematologie a onkologie 2. LF UK a FN Motol
1; Ústav biologie a lékařské a genetiky, 1. LF UK a VFN V Praze
2
Published in:
Čes.-slov. Patol., 50, 2014, No. 1, p. 25-29
Category:
Reviews Article
Overview
Nové molekulárně biologické metody umožnily zpřesnění průkazu chromozomálních změn v nádorové tkáni. Tyto změny lze prokázat u většiny maligních nádorů. Rychlý rozvoj celogenomových molekulárně biologických metod napomohl zlepšení poznání genetického pozadí nádorů. V současné době je jejich průkaz součástí diagnostických nebo prognostických schémat používaných u jednotlivých nádorových onemocnění. Jako první bylo v klinické praxi využíváno klasické cytogenetické vyšetření karyotypu, které je využíváno dodnes, i když s nástupem nových molekulárně biologických technik se jeho význam zmenšil. Metodami dnes nejčastěji používanými ke stanovení chromozomálních delecí nebo zmnožení při celogenomovém vyšetření jsou komparativní genomová hybridizace (CGH), array-CGH a SNP array. První dvě jsou založeny na principu porovnání nádorové DNA a normální, kontrolní DNA. Princip SNP array využívá přítomnosti jednonukleových polymorfismů rozložených po celém genomu u každého jedince. SNP array prokazuje nejen delece nebo zmnožení chromozómů, tak jak je tomu u CGH technik, ale je schopna prokázat i ztrátu heterozygozyty nebo unipaternální dizomii.
Vyšetření chromozomálních změn se dnes stává rutinním a v některých případech také nezbytným vyšetřením pro stanovené diagnózy a prognózy nádorového onemocnění a správného výběru onkologické terapie.
Klíčová slova:
cytogenetika – CGH – array-CGH – SNP array – genetická změna
Ve tkáni většiny nádorů je možné prokázat chromozomální změny, které vznikají primárně nebo sekundárně v důsledku genomové instability. Chromozomální abnormality, které se v nádorech prokazují, jsou jednak početní změny celých chromozómů (delece, nebo zmnožení chromozómů) nebo změny části chromozómu (zmnožení (amplifikace) a delece), takové změny označujeme jako segmentální. Druhou skupinou jsou strukturální chromozomální změny, mezi které řadíme translokace (balancované i nebalancované), inverze, inzerce a další. Mnohé z nich jsou nalézané opakovaně u stejného typu nádorových onemocnění a mohou tak napomoci při diagnostice těchto nádorů (např. t(11;22) u Ewingova sarkomu (1,2)). Jiné je možné využít jako biologický prognostický znak pro zařazení pacienta do příslušné prognostické skupiny a umožnit odpovídající způsob terapie (např. amplifikace MYCN onkogenu je významným negativním prognostickým znakem u neuroblastomu) (3).
První metodou, která byla k průkazu chromozomálních změn využívána, bylo klasické karyotypické vyšetření, které je využíváno dodnes, i když s nástupem nových molekulárně biologických technik se jeho význam zmenšil. Mezi takovéto nové techniky, které jsou používány k vyšetření celého genomu, řadíme komparativní genomovou hybridizaci (CGH), její variantu array-CGH a Single nucleotide polymorphism (SNP) array. Pomocí těchto technik jsme schopni prokázat delece, zmnožení a amplifikace na všech chromozómech při jednom vyšetření. Charakteristika jednotlivých vyšetření je shrnuta v tabulce 1.

KOMPARATIVNÍ GENOMOVÁ HYBRIDIZACE - CGH
Komparativní genomová hybridizace, byla poprvé popsána v roce 1992 Kallioniemi et al. (7). Jedná se o molekulárně cytogenetickou metodu, která umožňuje stanovení relativního počtu kopií (balance, zmnožení, delece) jednotlivých sekvencí DNA mezi dvěma genomy (DNA nádoru versus DNA zdravého jedince jako kontrolní). Hlavním přínosem je, že pro vyšetření není potřeba získat kvalitní mitózy, tak jak je tomu u klasického cytogenetického vyšetření, ale pouze 1 - 2 µg nádorové DNA. Metoda naopak vyžaduje, aby zastoupení nádorových buněk ve vzorku, ze kterého je DNA izolována, bylo aspoň 50 %. Zde je úloha patologa zcela nezastupitelná, protože vyšetřením vzorku, který má méně než 50 % nádorových buněk, získáme zkreslený nebo nepravdivý výsledek vyšetření. Kvalita vyšetření je také závislá na fixaci materiálu. Z tohoto pohledu je nejvhodnější materiál nativní, nebo zamražený. Vyšetření DNA z parafinových bloků jsou často nekvalitní a v některých případech nemožná i přesto, že jsou zaváděny nové izolační a recovery kity, které umožňují zlepšení výsledku těchto vyšetření.
Princip metody je založený na kvantitativním porovnání nádorové a kontrolní DNA. Po izolaci DNA je nádorová i kontrolní DNA fluorescenčně označena. V případě CGH je referenční DNA značená červeně a testovaný vzorek od pacienta zelenou barvou. Po obarvení jsou vzorky ve stejném množství smíchány a směs je nanesena na sklíčko s normálními metafázickými chromozómy získanými od zdravého dárce. Následuje hybridizace, při které kompetují jednotlivé značené úseky zdravé a nádorové DNA o párování se shodným místem na chromozómech přichycených na sklo. Chromozómy jsou dobarveny pomocí modrého fluorochromu DAPI (4‘,6‑diamidino‑2‑phenylindol), který na chromozómech vytváří pruhování, a umožňuje tak jejich karyotypování (8). Princip metody ukazuje obr. 1. Takto připravené mitózy jsou snímány pomocí černobílé kamery v jednotlivých fluorescenčních filtrech (FITC (zelená barva), rhodamin (červená barva), DAPI (modrá barva)). Po nasnímání do systému jsou mitózy počítačově zpracovány, je detekovaný kvantitativní poměr fluorescenčních intenzit jednotlivých chromozómu (viz obr. 1). Takto odhalíme místa s vyšší nebo nižší intenzitou červené a zelené barvy podél osy chromozómu, což odpovídá lokalizaci chromozómových oblastí se ziskem nebo ztrátou nádorové DNA. Pokud testovaný vzorek obsahuje nadbytečný materiál, objeví se silnější zelený signál v cílových oblastech chromozómů, naopak v případě ztráty genetického materiálu má signál v těchto oblastech intenzivnější červenou barvu (obr. 2A-C). Technikou CGH detekujeme pouze nebalancované přestavby v genomu, tj. zmnožení nebo ztráty, ale nedokážeme odhalit balancované chromozomální změny (translokace, inverze) (8). Dále není CGH nejvhodnější technikou pro hodnocení polyploidie, protože určuje pouze změny počtu kopií chromozómů, ale není schopna určit jejich absolutní počet. Například tetraploidní buněčná populace dává v genomu konstantní poměr a je nerozlišitelná od výsledku získaného z diploidní testované populace.



ARRAY-CGH
V druhé polovině devadesátých let Solinas-Toldo et al. (9) představili novou metodu CGH techniky probíhající na čipu, tzv. array-CGH. Tato metoda opět porovnává referenční genom proti genomu pacienta a identifikuje rozdíly. Princip array-CGH je velmi podobný CGH, referenční a zkoumaná DNA jsou fluorescenčně značeny a hybridizovány ke specifickým fragmentům normální DNA uchyceným na čipu (sklíčku) (10). Následuje přečtení hotového čipu na scanneru, softwarové zpracování a vyhodnocení. U array-CGH je opačné značení kontrolní a sledované DNA než u CGH (obr. 1.). Zásadní rozdíl je v citlivosti obou metod. Rozlišovací schopnost detekce chromozomálních alterací je u chromozomální CGH přibližně 5-10Mb (11,12). U array-CGH se velikost fragmentů pohybuje od desítek bází až po kilobáze.
Rozeznáváme několik typů čipů, jako jsou oligonukleotidové čipy nebo BAC čipy. U oligonukleotidových čipů jsou sekvence DNA nanášené na sklíčko velice krátké (25 – 85 bazí) (13), naproti tomu BAC čipy využívají DNA získanou z jednotlivých BAC (Bacterial Artificial Chromosome) což jsou plazmidy do kterých je možné vkládat a následně v E. coli namnožit specifickou lidskou DNA délky 150 – 200 kb.
Rozlišovací schopnost array-CGH závisí na dvou faktorech - na délce a množství nanášených fragmentů a na jejich skutečném zastoupení v rámci genomu (10). Rozlišovací schopnost stoupá se zvyšováním počtu co nejkratších fragmentů, a to tak, aby souvisle pokryly celý genom nebo sledovanou oblast. Právě proto je výhodnější používat čipy vytvořené metodou oligonukleotidů, jejich délka menší (13), a tím poskytuje přesnější výsledek, než je tomu u BAC čipů (viz obr. 1). Nevýhodou této metody, stejně jako při CGH, je nemožnost detekce balancovaných změn.
SINGLE NUCLEOTIDE POLYMORPHISM (SNP) ARRAY
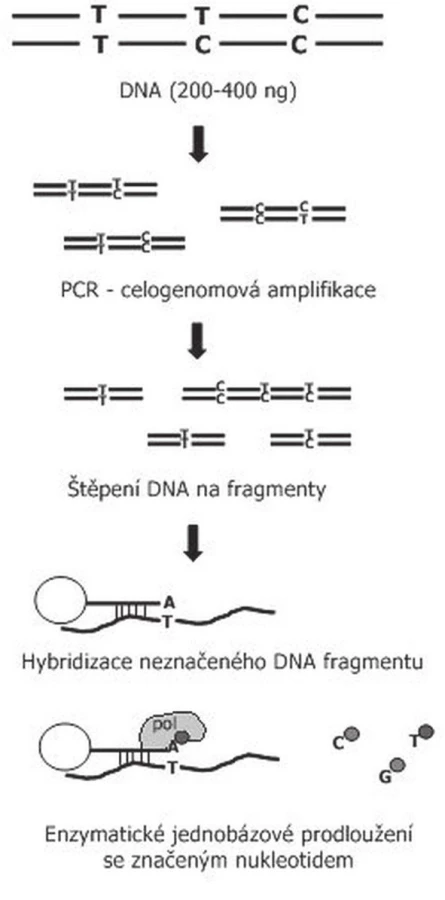
Zavedení SNP array znamenalo další pokrok v analýze genomických abnormalit u nádorových onemocnění. Jedná se o metodu s vysokým rozlišením, pomocí které jsme schopni prokazovat početní změny podobně jako u array-CGH, ale také ztrátu heterozygozity (LOH) a uniparentální dizomii. SNP tak poskytuje více informací při jednom vyšetření než aCGH a podle množství vyšetřovaných polymorfismů obdobnou nebo vyšší citlivost. (15). Metoda je založena na principu vyšetření jednonukleotidových polymorfismů SNP, které jsou nejčetnější genetickou variací genomu. O SNP mluvíme tehdy, pokud je nukleotid A, T, C nebo G rozdílný v jednotlivých alelách chromozómu. Například sekvence AAGCCTA a AAGCTTA se liší tím, že v jedné alele je C, ve druhé T. Tyto bodové mutace se přirozeně vyskytují v průběhu evoluce a jsou definovány přítomností minoritní alely minimálně v 1% případů sledované populace. V současné době bylo prokázáno přibližně 15 miliónů SNP rozložených po celém lidském genomu (16).
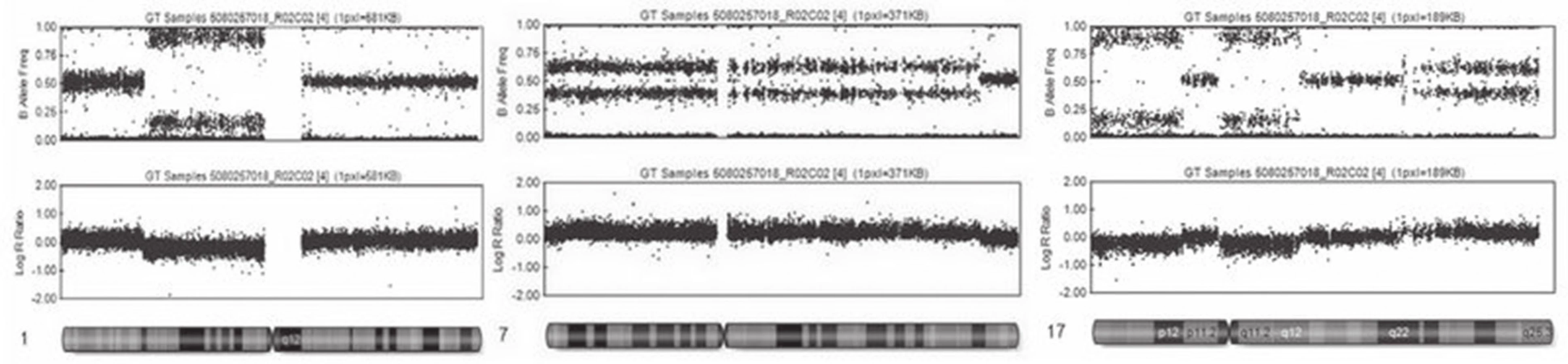
Metodika vyšetření je u jednotlivých systémů modifikována, u nás používáme SNP čipy firmy Illumina. Při tomto vyšetření je DNA (genomická DNA nebo DNA nádoru) nejdříve amplifikována, následně rozštěpena restrikčními enzymy a hybridizovana ke krátkým úsekům DNA přichyceným na sklíčko čipu. Tím je zajištěna specificita vyšetřovaného SNP. Následuje jednonukleotidová extenze značeným nukleotidem (obr. 3). Jednotlivé SNP spoty, které se podobně jako u aCGH barevně zvýrazní (červená, zelená, žlutá), jsou snímány pomocí skeneru a následně softwarově zpracovány (obr. 3). Výsledek vyšetření poskytne informaci o intenzitě SNP na základě intenzity fluorescence (obr. 4). Tak můžeme stanovit homozygotní, nebo heterozygotní deleci, případně duplikaci nebo amplifikaci genetického materiálu. Pomocí tohoto vyšetření je také možné určit, zda je vyšetřovaný vzorek pro daný SNP homozygot AA (obě alely mají v místě SNP stejný nukleotid), nebo BB, případně heterozygot AB (každá alela má v místě SNP jiný nukleotid). Pokud kombinujeme data o intenzitě a alelickém rozložení, jsme schopni prokazovat LOH i unipaternální dizomii (17).
PŘÍKLADY VYUŽITÍ CELOGENOMOVÝCH TECHNIK V DĚTSKÉ ONKOLOGII
Meduloblastom
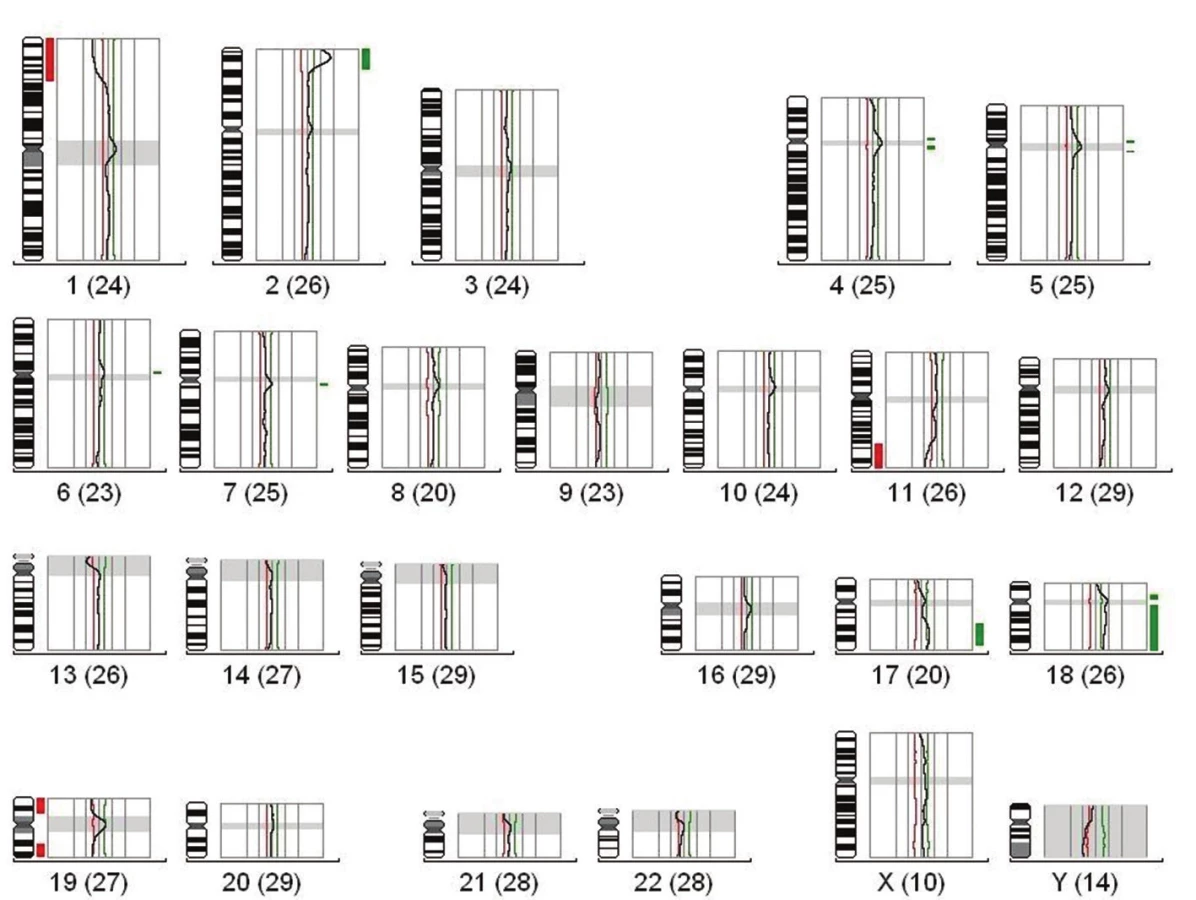
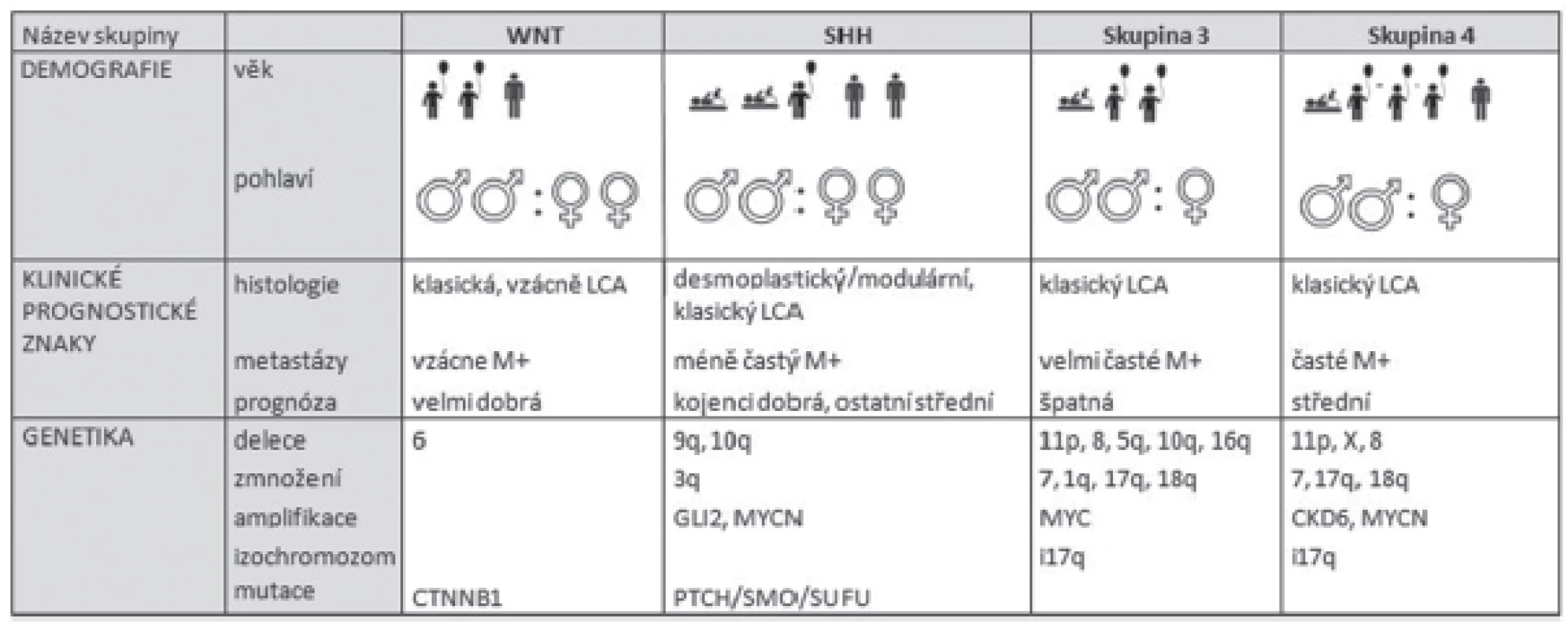
Na základě klinických a biologických faktorů je nově meduloblastom rozčleněn do 4 podskupin (tabulka 2). První podskupina nese název podle signální dráhy WNT, která je u této skupiny změněna. Charakteristická je pro ni delece 6. chromozómu a mutace CTNNB1 genu. Jedná se především o pacienty dětského věku. Pacienti WNT skupiny mají dobrou prognózu. Druhou podskupinou je SSH odvozena od signální dráhy SHH (Sonic Hedgehog), která se vyznačuje zmnožením 3q, delecí na 9q, 10q a amplifikací GLI2 a MYCN onkogenů. Pacienti s diagnostikovaným SHH meduloblastomem jsou kojeneckého věku, nebo dospělí a mají průměrnou prognózu. Meduloblastom třetí skupiny nebo tzv. klasicky meduloblastom, je charakteristický zmnožením 7, 1q, 17q, 18q a delecí na 8, 11p, 5q, 10q,16q, inverzí 17q chromozómu a MYCC amplifikací. Typický je pro pacienty dětského věku a má velmi špatnou prognózu. Poslední skupinou jsou meduloblastomy čtvrté skupiny, které se vyznačují chromozomálními změnami, jako jsou zmnožení 7, 17q, 18q nebo delecí 11p, 8, X chromozómů, inverze 17q a také amplifikace CDK6 a MYCN onkogenů (18). Vyskytují se hlavně u dětských pacientů a z prognostického hlediska patří mezi středně závažné.
Neuroblastom
V roce 1997 Brodeur et al. (20) navrhli klinicko-genetickou klasifikaci neuroblastomu na základě retrospektivní analýzy. Podle tohoto schématu se pacienti dělí do tří rizikových skupin (obr. 5). Pokud se nádorová buňka vydá cestou mitotické dysfunkce, následkem které je porucha separace chromozómů, vznikají změny celých chromozómů. Delece nejčastěji prokazujeme u chromozómů 1, 3 a 11; naopak zmnožení je nejčastější u chromozómu 2, 7 a 17 (20). Tyto pacienty řadíme do skupiny s nízkým rizikem neuroblastomu. Pacienti nízkého rizika jsou buď pouze sledováni pomocí zobrazovacích metod v pravidelných intervalech po předešlé biopsii nádoru (část těchto nádorů sama zaniká apoptózou, nebo se změní na benigní formy nádorů), nebo jsou léčení chirurgicky radikálním odstraněním nádoru, nevyžadují však další onkologickou léčbu. Celkové přežití pacientů nízkého rizika neuroblastomu se blíží téměř 100 %. Pokud při vyšetření prokážeme kromě změn celých chromozómů i segmentální změny, jde o pacienty středního nebo vysokého rizika neuroblastomu. Mezi nejčastěji prokazované změny v této skupině patří delece 1p, 3p, 4p a 11q a zmnožení 1q, 2p, a především 17q (21,22). Zmnožení 17q je nejčastější změnou u neuroblastomu středního a vysokého rizika. Pacienti středního rizika jsou již léčeni pomocí chemoterapie a často jsou indikováni k chirurgickému výkonu. Pacienti vysokého rizika jsou z biologického hlediska charakterizováni amplifikací MYCN onkogenu. Tito pacienti jsou léčení pomocí chemoterapie, chirurgické léčby, megachemoterapie s autologní transplantací hematopoetických progenitorových buněk, radioterapie, bioterapie (23,24), a za použití specifických protilátek anti-GD2 (25). I přesto 60 – 70 % pacientů vysokého rizika zemře.

Kromě výše uvedených nádorů se celogenomové techniky využívá i u nádorů vaječníků (26), nádoru prsů (27), rektálního adenomu a karcinomu (28), nádorů prostaty (29), karcinomu plic (30), ale i u mnoha dalších nádorových onemocnění.
ZÁVĚR
Rychlý rozvoj celogenomových molekulárně biologických metod napomohl zlepšení poznání genetického pozadí nádorů. Tyto poznatky jsou stále častěji využívány v onkologii při průkazu genetických abnormalit, které mohou být spojeny s jednotlivými nádorovými onemocněními. Detekce specifických genetických změn je využívána ke zlepšení klasifikace nádorů nebo stanovení prognostických znaků daného nádorového onemocnění. V neposlední řadě pak mohou napomoci k určení nových terapeutických cílů využitelných při onkologické léčbě.
PODĚKOVÁNÍ
Práce byla podpořena MZ ČR – RVO, FN v Motole 00064203 a PRVOUK-P27/LF1/1
Adresa pro korespondenci:
MUDr. Aleš Vícha, PhD.
Klinika dětské hematologie a onkologie 2. LF UK a FN Motol
V úvalu 84, Praha 5 - Motol, 150 08
tel.: +420-224436470, 6494, fax: +420-224436417
e-mail: ales.vicha@lfmotol.cuni.cz, avicha@yahoo.com
Sources
1. Delattre O, Zucman J, Melot T, et al. The Ewing family of tumors--a subgroup of small-round-cell tumors defined by specific chimeric transcripts. N Engl J Med 1994; 331 : 294-249.
2. Zoubek A, Pfleiderer C, Salzer-Kuntschik M, et al. Variability of EWS chimaeric transcripts in Ewing tumours: a comparison of clinical and molecular data. Br J Cancer 1994; 70 : 908-913.
3. Domingo-Fernandez R, Watters K, Piskareva O, et al. The role of genetic and epigenetic alterations in neuroblastoma disease pathogenesis. Pediatr Surg Int 2013; 29 : 101-119.
4. Imataka G, Arisaka O. Chromosome analysis using spectral karyotyping (SKY). Cell Biochem Biophys 2012; 62 : 13-17.
5. Kearney L. Multiplex-FISH (M-FISH): technique, developments and applications. Cytogenet Genome Res 2006; 114 : 189-198.
6. Mackinnon RN, Chudoba I. The use of M-FISH and M-BAND to define chromosome abnormalities. Methods Mol Biol 2011; 730 : 203-218.
7. Kallioniemi A, Kallioniemi OP, Sudar D, et al. Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science 1992; 258 : 818-821.
8. Weiss MM, Hermsen MA, Meijer GA, et al. Comparative genomic hybridisation. Mol Pathol 1999; 52 : 243-251.
9. Solinas-Toldo S, Lampel S, Stilgenbauer S, et al. Matrix-based comparative genomic hybridization: biochips to screen for genomic imbalances. Genes Chromosomes Cancer 1997; 20 : 399-407.
10. Ylstra B, van den Ijssel P, Carvalho B, et al. BAC to the future! or oligonucleotides: a perspective for micro array comparative genomic hybridization (array CGH). Nucleic Acids Res 2006; 34 : 445-450.
11. Kirchhoff M, Gerdes T, Rose H, et al. Detection of chromosomal gains and losses in comparative genomic hybridization analysis based on standard reference intervals. Cytometry 1998; 31 : 163-173.
12. Lichter P, Joos S, Bentz M, et al. Comparative genomic hybridization: uses and limitations Semin Hematol 2000; 37 : 348-357.
13. Costa JL, Meijer G, Ylstra B, et al. Array comparative genomic hybridization copy number profiling: a new tool for translational research in solid malignancies. Semin Radiat Oncol 2008; 18 : 98-104.
14. Fiegler H, Gribble SM, Burford DC, et al. Array painting: a method for the rapid analysis of aberrant chromosomes using DNA microarrays. J Med Genet 2003; 40 : 664-670.
15. Nowak D, Hofmann WK, Koeffler HP. Genome-wide Mapping of Copy Number Variations Using SNP Arrays. Transfus Med Hemother 2009; 36 : 246-251.
16. Botstein D, Risch N. Discovering genotypes underlying human phenotypes: past successes for mendelian disease, future approaches for complex disease. Nat Genet 2003; 33 Suppl: 228-237.
17. Bacolod MD, Schemmann GS, Giardina SF, et al. Emerging paradigms in cancer genetics: some important findings from high-density single nucleotide polymorphism array studies. Cancer Res 2009; 69 : 723-727.
18. Taylor MD, Northcott PA, Korshunov A, et al. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol 2012; 123 : 465-472.
19. Van Mater D, Knelson EH, Kaiser-Rogers KA, et al. Neuroblastoma in a pediatric patient with a microduplication of 2p involving the MYCN locus. Am J Med Genet A 2013 161A(3): 605-610.
20. Brodeur GM, Maris JM, Yamashiro DJ, et al. Biology and genetics of human neuroblastomas. J Pediatr Hematol Oncol 1997; 19 : 93-101.
21. Schleiermacher G, Janoueix-Lerosey I, Ribeiro A, et al. Accumulation of segmental alterations determines progression in neuroblastoma. J Clin Oncol 2010; 28 : 3122-3130.
22. Schleiermacher G, Michon J, Ribeiro A, et al. Segmental chromosomal alterations lead to a higher risk of relapse in infants with MYCN-non-amplified localised unresectable/disseminated neuroblastoma (a SIOPEN collaborative study). Br J Cancer 2011; 105 : 1940-1948.
23. Matthay KK, Reynolds CP, Seeger RC, et al. Long-term results for children with high-risk neuroblastoma treated on a randomized trial of myeloablative therapy followed by 13-cis-retinoic acid: a children‘s oncology group study. J Clin Oncol 2009; 27 : 1007-1013.
24. Matthay KK, Villablanca JG, Seeger RC, et al. Treatment of high-risk neuroblastoma with intensive chemotherapy, radiotherapy, autologous bone marrow transplantation, and 13-cis-retinoic acid. Children‘s Cancer Group. N Engl J Med 1999; 341 : 1165-1173.
25. Parsons K, Bernhardt B, Strickland B Targeted immunotherapy for high-risk neuroblastoma--the role of monoclonal antibodies. Ann Pharmacother 2013; 47 : 210-218.
26. Gunn S, Reveles X, Weldon K, et al. Molecular cytogenetics as a clinical test for prognostic and predictive biomarkers in newly diagnosed ovarian cancer. J Ovarian Res 2013; 6 : 2.
27. Sapkota Y, Ghosh S, Lai R, et al. Germline DNA copy number aberrations identified as potential prognostic factors for breast cancer recurrence. PLoS One 2013; 8: e53850.
28. Shi ZZ, Zhang YM, Shang L, et al. Genomic profiling of rectal adenoma and carcinoma by array-based comparative genomic hybridization. BMC Med Genomics 2012; 5 : 52.
29. Vainio P, Wolf M, Edgren H, et al. Integrative genomic, transcriptomic, and RNAi analysis indicates a potential oncogenic role for FAM110B in castration-resistant prostate cancer. Prostate 2012; 72 : 789-802.
30. Swarts DR, Claessen SM, Jonkers YM, et al. Deletions of 11q22.3-q25 are associated with atypical lung carcinoids and poor clinical outcome. Am J Pathol 2011; 179 : 1129-1137.
Labels
Anatomical pathology Forensic medical examiner ToxicologyArticle was published in
Czecho-Slovak Pathology

2014 Issue 1
Most read in this issue
- Lynchův syndrom v rukách patologa
- Tkáňové kultury
- Průkaz chromozomálních změn u nádorových onemocnění pomocí CGH, array-CGH a SNP array
- Uterine tumors resembling ovarian sex cord tumors (UTROSCT) - popis případu s metastázou do lymfatické uzliny