
Idiopatická plicní fibróza - problematika multidisciplinární diagnostiky a léčby ve světle nových poznatků
Idiopatic pulmonary fibrosis – news in multidisciplinary diagnostic and therapeutic approaches
Idiopathic pulmonary fibrosis (IPF) is a primary fibrosing pulmonary process. Due to the ineffectiveness of current therapeutic strategies and unfavorable prognosis, IPF is the most serious example of idiopathic interstitial lung diseases (ILD). Etiology and pathogenesis of this disorder have not been fully clarified yet; but it is anticipated, that the fibroproliferation is caused by the imbalance of reparative and immunologic processes in the genetically predisposed patients. Radiologically and histopathologically, IPF is characterized by specific pattern called usual interstitial pneumonia (UIP), however, this pattern is not fully typical in all cases, and, moreover, it could be seen in other ILD´s, e.g. chronic hypersensitivity pneumonitis, asbestosis, autoimmune connective tissue diseases and many others as well. The final diagnosis of IPF is a consensual result of multidisciplinary team composed of pulmologist, pathologist and radiologist. IPF was an incurable disease with prognosis worse than cancer till the year 2011, when antifibrotic drugs decelerating a progression of this disease have been introduced. Earlier and correct diagnosis of IPF is the most important issue for the patients because they could be effectively treated and thus, prolonging their survival as much as possible.
Keywords:
fibrosis – pulmonary – histopathology – diagnosis
Authors:
Martina Vašáková 1; Radoslav Matěj 2,3
Authors‘ workplace:
Pneumologická klinika 1. LF UK, Thomayerova nemocnice, Praha
1; Oddělení patologie a molekulární medicíny, Thomayerova nemocnice, Praha
2; Ústav patologie 1. LF UK a VFN, Praha
3
Published in:
Čes.-slov. Patol., 52, 2016, No. 2, p. 85-92
Category:
Idiopatická plicní fibróza (IPF) je primárně fibrotizujícím plicním procesem a je vzhledem k refrakternosti na dosud známou léčbu a špatné prognóze nejzávažnějším reprezentantem idiopatických intersticiálních pneumonií (IIP). Etiologie a patogeneze onemocnění není zatím plně objasněna, ale předpokládá se, že příčinou fibroproliferace jako odpovědi na neznámý inzult může být nerovnováha reparačních a imunitních dějů u geneticky disponovaného jedince středního a staršího věku.
Overview
Idiopatická plicní fibróza (IPF) je primárně fibrotizujícím plicním procesem a je vzhledem k refrakternosti na dosud známou léčbu a špatné prognóze nejzávažnějším reprezentantem idiopatických intersticiálních pneumonií (IIP). Etiologie a patogeneze onemocnění není zatím plně objasněna, ale předpokládá se, že příčinou fibroproliferace jako odpovědi na neznámý inzult může být nerovnováha reparačních a imunitních dějů u geneticky disponovaného jedince středního a staršího věku. Radiologicky a histopatologicky je onemocnění charakterizováno obrazem obvyklé intersticiální pneumonie (usual interstitial pneumonia - UIP). Radiologický a histopatologický nález však nemusí být ve všech případech typický, navíc se obraz UIP může vyskytovat i v rámci jiných plicních postižení, např. u systémových nemocí pojiva, chronické exogenní alergické alveolitidy, azbestózy a dalších. Pro diagnózu IPF je tedy zcela zásadní konsensus multidisciplinárního týmu, klinika, radiologa a patologa. IPF byla do začátku našeho tisíciletí chorobou neléčitelnou s prognózou horší než řada karcinomů. Od roku 2011 však máme již dostupné antifibrotické léky, které dokáží zpomalit progresi nemoci. Důležité je tedy pacienty s IPF diagnostikovat správně a včas, abychom jim prodloužili život, jak nejvíce to jde.
Klíčová slova:
fibróza – plicní – histopatologie – diagnóza
Poprvé byla idiopatická plicní fibróza (IPF) popsána v roce 1892 Williamem Oslerem, který sledoval klinický průběh onemocnění a následně popsal i sekční nález u jednoho pacienta. V učebnicích tradovaný Hammanův-Richův syndrom, který je považován za historický záznam IPF u 5 pacientů s progredující dušností a difúzními infiltráty na skiagramu hrudníku, kteří zemřeli do 6 měsíců, odpovídal pravděpodobně spíše akutní intersticiální pneumonii (AIP) než IPF (1). Znalost této nemoci se vyvíjela velmi pozvolna, což bylo poznamenáno také tím, že nebyla známa žádná účinná léčba této nemoci a navíc nebyla ani jednoznačná klasifikace intersticiálních plicních procesů (IPP) a zvláště idiopatických intersticiálních pneumonií (IIP), která by jasně odlišila IPF od ostatních IIP. Koneckonců správná diagnóza až do začátku tohoto století znamenala stejně pouhé konstatování závažné nemoci s infaustní prognózou a s jedinou léčebnou možností, transplantací plic. Kortikoidy ani jiná imunosupresiva totiž nedokázala ovlivnit průběh IPF a pouze pacienty ohrožovala vedlejšími účinky. Vzhledem k tomu byla IPF nemocí opomíjenou lékaři i společností. Přelom nastal až s objevem antifibroticky působících léků, které dokáží ovlivnit fibroproliferativní proces u IPF a mění tak prognózu nemocných. Tím se dostává IPF po letech zasloužené pozornosti a její poznání, diagnostikovanost a léčitelnost se k potěše lékařů i pacientů rozvíjí a stoupá (2).
Definice
Idiopatická plicní fibróza (IPF) je definována jako specifická forma chronického fibrotizujícího intersticiálního plicního procesu dospělých nejasné etiologie s histologickým a radiologickým obrazem obvyklé intersticiální pneumonie (usual interstitial pneumonia, UIP). Vyskytuje se výhradně u dospělých, a to středního a staršího věku.
Epidemiologie
IPF postihuje asi 5 milionů lidí na celém světě. Prevalence této nemoci je celosvětově odhadována podle výsledků epidemiologických studií na 13 – 20/100 000 a incidence na 6,8 –16,3/100 000 obyvatel. Údaje americké a evropské se značně liší. V USA je dle klinických studií prevalence udávána 14 – 27,9 případů/100.000 obyvatel při užití volnějších kritérií, při užití přísnějších kritérií 42,7 – 63/100.000. Obdobně incidence je v USA udávána 16,3 – 17,4/100.000 při aplikaci volnějších kritérií a 6,8 – 8,8 při užití přísnějších kritérií. V Evropě je prevalence udávána 1,25 – 25,6/100.000 a incidence 0,22 – 7,56/100.000 obyvatel. IPF se vyskytuje s o něco vyšší četností u mužů než u žen. Incidence onemocnění stoupá s věkem. IPF pacienti jsou nejčastěji ve středním věku, v rozmezí od 45 do 70 let. Přibližně dvě třetiny pacientů jsou starší 60 let (3).
IPF se obvykle vyskytuje sporadicky, familiární případy jsou vzácné. Doposud byla objevena řada faktorů majících pravděpodobný vliv na vznik a vývoj IPF. V kontrolovaných studiích bylo prokázáno, že kouření je potenciálním rizikovým faktorem. Pravděpodobnost vzniku onemocnění stoupá s narůstajícím počtem let kouření. Mezi rizikové pro vznik IPF patří i expozice prachům obsahujícím ocel, mosaz, olovo a částice z borovicového dřeva, a to bez závislosti na tom, zda pacient je kuřákem či nikoliv. Riziko vzniku IPF pozitivně koreluje s dobou expozice. V některých studiích byl prokázán vliv profesní expozice antigenům zevního prostředí na vznik IPF. Jednalo se hlavně o expozici prachu z březového a tvrdého dřeva a expozici ptačím antigenům. Nicméně studie byla realizována na pacientech vybraných z registru uživatelů dlouhodobé domácí oxygenoterapie (DDOT) s diagnózou plicní fibróza, čili není jasné, zda šlo skutečně o IPF (4,5). Na patogenezi IPF má vliv také řada virových patogenů. U pacientů s IPF byla zjištěna vyšší incidence EBV, chřipkového viru, CMV a viru hepatitidy C. Další mikroorganismy, které by se mohly účastnit patogeneze IPF, jsou virus parainfluenzy, Human imunodeficiency virus-1 (HIV-1), virus spalniček, herpesviry a Mycoplasma spp. (6-8).
Etiopatogeneze IPF
Etiopatogeneze IPF není zatím zcela prozkoumaná, je však pravděpodobné, že podkladem nemoci je uniformní patologická odpověď plicní tkáně na různá infekční i neinfekční agens. Dříve se předpokládalo, že IPF vzniká jako reakce na zánětlivý proces, jenž přešel do chronického stadia. Podle posledních poznatků je ale zřejmé, že IPF vzniká pravděpodobně v nezánětlivém terénu jako odpověď na různé stimuly, jež způsobují opakované poškození výstelky plicních sklípků, vyúsťující v nekontrolovatelné a progredující jizvení. Zánětlivá reakce se někdy může vyskytnout až sekundárně. Alveolární makrofágy jsou u IPF prostřednictvím Th2 cytokinů pravděpodobně tzv. alternativně aktivovány a zvyšují pak produkci fibronektinu a tím indukují fibrogenezi ve fibroblastech. Na patologickém hojení alveolárních lézí se podílí nepochybně i genetické faktory a faktor stárnutí organismu, neboť IPF se nevyskytuje u jedinců pod 45 let. Z vyvolávajících faktorů, které způsobují léze alveolární výstelky, je důležité zmínit hlavně virové infekce (viz výše) a také gastroeofageální reflux, který je v subklinické podobě přítomen až u 90 % pacientů s IPF (9).
Přirozený průběh nemoci je u jednotlivých pacientů obtížně klinicky mapovatelný, neboť není možné klinické a radiologické nálezy v čase doložit opakovanými histologickými nálezy. Nicméně dle typického vzhledu radiologického obrazu na HRCT hrudníku je patrné, že IPF je časově značně heterogenní proces, s okrsky normální plicní tkáně, okrsky s aktivní fibrotizací a okrsky konečného stadia fibrózy s obrazem voštinovité plíce. Tradiční pohled na průběh IPF je založen na představě o pomalém poklesu plicních funkcí, vedoucím k respiračnímu selhání a smrti. Pravděpodobněji však vypadá teorie o mnohočetných inzultech způsobujících tzv. akutní exacerbace nemoci s rychlejším poklesem plicních funkcí.(10,11)
Jednotlivé součásti patogenetického procesu od alveolární léze až po fibrotizaci jsou přehledně rozděleny a popsány níže. Nicméně je třeba si uvědomit, že jednotlivé složky imunopatologického procesu v plicích probíhají ve stejnou dobu v různých částech plic simultánně a podílejí se tak na heterogennosti postižení u IPF.
Genetický podklad IPF
Na genetické dispozici k IPF se podílí zřejmě celá řada genů. Jsou to jednak geny, jejichž mutace způsobují familiární intersticiální pneumonie/fibrózy, které se mohou projevit již v dětství, a pak geny, které jsou vázány převážně se sporadickou formou dospělých. Z genů podílejících se jak na dětském typu plicní fibrózy, tak na sporadické IPF je nejčastěji mutován gen pro surfaktantový protein C (SP-C); 5 z 20 pacientů s familiární plicní fibrózou (FPF) jsou nositeli mutovaného genu. Mutace surfaktantových genů vede k produkci vadného surfaktantového proteinu, což vede k dysfunkci alveolárních epiteliálních buněk II. typu. Z dalších genů, jejichž mutace vede k poškození epiteliálních buněk, a to mechanismem jejich předčasného stárnutí, jsou geny pro telomerázy (hTERT, hTER). Mutace genů pro telomerázy se uplatňují hlavně u FPF. Mezi geny, jejichž polymorfismy se uplatňuje hlavně u sporadické IPF, je v poslední době nejdiskutovanějším gen pro mucin, a to jmenovitě jeho variace v promotorové oblasti MUC5B. V souvislosti s tím byla prokázána i vyšší exprese MUC5B v bioptických materiálech plic u pacientů s IPF. Genetické variace genu pro TOLLIP, které ovlivňují mechanismy vrozené imunity, také zřejmě zvyšují pravděpodobnost vzniku IPF (12-14).
Mnohočetné léze alveolární výstelky
Na začátku patogenetického procesu pravděpodobně dochází k mnohočetnému poškození alveolárního epitelu, kdy je obnažena bazální membrána (BM) a spuštěna kaskáda produkce cytokinů, chemokinů a enzymů, které se pak podílejí na dalším rozvoji a udržování procesu fibroprodukce a jizvení plicní tkáně. Samotná BM je procesem poškozena také, a tak dochází k proliferaci pneumocytů II. typu a endoteliálních buněk v terénu „nedokonalé“ extracelulární matrix (ECM) a nemůže tak dojít k restituci původní architektoniky alveolu. Dalším krokem pak je nábor („recruitment“) fibroblastů a myofibroblastů s další depozicí ECM a progrese k nevratné fibróze alveolu. Vyvíjejí se tzv. fibroblastické fokusy, které jsou navzájem mezi sebou spojeny pojivovou tkání a vytvářejí jakousi trojrozměrnou síťovitou strukturu, která vytlačuje a nahrazuje spojitou architektoniku respiračních lobulů v plicích. Množství fibroblastických fokusů v plicích pravděpodobně ovlivňuje prognózu pacientů s IPF (15,16).
Porucha oxidačně redukčních dějů
Oxidativní stres se v patogenezi IPF uplatňuje hlavně při vzniku iniciálních mikroskopických lézí alveolárního epitelu a podílí se na nemožnosti úspěšného zhojení těchto lézí bez významné fibroprodukce. Pravděpodobnou roli hraje nerovnováha mezi redukčními a oxidačními systémy v plicích, které zahrnují celou škálu mechanismů ke kontrole tvorby a odbourávání reaktivních kyslíkových radikálů (ROS). Navíc se zdá, že oxidačně redukční systémy v plicích přímo ovlivňují fibroprodukci cestou aktivace exprese TGF beta a naopak myofibroblasty diferencované pod vlivem TGF beta jsou samy zdrojem ROS (17,18).
Role koagulační kaskády
Koagulačni kaskáda hraje u IPF významnou roli v patologickém hojení alveolárních lézí. Koagulační systém je zřejmě aktivován při poranění alveolů zevní cestou, tkáňovým faktorem. U IPF je prokázána vyšší exprese aktivovaného faktoru X (Stuarta-Prowerové), který již přímo aktivuje protrombin. Vliv koagulačních proteáz na buněčnou signalizaci a aktivaci alveolárních buněk a makrofágů je zprostředkován přes proteinázou aktivované receptory (PAR). PAR je tedy zřejmě spojka mezi koagulační kaskádou spuštěnou zevním způsobem, tkáňovým faktorem, a fibroproliferativním hojením alveolárních lézí u IPF. Otazné je, proč tedy antikoagulační léčba warfarinem má negativní vliv na průběh IPF. Je to zřejmě tím, že warfarin ovlivňuje hlavně vnitřní koagulační kaskádu a neovlivňuje signalizaci přes PAR (19,20).
Nerovnováha proteolytických enzymů
Nerovnováha systému proteázy-antiproteázy může být jednou z příčin udržování bludného kruhu nekontrolované fibroprodukce. Hlavními reprezentanty proteázového-antiproteázového systému jsou matrix-metaloproteázy (MMP) a tkáňové inhibitory matrix-metaloproteáz (TIMP). Tradiční rolí MMP je štěpení komponent ECM, zejména kolagenu I, elastinu a lamininu. Navíc dokáží štěpit i substráty, které významně zasahují do buněčných dějů, jako je decorin a povrchově vázaný Fas ligand. Kromě toho MMP dokáží štěpením aktivovat v plicní tkáni růstové faktory, mj. i TGF beta. Z MMP se na patogenezi IPF podílejí zejména MMP-2, MMP-7 a MMP-9. Nejvýznamnější roli u IPF hraje MMP-7, která se pravděpodobně přímo účastní procesu fibroproliferace, jak bylo prokázáno ve studiích na tkáňových kulturách a in vivo pokusech na geneticky modifikovaných myších. Navíc je MMP-7 detegována ve zvýšeném množství v BALTe a tkáních u pacientů s IPF ve srovnání se zdravými kontrolami. Podobně je ale zvýšena v BALT i u pacientů s COP, což svědčí o přítomnosti dalších faktorů, které zasahují do procesu fibrogeneze, neboť COP má na rozdíl od IPF prognózu dobrou. Z TIMP se účastní v patogenezi IPF TIMP-1, TIMP-2, TIMP-3 a TIMP-4 (21).
Nerovnováha cytokinového spektra
Prozánětlivé cytokiny
TNF alfa spolu s IL-1 hrají roli spíše v iniciálních fázích poškození plicního parenchymu než u pokročilého onemocnění. TNF alfa stimuluje hlavně zánětlivý typ odpovědi, hraje roli v mezibuněčných adhezích a v transendoteliální migraci buněk a spouští časnou cytokinovou kaskádu vedoucí následně k procesu fibroproliferativního hojení. Má navíc zřejmě ambivalentní úlohu v patologickém hojení tkáňového poškození, neboť v experimentu na zvířatech dokáže vyvinout fenotyp plicního postižení s obrazem emfyzému spolu s fibrotickými změnami v plicním parenchymu (22).
Th2 cytokiny
Hlavními cytokiny, které určují vzorec patologického hojení plicní tkáně směrem k fibróze, jsou IL-4 a IL-13. Oba tyto cytokiny podporují růst fibroblastů a produkci kolagenu a zvyšují produkci TGF beta 2 lidskými bronchiálními epiteliemi. IL-13 navíc indukuje tvorbu řady tzv. CC chemokinů: MCP, MIP-1 alfa, MIP-1 beta, MIP-2, MIP-3 alfa, „thymus-and activation-regulated chemokine“, „thymus expressed chemokine“, eotaxinu a eotaxinu 2. Tyto chemokiny působí převážně přes receptor CCR2 a mají profibrotický účinek. Jedním z cytokinů, který patří též do Th2 spektra a patří mezi protizánětlivé cytokiny, je IL-10. Zvýšená exprese IL-10 v experimentu na myším modelu s křemíkem indukovanou plicní fibrózou způsobuje exacerbaci fibrotických lézí, zvyšuje zastoupení lymfocytů a IgG1 v BAL a zároveň zvyšuje i expresi IL-4 a IL-13 (23).
Th1 cytokiny
IFN-gama v experimentu dokáže tlumit vývoj bleomycinem indukované fibrózy u myší cestou regulace produkce TGF beta 1. U IPF je jeho regulační role potlačena, neboť alternativní aktivace makrofágů zvyšuje sekreci IL-13 a nikoli IFN-gama. IL-12 je naopak cytokinem působícím antifibroticky cestou indukce IFN-gama. V pokusu na myších byl totiž prokázán vliv IL-12 na zvýšení hladiny IFN gama v BALTe, což mělo protektivní vliv proti vzniku bleomycinem indukované plicní fibrózy u pokusných zvířat (24,25).
Profibrogenní cytokiny
Jedním z cytokinů, který hraje kruciální roli v indukci fibrogeneze, je TGF beta. Indukuje transkripci kolagenu typu I a fibronektinu ve fibroblastech. V endotelu navíc reguluje produkci a aktivitu růstového faktoru fibroblastů (FGF)-2, který má výrazný mitogenní vliv na pneumocyty II. typu. To může mít za následek poruchu fyziologické reepitalizace drobných alveolárních lézí s následným vznikem prefibrotických okrsků. TGF beta je exprimován převážně hyperplastickými alveolárními epiteliálními buňkami, a to spolu s IL-10, destičkovým růstovým faktorem (PDGF) a růstovým faktorem keratinocytů (KGF). Další ze zkoumaných faktorů fibrogeneze je vliv hepatocytového růstového faktoru – hepatocyte growth factor (HGF) – na fibroblasty. Produkce HGF z prekurzoru pro-HGF je regulována aktivátorem – serinovou proteázou (HGFA) – a inhibována specifickými inhibitory (HAI-1, HAI-2). Předpokládá se, že HGF má protektivní vliv proti vzniku plicní fibrózy, ale exprese HGFA je ve fibroblastech u jedinců s IPF snížena (18,26).
Porucha angiogeneze
Patogenetický proces u IPF je komplexní a přestavuje nejen plicní parenchym, ale i vaskulaturu. Za normálních podmínek jsou v plicích angiogenní a angiostatické stimuly v rovnováze. V případě IPF dochází k posunu této rovnováhy a aberantní angiogenezi. Hlavními faktory ovlivňujícími angiogenezi ve smyslu pozitivním jsou bazický fibroblastový růstový faktor (bFGF), vaskulární endoteliální růstový faktor (VEGF) a angiogenní CXC chemokiny obsahující motiv ELR (kyselina glutamová, leucin, arginin); ve smyslu negativním je angiogeneze ovlivněna angiostatinem a angiostatickými, interferon-inducibilními CXC chemokiny. Sekrece angiogenních CXC chemokinů je pozitivně stimulována lipopolysacharidy, TNF alfa a IL-1 beta; IFN-gama angiogenezi inhibuje. Exprese CXC chemokinů, angiogenního interleukinu-8 oproti angiostatickému IFN-gama inducibilnímu proteinu IP-10 je výrazně zvýšena v plicní tkáni nemocných s IPF ve prospěch angiogenního IL-8 oproti zdravým kontrolám a lokalizace zvýšené exprese IL-8 koreluje s nakupením fibroblastů a extracelulární matrix. Angiogenní chemokiny tedy pravděpodobně přímo zasahují do procesu fibrogeneze u IPF (5,27).
Vliv osy renin-angiotensin (RAS)
Aktivace RAS se velmi pravděpodobně účastní v patogenezi plicní fibrózy a plicní hypertenze, a to svým účinkem vazokonstrikčním, proliferačním a profibrogenním. V nedávné době byla objevena osa působící regulačně na RAS, zahrnující angiotensin konvertující enzym-2 a angiotensin 1–7 [ACE 2-Ang-(1–7)-Mas]. V pokusech na myších byl prokázán protektivní efekt ACE 2 jak na bleomycinem indukovanou plicní fibrózu, tak na monocrotalinem indukovanou plicní hypertenzi (28).
Původ fibroblastů a jejich „nesmrtelnost“
Existuje několik teorií o původu fibroblastů a myofibroblastů v plicní tkáni u pacientů s IPF. Jedna z nich předpokládá, že jde o rezidentní kmenové buňky, které se aktivují a proliferují v odpověď na poranění. Je možný i jejich původ z buněk alveolárního epitelu, které projdou takzvanou epitelo-mezenchymální transdiferenciací (EMT). Je pravděpodobné, že na procesu EMT se účastní přímo alveolární epitel, a to produkcí endotelinu-1, který způsobuje aktivací endotelinového receptoru A zvýšenou produkci TGF beta 1. Dalším možným zdrojem myofibroblastů jsou mesoteliální buňky, které mohou vcestovávat do plíce a vykazovat rysy myofibroblastické diferenciace. Svým způsobem by to vysvětlovalo i subpleurální predominanci změn u IPF. Některé teorie a studie však podporují spíše hypotézu o mimoplicním původu fibroblastů, které jsou původem z kostní dřeně, chovají se jako mezenchymální kmenové buňky a jsou chemotakticky atrahovány do míst tkáňového poranění, kde přispívají k fibrotizaci plicní tkáně. Je pravděpodobné, že fibroblasty u jedinců s IPF mají i sníženou schopnost apoptózy a jsou chráněny proti apoptóze mediované Fas receptory antiapoptotickými faktory ze skupiny inhibitorů apoptózy (IAP) (9,10,29).
Role senescence
V patogenezi sporadické IPF nepochybně hraje roli i stárnutí, neboť IPF se nevyskytuje u jedinců do 45 let věku. Vliv senescence lze vysvětlit ztrátou epiteliální integrity na podkladě opakovaných poranění epiteliálních buněk, sníženou schopností normální regenerace alveolárního epitelu, kumulací oxidativního stresu a sklonem k fibroproliferativnímu hojení ve stáří. Ztrátu schopnosti regenerace epitelu lze vysvětlit poruchou autofagie, která je důležitá pro normální obrat subcelulárních organel a proteinů, a dysfunkcí mitochondrií, které mohou mediovat apoptózu alveolárních epiteliálních buněk u IPF. Z genetických faktorů hraje zřejmě roli v patogeneze IPF instabilita mikrosatelitů DNA, ztráta heterozygozity a zkrácení telomer, dána mutací genů pro telomerázy. Mezi genetické faktory přispívající ke ztrátě integrity epitelu patří hlavně geny pro desmoplakin, katenin, kadherin asociovaný protein alfa 3 a dipeptidyl peptidáza 9. Význam má i imunosenescence, kdy s věkem klesá imunokompetence jedince, objevuje se zvýšené zastoupení CD4+CE8+ T lymfocytů a zvyšuje se tvorba autoprotilátek, které se pravděpodobně mohou také účastnit patogeneze plicní fibrózy. Svou úlohu jistě má i dlouhotrvající mechanické namáhání plicní tkáně (komprese, relaxace, střižní síly) při dechových exkurzích, kdy nejvíce namáhané části plíce - tedy partie v zadních kostofrenických úhlech - jsou fibrózou postiženy nejvíce (30-32).
Klinický obraz
IPF se klinicky se projevuje progredující námahovou a posléze klidovou dušností, snadnou unavitelností, kašlem a v pozdějších fázích při nastupující hypoxemii i cyanózou. I když je pro IPF typický pozvolný a plíživý nástup dušnosti a její pozvolná progrese, u některých pacientů se vyskytnou epizody tzv. akutní exacerbace IPF, kdy dojde k náhlému klinickému zhoršení s poklesem plicních funkcí a radiologickým obrazem tzv. mléčného skla svědčícím pro probíhající floridní alveolitidu. IPF má obvykle nevyhnutelně progredující průběh navzdory jakékoli léčbě a střední přežití pacientů není delší než 2,5 – 3 roky (2,33-35).
Fyzikální nález
U cca 75 % pacientů se vyskytují fenotypové projevy, jako jsou paličkovité prsty s nehty tvaru hodinového sklíčka a poslechový fenomén krepitu slyšitelný nad plicními bazemi.
Funkční vyšetření plic
Typickou funkční poruchou je u pacientů s IPF restrikční ventilační porucha (RVP) s poruchou difúzní kapacity a snížením plicní poddajnosti. Reziduální objem (RV) je u IPF obvykle zachován a poměr RV/TLC je často zvýšen. Funkce dýchacích cest je u IPP obvykle dobře zachována. Spirometrické hodnoty včetně usilovné vitální kapacity (FVC), usilovné vitální kapacity za 1 vteřinu (FEV1) a jejich poměru jsou typicky normální nebo je zachován jejich normální poměr i při absolutně redukovaných hodnotách. Vzácně se u pacientů s IPF setkáme i s obstrukční ventilační poruchou (OVP), a to u podskupiny nemocných s kombinací plicní rozedmy a plicní fibrózy. Difúzní kapacita je významně snížena a to ve větší míře, než by odpovídalo redukci TLC. Pro IPF je typické výrazné zhoršení dušnosti již při submaximální zátěži a je snížená vrcholová spotřeba kyslíku a ventilační rezerva.
Nedílnou součástí je i vyšetření krevních plynů, kdy je u pacientů s IPF v úvodu onemocnění patrný pokles saturace krve kyslíkem (SaO2) a parciálního tlaku kyslíku (paO2) v tepenné krvi pouze při zátěži a teprve s progresí onemocnění dochází i ke klidové hypoxemii. Dobrou informaci o funkčním stavu pacienta nám dá šestiminutový test chůzí (6-MWT), který je také nedílnou součástí vyšetření pro případnou indikaci přidělení kapalného kyslíku (2,33-35).
Zobrazovací metody
Skiagram hrudníku
V časném stadiu se zjišťuje jemná oboustranná retikulace, zvláště v dolních plicních polích a v axilárních částech plic. V této fázi může být nemocný asymptomatický, ale jsou i případy, kdy i při respiračních potížích je snímek hrudníku ještě normální. S postupujícím onemocněním se zvýrazňuje retikulace až retikulonodulace s cystami 2 – 20 mm a s obrazem voštinovité plíce. Současně dochází i ke zmenšení objemu plic s vyšším uložením bránice. Bývá i mírné zvětšení nitrohrudních uzlin. Ojedinělým nálezem je pleurální výpotek.
CT hrudníku s vysokou rozlišovací schopností (HRCT)
HRCT je u IPF zásadním diagnostickým vyšetřením. IPF je charakterizována dle HRCT obrazem obvyklé intersticiální pneumonie (UIP). Podmiňuje subpleurální a bazální retikulaci s tlustostěnnými cystami různé velikosti a voštinovitou plíci, která bývá u více než 90 % nemocných. Častým nálezem jsou trakční bronchiektazie, popřípadě i bronchioloektazie. Trakční bronchiektazie jsou jedním ze základních předpokladů pro stanovení diagnózy IPF. Mezi cystami je fibrózní tkáň, vlivem retrakce dochází ke zmenšení objemu plic. Vyznačena je obvykle distorze plicní architektoniky. Opacita mléčného skla je nespecifickým nálezem, může odpovídat fibróze intersticia uvnitř sekundárních lobulů, ale může jít i o příznak akcelerující fáze onemocnění nebo o pneumonii. Rozlišení umožní porovnání nálezů s dřívějším HRCT (33-37).
Bronchoalveolární laváž
V případě IPF má BAL význam pouze diferenciálně diagnostický. Pro IPF je typické zmnožení granulocytů obvykle s malou příměsí eozinofilů, lymfocyty bývají zvýšeny minimálně. Výrazně zvýšené lymfocyty - nad 30 % - již indikují, že by se mohlo jednat o jinou diagnózu, navzdory tomu, že HRCT nález odpovídá UIP. V tomto případě bychom měli vždy pečlivým pátráním vyloučit hlavně exogenní alergickou alveolitidu a systémové nemoci pojiva. Co se týče vyšetření povrchových markerů exprimovaných na lymfocytech a makrofázích, žádný z nich zatím nemá prokázaný význam pro diagnostiku IPF, mají význam pouze diferenciálně diagnostický (2,33-35).
Plicní biopsie
Otázka, do jaké míry jsou plicní biopsie a její histopatologická interpretace nutné pro definitivní stanovení diagnózy IPF, byla opakovaně diskutována. Chirurgická biopsie, převážně videothorakoskopickou cestou, byla považována dlouho za zlatý standard diagnostiky IPP, IPF nevyjímaje. Změnu postoje k nutnosti podpořit diagnózu IPF chirurgickou plicní biopsií přinesl konsensus Americké hrudní společnosti, Evropské respirační společnosti, Japonské respirační společnosti a Hrudní asociace Latinské Ameriky (ETS/ERSJRS/ALAT) o diagnostice a léčbě IPF z roku 2011 (33-35). V zásadě pokud je klinický obraz kompatibilní s IPF, HRCT nález odpovídá UIP a vyloučili jsme exogenní příčiny IPP a systémové nemoci pojiva, není plicní biopsie indikována. Je třeba si navíc uvědomit, že chirurgická plicní biopsie u pacientů s IPF starších 65 let a s transfer faktorem nižším než 35 % je zatížena signifikantní mortalitou a měli bychom se jí tedy pokud možno vyhnout. Nově přináší bezpečnější alternativu získání vzorku plicní tkáně pro diagnostiku IPF transbronchiání kryobiopsie, která má slušnou výtěžnost a pravděpodobně menší riziko akutní excarebace spojené s výkonem (38).
Histopatologický nález
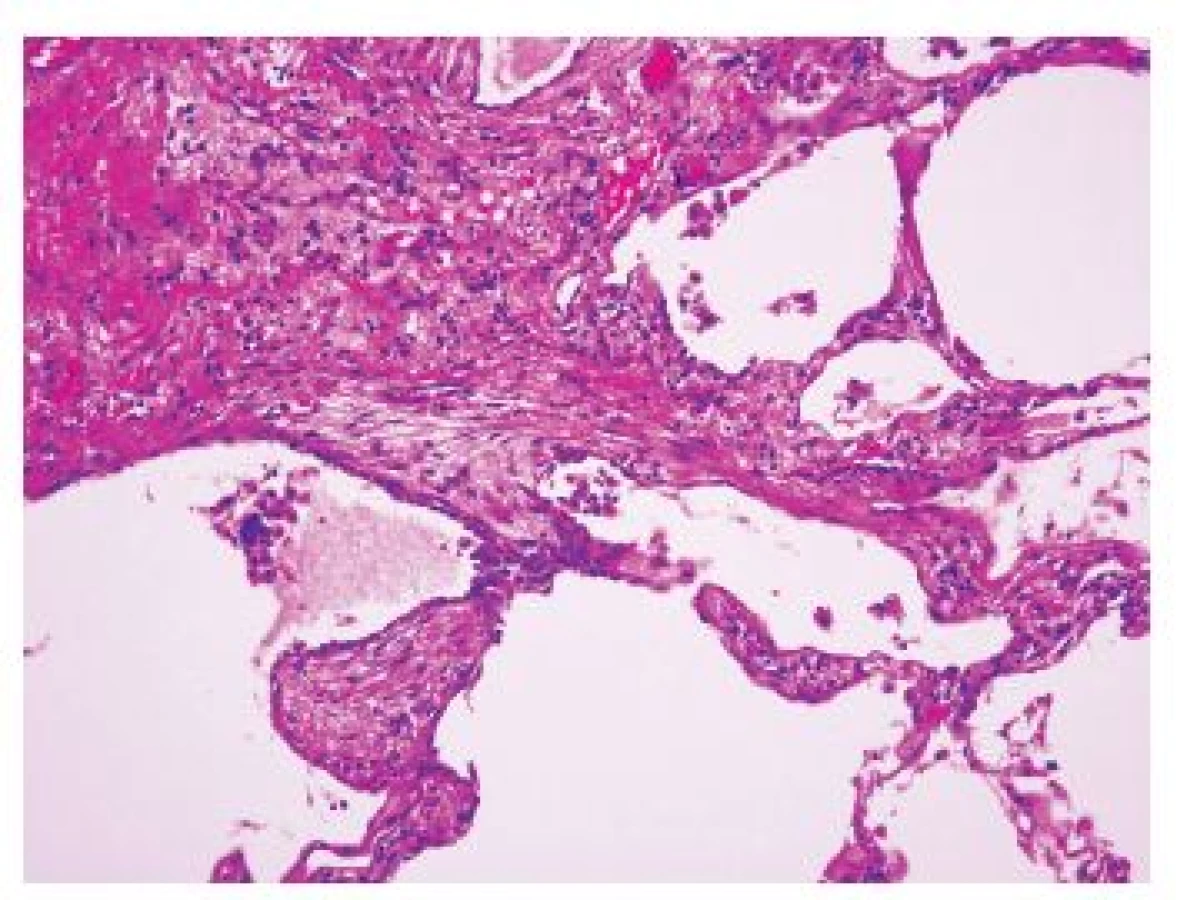
Základním histomorfologickým rysem při malém zvětšení je ložiskové postižení plicního parenchymu. Oblasti postižené chronickým intersticiálním zánětlivým procesem a fibrózou, v níž jsou přítomná fibroblastická ložiska, se střídají s částmi relativně normálního nepostiženého plicního parenchymu. Fibroticky změněná plicní tkáň je prostoupena poměrně dobře formovanou fibrózou, která je tvořena jen velmi málo buněčným, hyalinně transformovaným hustým kolagenním pojivem. Bezprostředně na tuto starou fibrotizaci však navazují oblasti granulační tkáně s nakupením fibroblastů s myxoidním stromatem (obr. 1). Právě výrazná proměnnost stáří fibrotických změn popisovaná jako „časová heterogenita“ je typická pro tento typ postižení. Fibroblastická ložiska reprezentují nejspíše cílová místa opakovaných postižení plicního parenchymu. Některé literární údaje uvádějí, že jejich počet koreluje s mortalitou a rychlejší progresí onemocnění, naopak přítomnost lymfoplasmocelulární zánětlivé infiltrace může mít lepší prognostický význam. Jiné literární odkazy však jednoznačnou souvislost mezi počtem fibroblastických ložisek a prognózou neprokázaly. I přesto, že jde o chronický zánětlivý proces, je lymfoplasmocelulární zánětlivá celulizace pouze nevýrazná. Nacházíme-li výraznější zánětlivou celulizaci či jsou přítomné dokonce lymfoidní folikuly, je vždy třeba myslet na postižení v rámci systémové nemoci pojiva.
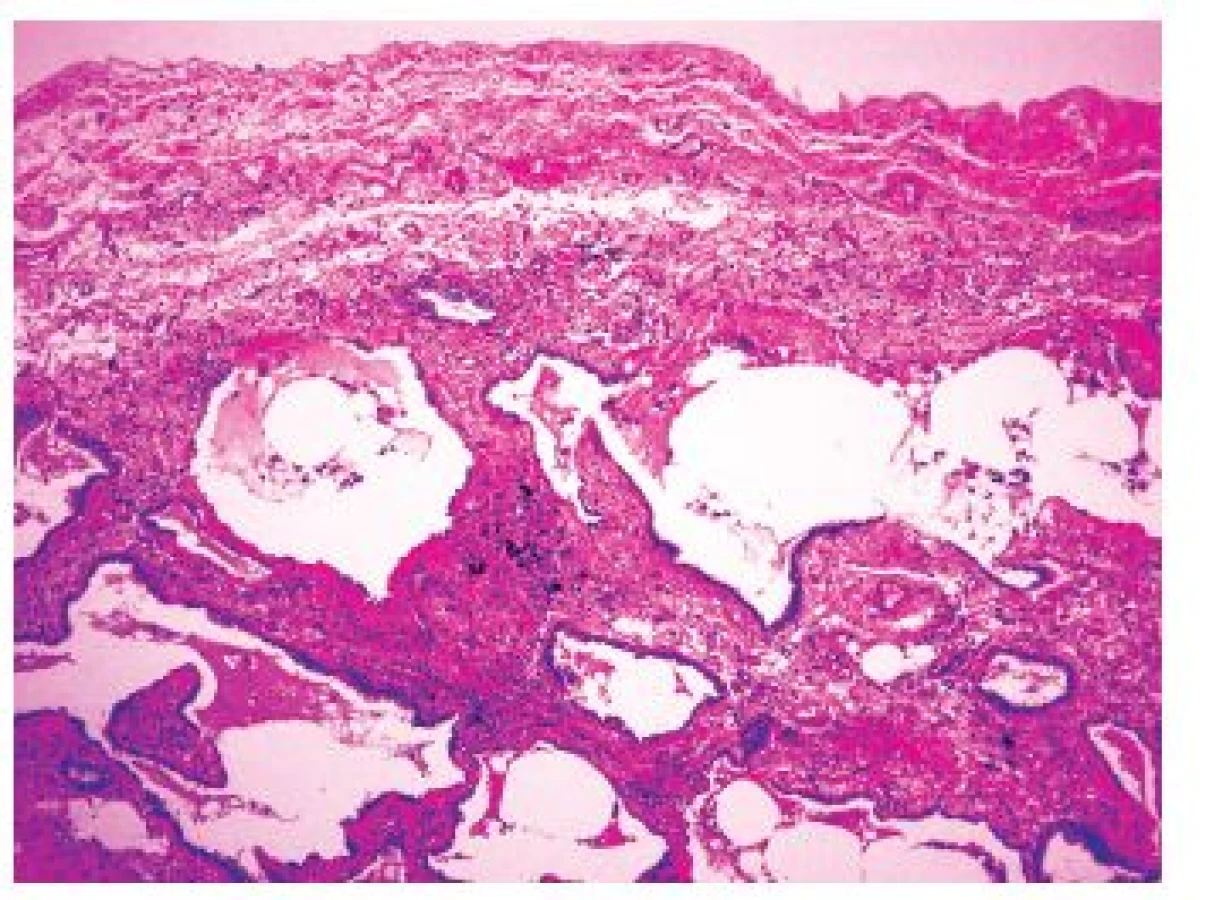
Když je proces již výrazně pokročilý, dochází ke ztrátě alveolární architektoniky s nápadnou remodelací tkáně, subpleurálně se formují poměrně velké cysty obklopené pruhy husté fibrotizace, pro které se používá historický název „voštinová plíce“ (obr 2.). Tato změna je sice poměrně charakteristická pro IPF-UIP, není však zcela specifická. Přítomnost takového obrazu postižení může zvýšit pravděpodobnost odlišení terminálního stadia UIP od NSIP, avšak i v případě NSIP můžeme v konečných stadiích nacházet obraz voštinové plíce. Je však zřejmé, že výskyt četných voštin svědčí spíše pro postižení v rámci IPF-UIP. Charakteristický obraz těchto změn je navíc důležitý z klinického hlediska pro chirurga provádějícího odběr plicní biopsie, protože by se měl právě oblastem voštin při odběru vyhnout. V těchto úsecích je totiž již vytvořen konečný obraz „end-stage“ postižení plicního parenchymu a odlišení UIP od NSIP či jiných typů IPP je velmi obtížné, či zcela nemožné. Kromě těchto změn jsou přítomny v rámci UIP-IPF další histopatologické změny mající jen omezenou diagnostickou výtěžnost, protože mohou být zastoupeny v různém stupni i v rámci dalších jednotek IPP.
Nejvýznamnější je zřejmě hyperplázie bronchiolárního epitelu, v níž mohou být různě rozsáhlé úseky skvamocelulární metaplázie či proliferace pohárkových buněk. Rovněž často nacházíme rozšířené cystické prostory obsahující mucin, buněčný detritus, cholesterolové hlatě a makrofágy či jiné zánětlivé elementy. Cévní stěny jsou někde ztluštělé se zřetelnou proliferací intimy. Reaktivní hyperplázie hladké svaloviny může dosahovat velmi nápadných rozměrů a v některých případech až odpovídá historickému termínu „svalová cirhóza“ plic. Původ hyperplastické hladké svaloviny je nejistý, zřejmě však zbytnělá svalovina pochází ze stěny bronchiolů a malých cévních struktur spíše než z myofibroblastické transformace mezenchymových elementů mladého kolagenního pojiva.
Do obrazu UIP nepatří nález výraznější zánětlivé celulizace, nápadného zánětu pleury a zejména přítomnost granulomatózních formací, které vždy musí budit podezření z postižení plicního parenchymu v rámci sarkoidózy, EAA či jiných onemocnění, jejichž obraz v konečném stádiu může být od UIP v rámci IPF téměř nerozlišitelný.
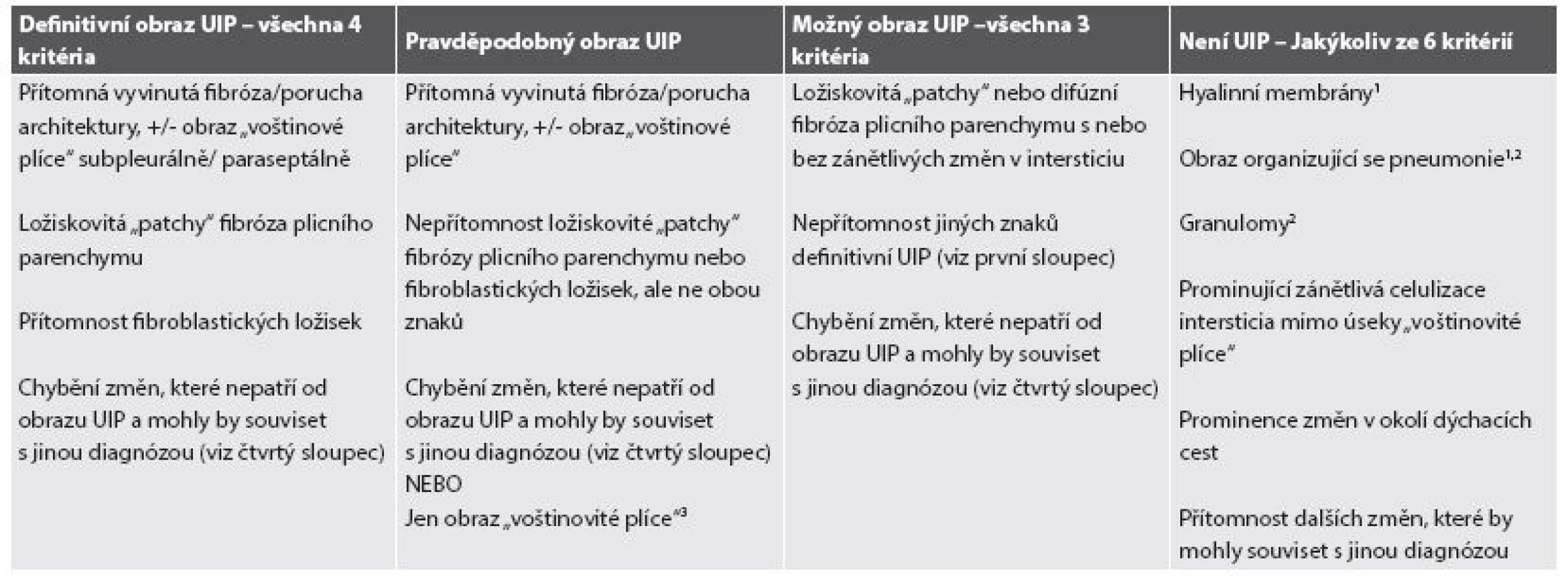
Souhrou inkluzivních a exkluzivních kriterií je možné stanovit histopatologickou diagnózu UIP na úrovni definitivní, pravděpodobné či možné, eventuálně diagnózu UIP vyloučit, což je rovněž zcela zásadní pro diagnózu intersticiálního plicního postižení. Přehled kriterií je shrnut v tabulce 1 (33-35,38-40).
Diagnóza
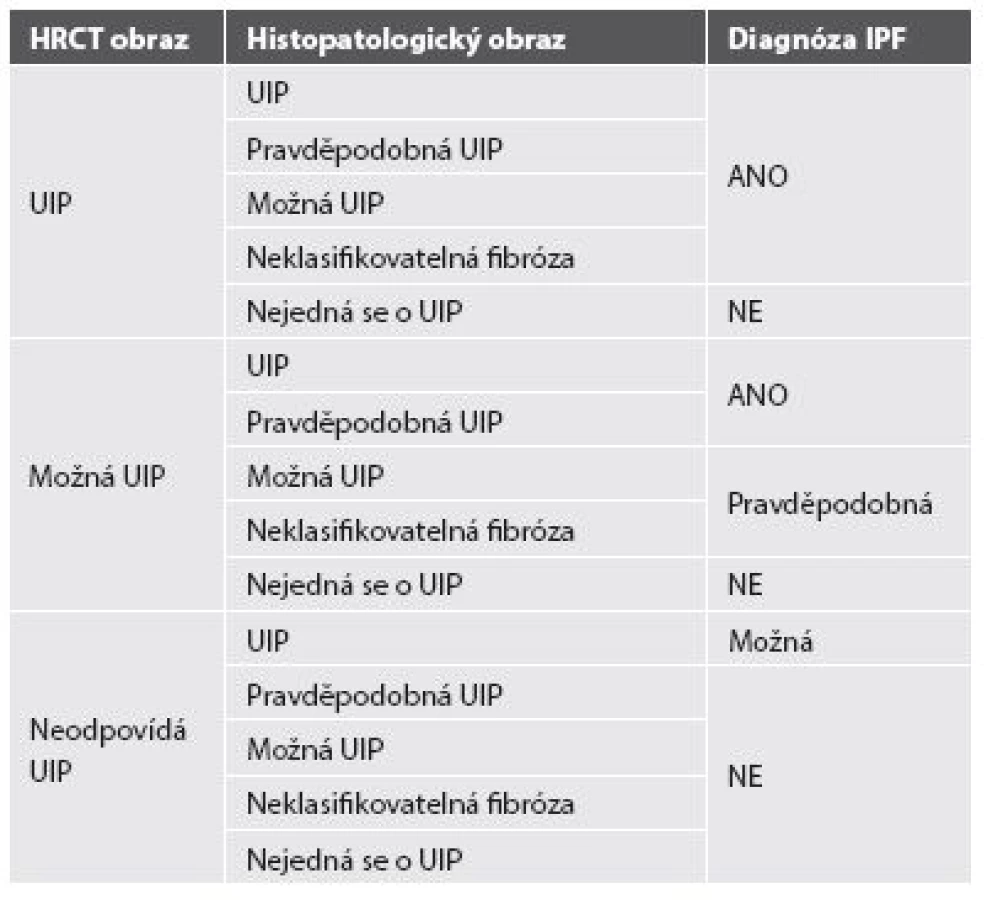
Pravidla pro diagnózu IPF se řídí konsensem Americké hrudní společnosti, Evropské respirační společnosti, Japonské respirační společnosti a Hrudní společnosti Latinské Ameriky (ATS/ERS/JRS/ALAT) z roku 2011. Diagnóza IPF by měla být stanovena na základě multidisciplinárního konsensu pneumologa, radiologa znalého problematiky intersticiálních plicních procesů a případně zkušeného pneumopatologa, je-li provedena plicní biopsie. Kruciální roli hraje v diagnóze IPF klinický obraz s nálezem námahové dušnosti, krepitu a paličkovitých prstů u pacienta středního a staršího věku s HRCT obrazem obvyklé intersticiální pneumonie - UIP. Plicní biopsie s histomorfologickým potvrzením diagnózy není striktně vyžadována u pacientů, kde klinický obraz a radiologický nález korespondují s diagnózou IPF, pouze v situacích, kdy je klinický a radiologický obraz nejednoznačný, je indikována chirurgická plicní biopsie, případně kryobiopsie. Nutné je vyloučení ostatních příčin (domácí a profesní expozice, systémové nemoci pojiva, léková toxicita), které by mohly vést k HRCT a histopatologickému obrazu UIP, a to především exogenní postižení plicního intersticia - exogenní alergické alveolitidy a azbestóza (anamnéza expozice anorganickým a organickým prachům) a systémové nemoci pojiva (podle mimoplicních příznaků a imunologických parametrů). Specifické kombinace HRCT a histopatologického (chirurgická plicní biopsie) UIP vzorce u pacientů s plicní biopsií uvádí tabulka 2 (33-35).
Diferenciální diagnóza
V rámci diferenciální diagnostiky je nejobtížnější odlišení fibrotické NSIP, chronické exogenní alergické alveolitidy, sarkoidózy IV. stadia a plicní fibrózy typu UIP v rámci systémové nemoci. V případě systémové nemoci pomůže obvykle při diagnóze nález mimoplicních známek postižení a typických autoprotilátek, při odlišení chronické EAA nám napoví anamnéza expozice, případně pozitivita specifických IgG proti podezřelému antigenu. V ostatních případech se většinou neobejdeme bez plicní biopsie, pro odlišení sarkoidózy IV. stadia by mohla stačit biopsie transbronchiální, jinak je indikována biopsie chirurgická (33-35).
Léčba
V průběhu posledních 10 let zaznamenal přístup nejen k diagnostice, ale i k léčbě IPF zcela zásadní změny. Původně byli pacienti léčení kortikoidy nebo kombinací kortikoidů s imunosupresivy, popřípadě ještě s přidáním N-acetylcysteinu ke kombinaci kortikoidů a azathioprinu. Kritickou metaanalýzou studií z posledních 10 let, která byla základem pro prohlášení světových respiračních společností o diagnostice a léčbě IPF vydaného v roce 2011 a posléze ještě na základě interim analýzy studie PANTHER z jara 2012, byly všechny tyto léčebné modality shledány neúčinné a dokonce v řadě případů způsobující vyšší mortalitu pacientů. U pacientů s IPF tedy není doporučena ani léčba kortikoidy, ani imunosupresivy, ani jejich kombinacemi. Navíc bylo zjištěno, že prognosticky nepříznivý efekt pro pacienty s IPF má i antikoagulační léčba. Novou cestou v léčbě IPF se stala léčba antifibrotická, která byla iniciována objevením antifibrotického účinku malé molekuly, pirfenidonu, jenž byl registrován k léčbě IPF nejprve v Japonsku a následně i v Evropě a nyní i v Kanadě a USA. Nový přístup k léčbě IPF byl poprvé uveden v konsensu ATS/ERS/JRS/ALAT v roce 2011 a následně aktualizován v doporučení pro klinickou praxi ATS/ERS/JRS/ALAT v roce 2015, kde je poprvé zakotveno doporučení antifibrotické léčby IPF (33-35,41).
Antifibrotická léčba IPF
Pirfenidon (Esbriet, Roche) je prvním lékem, který zasahuje přímo do patogeneze IPF, i když mechanismus jeho účinku není doposud v detailech objasněn. Nicméně na zvířecím modelu a buněčných kulturách je prokázáno, že pirfenidon snižuje proliferaci fibroblastů a produkci s fibrózou asociovaných proteinů a cytokinů pravděpodobně inhibicí TGF-beta a destičkového růstového faktoru (platelet-derived growth factor - PDGF), a to cestou blokády nukleární translokace proteinu smad. Nintedanib (Ofev, Boehringer Ingelheim) je trikinázový inhibitor (inhibice receptoru pro růstový faktor fibroblastů - fibroblast growth factor - FGF, vaskulární endoteliální růstový faktor - vascular endothelial growth factor - VEGF a PDGF), který prokazatelně snižuje pokles plicních funkcí u pacientů s IPF.
Klinické studie s novými antifibrotickými léky. V současné době probíhají další klinické studie zkoušející léky, které by měly přímo zasahovat do patogenetických pochodů IPF. Pacienty s IPF, obzvláště ty, kteří nesplní kritéria pro léčbu pirfenidonem, případně ani nintedanibem, je vhodné pokud možno do těchto studií zařazovat. Ze zkoušených potenciálních nových léků pro léčbu IPF je vhodné zmínit: monoklonální protilátku proti IL-13 lebrikizumab a inhibitor molekuly lysyl oxidase - like 2 ( LOXL-2) a pentraxin (33-35,41,42).
Ostatní léky
U pacientů s IPF obvykle podáváme ještě N-acetylcystein (NAC), i když nebyl prokázán jeho vliv na zabránění poklesu plicních funkcí ve studii PANTHER, a to pro jeho antioxidační efekt, s cílem předcházet dalšímu poškozování alveolárního epitelu oxidačními ději, a tím bránit tzv. akutním exacerbacím. Je to dobře tolerovaný lék, některým pacientům vadí pouze zvýšená expektorace.
Otazné je podávání inhibitorů protonové pumpy (PPI) z důvodu prevence asymptomatického refluxu. PPI totiž indukují CYP1A2 (viz výše) a tím snižují efektivitu léčby pirfenidonem a navíc nebyl prokázán efekt podávání PPI na průběh IPF (pokles plicních funkcí).
Dušnost u pacientů v terminálních stadiích IPF obvykle tlumíme opiáty, a to většinou v náplasťových formách s pomalým uvolňováním (33-35,41,42).
Nefarmakologická léčba IPF
Dlouhodobá domácí oxygenoterapie. V případě pokročilého onemocnění s hypoxemií indikujeme pacientům, kteří splní kritéria České pneumologické a ftizeologické společnosti, dlouhodobou domácí oxygenoterapii, a to buď koncentrátorem kyslíku, nebo kyslíkem kapalným, který je indikován pro pacienty vyžadující vysoké průtoky kyslíku a pro pacienty mobilní, kdy umožňuje rehabilitaci i pohyb mimo domácí prostředí s předplněnou lahví kapalného kyslíku.
Transplantace plic. Pro některé přísně selektované pacienty je vhodná transplantace plic, a to preferenčně obou.
Rehabilitace. Jednou z možností, jak zlepšit kvalitu života pacienta, je zlepšení jeho funkční výkonnosti a zmírnění dušnosti. Rehabilitace musí být v případě IPF komplexní, zahrnující učení, poradu a behaviorální techniky ke zlepšení sebeobsluhy, dále redukci symptomů a optimalizaci funkční kapacity.
Umělá plicní ventilace. Při respiračním selhání u pacientů s IPF, ať už při přirozené pozvolné progresi nemoci nebo při její akutní exacerbaci či při infekci, se nekloníme k umělé plicní ventilaci, neboť pro pacienty většinou není žádným přínosem, dochází k při ní naopak k ještě rozsáhlejšímu poškození plicní tkáně s následným úmrtím pacienta. Umělou plicní ventilaci bychom tedy měli indikovat pouze u těch pacientů s IPF, kteří jsou již zařazeni na čekací listinu transplantace plic, a mají tedy alespoň nějakou šanci se darované plíce dočkat (33-35,42,43).
Prognóza
Prognóza IPF je obecně špatná, IPF má obvykle nezvratitelně progredující průběh, nicméně se zavedením antifibrotické léčby pirfenidonem a nintedanibem dochází u léčených pacientů s IPF ke zmírnění poklesu plicních funkcí a předpokládáme tedy, že střední přežití se bude u těchto pacientů prodlužovat. Před érou antifibrotické léčby bylo střední přežití pacientů 2,5 - 3 roky. Jak pirfenidon, tak nintedanib také prokazatelně snižují mortalitu na IPF i mortalitu ze všech příčin.
U pacientů s IPF je ve zvýšené míře pozorován bronchogenní karcinom a ischemická choroba srdeční a žilní trombóza. Antikoagulační léčba z jakýchkoli důvodů významně zhoršuje prognózu pacientů s IPF, a to pravděpodobně nezávisle na diagnóze, pro kterou byla zavedena. Nicméně mechanismus trohoto dysefektu není přesně znám.
Prognóza pacientů s novou diagnózou IPF je do značné míry závislá na pokročilosti a rozsahu fibrózních změn, rychlosti progrese a přítomnosti akutní exacerbace. Také charakter HRCT změn ovlivňuje prognózu. Ti pacienti, kteří mají HRCT změny zcela typické pro IPF, mají nejhorší prognózu, ti, kteří mají obraz HRCT spíše podobný NSIP nebo necharakteristický, mají prognózu lepší (33-35,44,45).
PODĚKOVÁNÍ
Práce vznikla za částečné podpory projektů PRVOUK P37/11 a P27/LF1/1, OPPK CZ.2.16/3.1.00/24509 a BBMRI LM2010004.
PROHLÁŠENÍ
Autor práce prohlašuje, že v souvislosti s tématem, vznikem a publikací tohoto článku není ve střetu zájmů a vznik ani publikace článku nebyly podpořeny žádnou farmaceutickou firmou. Toto prohlášení se týká i všech spoluautorů.
Adresa pro korespondenci:
doc. MUDr. Radoslav Matěj, Ph.D.
Oddělení patologie a molekulární medicíny
Thomayerova nemocnice
Vídeňská 800,
14059 Praha 4 - Krč,
e-mail: radoslav.matej@ftn.cz
tel.: +420 261 083 741
Sources
1. Olson J, Colby TV, Elliott CG. Hamman-Rich syndrome revisited. Mayo Clin Proc. 1990; 65 : 1538–1548.
2. Maffessanti M, Dalpiaz G. Diffuse Lung Diseases. Clinical Features, Pathology, HRCT. Springer, 2006.
3. Nalysnyk L, Cid-Ruzafa J, Rotella P, Esser D. Incidence and prevalence of idiopathic pulmonary fibrosis: review of the literature. Eur Respir Rev 2012; (21)126 : 355-361.
4. Spagnolo P, Sverzellati N, Rossi G et al. Idiopathic pulmonary fibrosis: an update. Ann Med 2015; 47(1): 15-27.
5. Helling BA, Yang IV. Epigenetics in lung fibrosis: from pathobiology to treatment perspective. Curr Opin Pulm Med 2015; 21(5): 454-462.
6. Molyneaux PL, Maher TM. The role of infection in the pathogenesis of idiopathic pulmonary fibrosis. Eur Respir Rev 2013; 22(129): 376-381.
7. Molyneaux PL, Cox MJ, Willis-Owen SA et al. The role of bacteria in the pathogenesis and progression of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2014; 190(8): 906-913.
8. Folcik VA, Garofalo M, Coleman J et al. Idiopathic pulmonary fibrosis is strongly associated with productive infection by herpesvirus saimiri. Mod Pathol 2014; 27(6): 851-862.
9. Spagnolo P, Rossi G, Cavazza A et al. Pathogenesis of idiopathic pulmonary fibrosis and its clinical implications. Expert Rev Clin Immunol 2014; 10(8): 1005-1017.
10. Borensztajn K, Crestani B, Kolb M. Idiopathic pulmonary fibrosis: from epithelial injury to biomarkers--insights from the bench side. Respiration 2013; 86(6): 441-452.
11. Wuyts WA, Agostini C, Antoniou KM et al. The pathogenesis of pulmonary fibrosis: a moving target. Eur Respir J 2013; 41 : 1207-1218.
12. Noth I, Zhang Y, Ma SF et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: a genome-wide association study. Lancet Respir Med 2013; 1(4): 309-317.
13. Putman RK, Rosas IO, Hunninghake GM. Genetics and early detection in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2014; 189(7): 770-778.
14. Hambly N Shimbori C, Kolb M. Molecular classification of idiopathic pulmonary fibrosis: personalized medicine, genetics and biomarkers. Respirology 2015; 20(7): 1010-1022.
15. Habiel DM, Hogaboam C. Heterogeneity in fibroblast proliferation and survival in idiopathic pulmonary fibrosis. Front Pharmacol 2014; 5 : 2.
16. Ley B. Brown KK, Collard HR. Molecular biomarkers in idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 2014; 307(9): 681-691.
17. Fernandez IE, Eickelberg O. The impact of TGF-β on lung fibrosis: from targeting to biomarkers. Proc Am Thorac Soc 2012; 9(3): 111-116.
18. Gu H, Mickler EA, Cummings OW, et al. Crosstalk between TGF-β1 and complement activation augments epithelial injury in pulmonary fibrosis. FASEB J 2014; 28(10): 4223-4234.
19. Vasakova M, Sterclova M, Matej R et al. IL-4 polymorphisms, HRCT score and lung tissue markers in idiopathic pulmonary fibrosis. Hum Immunol 2013; 74(10): 1346-1351.
20. Nishioka Y, Azuma M, Kishi M, Aono Y. Targeting platelet-derived growth factor as a therapeutic approach in pulmonary fibrosis. J Med Invest 2013; 60(3-4): 175-183.
21. Thannickal VJ, Henke CA, Horowitz JC et al. Matrix biology of idiopathic pulmonary fibrosis: a workshop report of the national heart, lung, and blood institute. Am J Pathol 2014; 184(6): 1643-1651.
22. Redente EF, Keith RC, Janssen W et al. Tumor necrosis factor-α accelerates the resolution of established pulmonary fibrosis in mice by targeting profibrotic lung macrophages. Am J Respir Cell Mol Biol 2014; 50(4): 825-837.
23. Jakubzick C, Choi ES, Carpenter KJ et al. Human pulmonary fibroblasts exhibit altered interleukin-4 and interleukin-13 receptor subunit expression in idiopathic interstitial pneumonia. Am J Pathol 2004; 164(6): 1989-2001.
24. Estany S, Vicens-Zygmunt V, Llatjós R et al. Lung fibrotic tenascin-C upregulation is associated with other extracellular matrix proteins and induced by TGFβ1. BMC Pulm Med 2014; 14 : 120.
25. Oruqaj G, Karnati S, Vijayan V et al. Compromised peroxisomes in idiopathic pulmonary fibrosis, a vicious cycle inducing a higher fibrotic response via TGF-β signaling. Proc Natl Acad Sci U S A 2015; 112(16): 2048-2057.
26. Marchand-Adam S, Fabre A, Mailleux AA et al. Defect of pro-hepatocyte growth factor activation by fibroblasts in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2006; 174(1): 58-66.
27. Hanumegowda C, Farkas L, Kolb M. Angiogenesis in pulmonary fibrosis: too much or not enough? Chest 2012; 142(1): 200-207.
28. Montes E, Ruiz V, Checa M et al. Renin is an angiotensin-independent profibrotic mediator: role in pulmonary fibrosis. Eur Respir J 2012; 39(1): 141-148.
29. Ajayi IO, Sisson TH, Higgins PD et al. X-linked inhibitor of apoptosis regulates lung fibroblast resistance to Fas-mediated apoptosis. Am J Respir Cell Mol Biol 2013; 49(1): 86-95.
30. Selman M, Pardo A. Revealing the pathogenic and aging-related mechanisms of the enigmatic idiopathic pulmonary fibrosis. An integral model. Am J Respir Crit Care Med 2014; 189 : 1161-1172.
31. Leung J, Cho Y, Lockey RF, Kolliputi N. The Role of Aging in Idiopathic Pulmonary Fibrosis. Lung 2015; 193(4): 605-610.
32. Yanai H, Shteinberg A, Porat Z et al. Cellular senescence-like features of lung fibroblasts derived from idiopathic pulmonary fibrosis patients. Aging (Albany NY) 2015; 7(9): 664-672.
33. Raghu G, Collard HR, Egan JJ et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011; 183(6): 788-824.
34. Travis WD, Costabel U, Hansell DM et al. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med 2013; 188(6): 733-748
35. Raghu G, Rochwerg B, Zhang Y et al. An official ATS/ERS/JRS/ALAT clinical practice guideline: treatment of idiopathic pulmonary fibrosis: executive summary. Am J Respir Crit Care Med 2015; 192(2): e3-19.
36. Gotway MB, Freemer MM, King TE Jr. Challenges in pulmonary fibrosis. The use of high resolution CT scanning of the lung for the evaluation of patients with idiopathic interstitial pneumonias. Thorax 2007; 62 : 546–553.
37. Stern EJ, Swensen JS, Kanne PJ. High-Resolution CT of the Chest. Lippincott Williams, Wilkins, 2009.
38. Tomassetti S, Wells AU, Costabel U et al. Bronchoscopic Lung Cryobiopsy Increases Diagnostic Confidence in the Multidisciplinary Diagnosis of Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med 2015; Nov 12. In press.
39. Katzenstein AL, Myers JL. Idiopathic pulmonary fibrosis: clinical relevance of pathologic classification. Am J Respir Crit Care Med 1998; 157 : 1301–1315.
40. Leslie KO. Historical perspective. A pathologic approach to the classification of idiopathic interstitial pneumonias. Chest 2005; 128(5): 513–519.
41. McGrath EE, Millar AB. Hot off the breath: triple therapy for idiopathic pulmonary fibrosis-hear the PANTHER roar. Thorax 2012; 67(2): 97-98.
42. Maher TM. Idiopathic pulmonary fibrosis: pathobiology of novel approaches to treatment. Clin Chest Med 2012; 33(1): 69-83.
43. Ferreira A, Garvey C, Connors GL, et al. Pulmonary rehabilitation in interstitial lung disease: benefits and predictors of response. Chest 2009; 135 : 442–447.
44. Mura M, Porretta MA, Bargagli E, et al. Predicting survival in newly diagnosed idiopathic pulmonary fibrosis: a 3-year prospective study. Eur Respir J 2012; 40 : 101-109.
45. Du Bois, RM. Albera C, Bradford WZ et al. 6-minute walk distance is an independent predictor of mortality in patients with idiopathic pulmonary fibrosis. Eur Respir J 2014; 43 : 1421-1429.
Labels
Anatomical pathology Pneumology and ftiseology General practitioner for adults Radiodiagnostics Forensic medical examiner ToxicologyArticle was published in
Czecho-Slovak Pathology

2016 Issue 2
- Advances in the Treatment of Myasthenia Gravis on the Horizon
- Memantine Eases Daily Life for Patients and Caregivers
- Memantine in Dementia Therapy – Current Findings and Possible Future Applications
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole vs. Tramadol in Postoperative Analgesia
Most read in this issue
- Diferenciální diagnostika granulomatózních procesů v plicích
- Intersticiální plicní onemocnění asociovaná s kouřením
- Idiopatická plicní fibróza - problematika multidisciplinární diagnostiky a léčby ve světle nových poznatků
- Histopatologické principy vyšetření intersticiálních plicních procesů