
Noonanovej syndróm z pohľadu fetopatológa
Noonan syndrome from a fetopathologist perspective
We present our experience with four cases of fetal autopsies with abnormal prenatal ultrasound findings and suspicion of Noonan syndrome. These were fetuses from the 17th to the 24th age of gestation (GA). In all cases, prenatal ultrasound examination recorded increased nuchal translucency (NT) and presence of lymphatic neck sacs. Some fetuses showed signs of fetal hydrops and polyhydramnion was found. Similar signs and congenital developmental defects were confirmed in the autopsy examination. These were primarily signs of developing fetal hydrops with increased nuchal edema, in some cases up to the character of cystic hygroma, pleural and abdominal effusions, congenital heart and kidney defects, skeletal defects and facial dysmorphism. A karyotype was examined in all cases without chromosome aneuploidy. The diagnosis of NS was confimed by subsequent genetic analysis of causal gene mutations (mainly PTPN11, KRAS, RAF 1,…). Our cases demonstrate a wide range of signs of prenatal presentation of this syndrome. Because of wide differential diagnosis, summarizing prenatal ultrasound findings, autopsy examination and molecular genetic testing is essential.
Keywords:
Noonan syndrome – prenatal testing – polyhydramnion – hydrops fetalis – RASopathies.
Authors:
Tatiana Stupková 1; Marta Ježová 2; Monika Matyášová 3; Pavel Vlašín 3
Authors‘ workplace:
Ústav patologie FN Brno
1, 2; Cytogenetická laboratoř Brno
3
Published in:
Čes.-slov. Patol., 55, 2019, No. 1, p. 48-52
Category:
Original Articles
Overview
Prezentujeme našu skúsenosť so štyrmi prípadmi pitiev plodov s abnormálnym prenatálnym sonografickým vyšetrením a suspekciou na Noonanovej syndróm (NS). Jednalo sa o pitvy plodov v rozmedzí 17. až 24. týždňa gestácie (t.g.). Prenatálne ultrazvukové vyšetrenie vo všetkých prípadoch zaznamenalo zvýšené hodnoty nuchálnej translucencie (NT) a lymfatické vaky na krku. Niektoré plody prejavovali známky fetálneho hydropsu a bol zistený polyhydramnion. Nekroptickými vyšetreniami sme potvrdili súbor podobných znakov a vrodených vývojových vad, ktoré spadajú do základných charakteristík NS. Jednalo sa predovšetkým o známky rozvíjajúceho sa fetálneho hydropsu s nuchálnym edémom, v niektorých prípadoch až charakteru cystického hygromu, pleurálne a abdominálne výpotky, vrodené vývojové vady srdca a obličiek, vady skeletu a faciálny dysmorfizmus. U všetkých plodov bol vyšetrený karyotyp, bez nálezu aneuploidií chromozómov. Následná genetická analýza mutácií kauzálnych génov (hlavne PTPN11, KRAS, RAF 1,...), túto diagnózu potvrdila. Prípady demonštrujú širokú škálu znakov prenatálnej prezentácie tohoto syndrómu. Z dôvodu rozsiahlosti diferenciálnej diagnózy, je sumarizácia prenatálneho skríningu, nekroptického nálezu a molekulárne genetického vyšetrenia nevyhnutná.
Klíčová slova:
Noonanovej syndróm – prenatálna diagnostika – polyhydramnion – fetálny hydrops – RASopathie.
Noonanovej syndróm (NS) bol prvýkrát popísaný doktorkou Jacquelin Noonanovou v roku 1963 (1). Udáva sa incidencia 1 : 1000 až 1 : 2500 živo narodených detí. K charakteristickým znakom patria predovšetkým nízký vzrast, typické črty tváre, vrodené vady srdca, abnormality skeletu, kryptorchizmus a známky oneskoreného vývoja. NS patrí do skupiny tzv. RASopathií, špecifickej skupiny vývojových vad vyvolaných germinálnymi mutáciami génov, kódujúcich proteíny participujúce v RAS/MAPK signalizačnej kaskáde, ktorá hraje základnú úlohu v kontrole bunkového cyklu (2).
Prípad č. 1
Na Ústave patológie Fakultnej nemocnice Brno (ÚPA FN Brno) bola vykonaná pitva plodu ženského pohlavia. Jednalo sa o tehotenstvo 32-ročnej sekundigravidy, ktoré bolo ukončené v 23. týždni gestácie (t.g.). Dôvodom bol patologický ultrazvukový nález v 13. t.g., kedy bola zistená zvýšená hodnota nuchálnej translucencie (NT) 3,2 mm a popisovaný obraz rozvíjajúcich sa lymfatických vakov na krku. Nosová kosť bola prítomná. Ultrazvukový skríning bol zopakovaný v 16., 19. a 23. t.g.. Nález v oblasti nuchálneho prejasnenia zostal stacionárny, ale stále zvýšený, pretrvávali lymfatické vaky na krku a bola zistená zdvojená pravá oblička s dilatovanou panvičkou. Kardiologické ultrazvukové vyšetrenie preukázalo miernu hypertrofiu ľavej srdcovej komory, perikardiálny výpotok a známky zvýšenej záťaže srdca. Ostatné biometrické parametre boli na úrovni normy a neboli zistené iné štrukturálne anomálie plodu. Vzhľadom k patologickému prenatálnemu ultrazvukovému nálezu a zvýšenému riziku chromozomálnej aberácie, pacientka podstúpila amniocentézu so stanovením karyotypu plodu. Súčasne sa rodičia rozhodli pre prerušenie tehotenstva z genetickej indikácie. V rode matky i otca nebola zistená žiadna genetická záťaž.
Pitva plodu, v zhode s prenatálnym ultrazvukom, preukázala známky rozvíjajúceho sa hydropsu s výpotkami v dutine brušnej, hrudnej a difúznym presiaknutím mäkkých tkanív. Bola potvrdená dilatácia obličkovej panvičky vľavo, zdvojenie obličky vpravo s dilatáciou panvičky a zdojeným močovodom. Ďalej bola vyslovená suspekcia na agenéziu venózneho duktu (obr. 1, tab. 1).
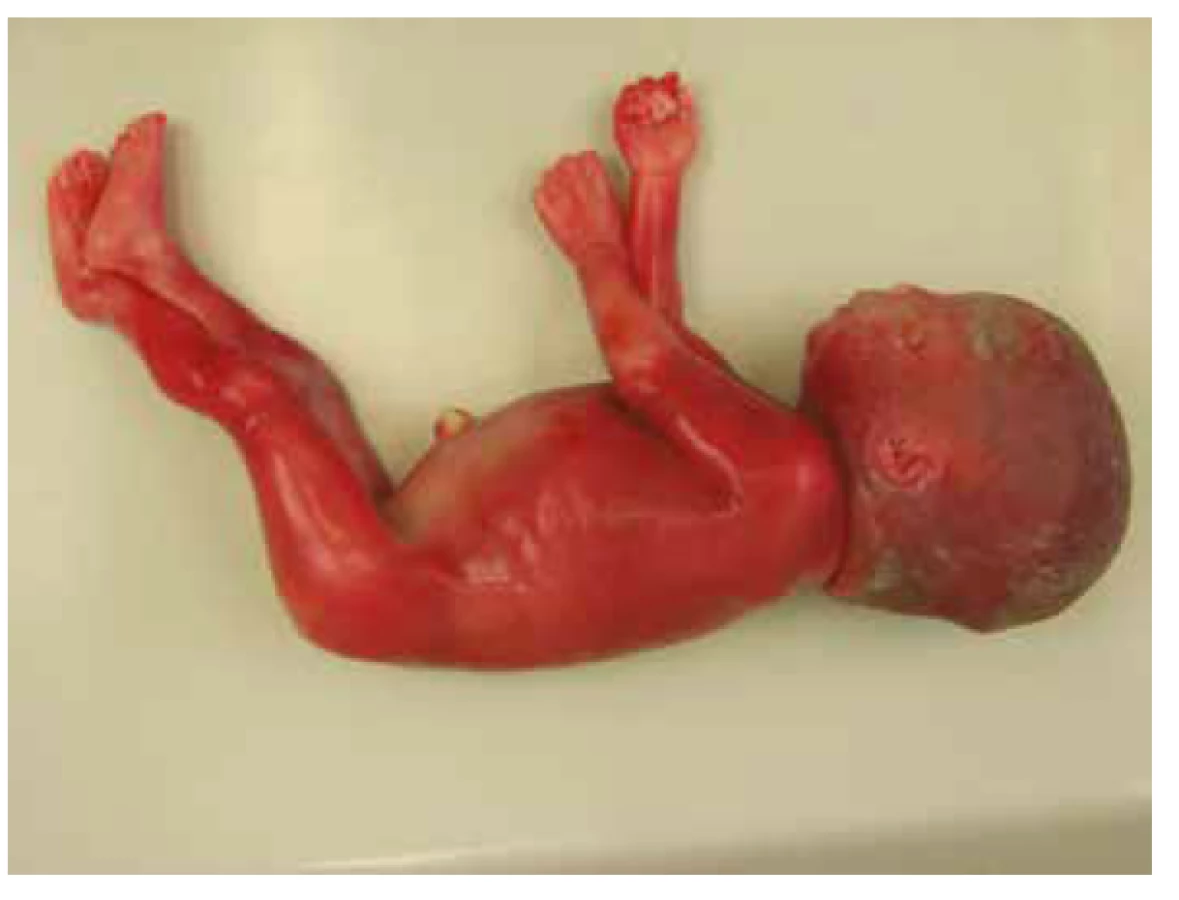
Molekulárne genetickým vyšetrením nebola potvrdená aneuploidia chromozómov 13, 18, 21, X a Y, karyotyp plodu bol normálny, ženský 46,XX. DNA analýza u plodu preukázala mutáciu génu PTPN11 – c853T>C; p.Phe285Leu (c.[853T>C];[853T=]). Vzhľadom k tomu, že rodičia vyjadrili nesúhlas so zverejnením výsledkov genetickej analýzy, nie je možné informáciu o tom, či sa jednalo o mutáciu vrodenú alebo de novo, publikovať.
Prípad č. 2
K prevedeniu pitvy bol na ÚPA FN Brno dodaný plod mužského pohlavia. Jednalo sa o graviditu po in vitro fertilizácii u 35-ročnej primigravidy. Tehotenstvo bolo ukončené v 17. t.g. Príčinou indukovaného potratu bol patologický nález na prenatálnom ultrazvukovom vyšetrení. V 13. t.g. toto vyšetrenie preukázalo cystický hygrom krku s hodnotou NT 9 mm. Nosová kosť bola prítomná. Opakovaný ultrazvuk v 16. t.g. potvrdil progresiu nálezu s nuchálnym prejasnením 12 mm, ďalším nálezom bola agenézia venózneho duktu s patologickým napojením pupočníkovej žily cez hepatálné žily a polydaktýlia pravej hornej a dolnej končatiny. Karyotyp plodu bol normálny mužský 46,XY, bez detekovaných aneuploidií chromozómov 13, 18, 21, X a Y. Anamnéza obidvoch rodičov nepreukázala žiadnu genetickú záťaž.
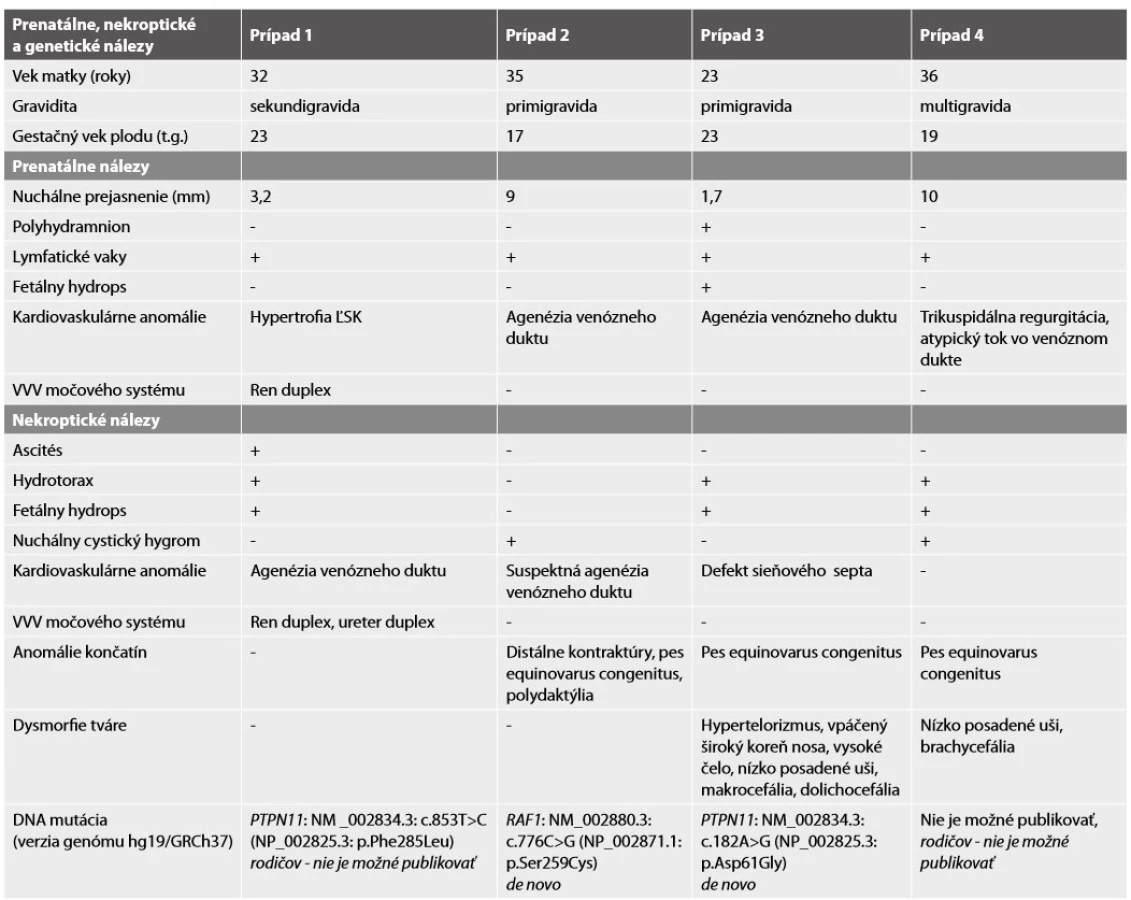
Pitva plodu potvrdila prítomnosť nuchálneho cystického hygromu, postaxiálnu polydaktýliu pravej hornej končatiny, agenéziu venózneho duktu a distálne kontraktúry končatín s flexiou horných končatín v zápestiach a pes equinovarus congenitus vľavo. Abnormálne prepojenie žil nebolo potvrdené (obr. 2). Na základe pitvy bolo vyslovené podozrenie na chromozomálnu aberáciu alebo neuromuskulárnu poruchu, artrogrypózu (tab. 1).
DNA analýza plodu potvrdila mutáciu génu RAF1 c.776C>G; p.Ser259Cys (c.[776C>G];[776C=]), rodičia neboli nosičmi tejto mutácie, jednalo sa o mutáciu de novo.
Prípad č. 3
K pitve, na ÚPA, bol dodaný plod ženského pohlavia. Tehotenstvo 23-ročnej primigravidy bolo ukončené v 24. t.g. Prenatálnym ultrazvukovým vyšetrením bola vyslovená suspekcia na NS vzhľadom na súbor zistených príznakov. Jednalo sa o prítomnosť polyhydramnionu, celkového hydropsu plodu a agenéziu venózneho duktu. Ultrazvukové vyšetrenia boli vykonané v 12., 16. a 23. t.g. s konštatovaním postupnej progresie nálezu. Genealógia rodov rodičov bola bez genetickej záťaže.
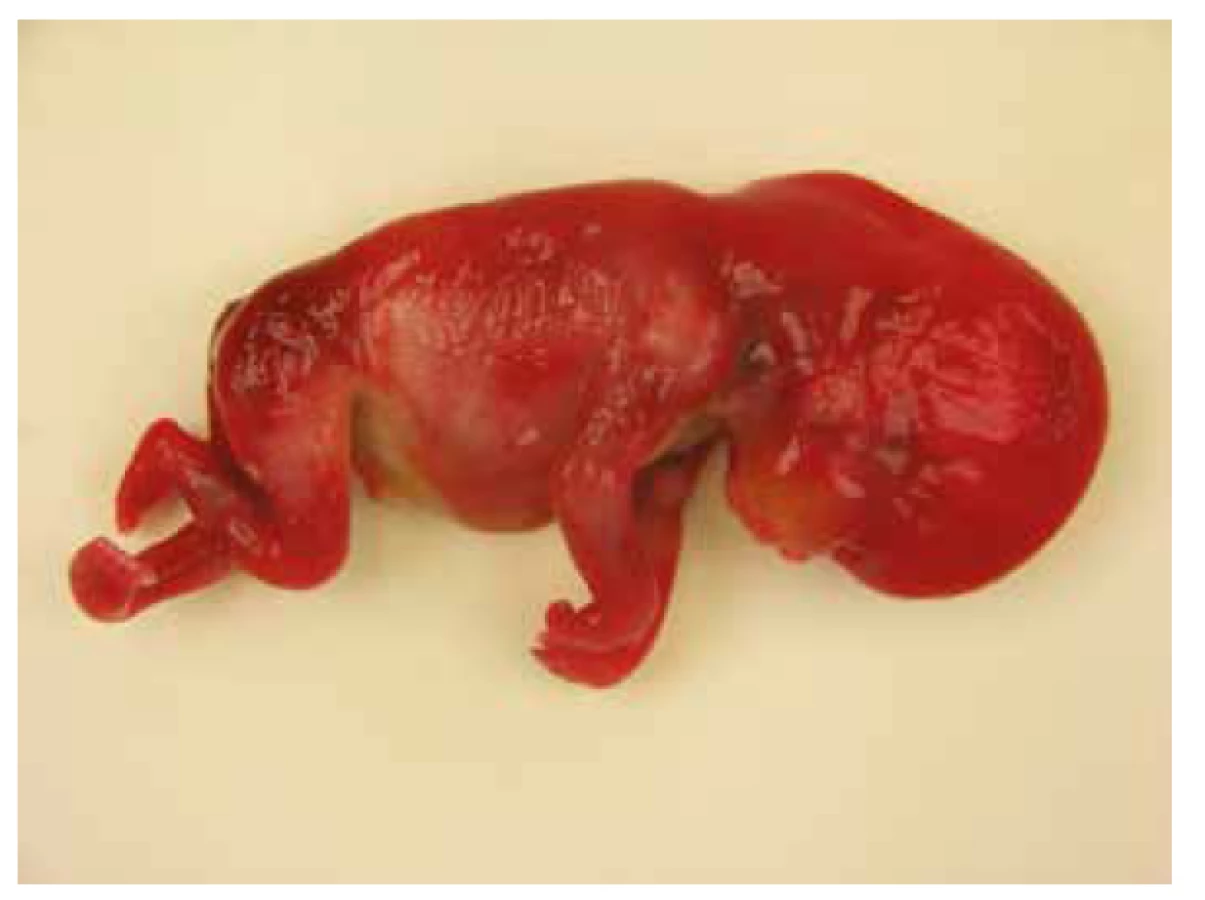
Nekropsia plodu, v zhode s prenatálnym ultrazvukom, potvrdila celkový hydrops plodu s akcentáciou v oblasti hlavy, masivný obojstranný hydrotorax s rozvojom pľúcnej hypoplázie. Bol prítomný dysmorfizmus tváre s hypertelorizmom, vysokým čelom, plochým nosom so širokým vpáčeným koreňom a nízko nasadajúcími ušami. Ďalej bola zistená relatívna makrocefália s dolichocefáliou. Súčasťou nálezu bola i vrodená vývojová vada srdca s defektom sieňového septa typu fossa ovalis a pridruženou agenéziou venózneho duktu. Pitvou bola zistená hepatomegália a pes equinovarus congenitus s obojstranným postihnutím (obr. 3, obr. 4, tab. 1).

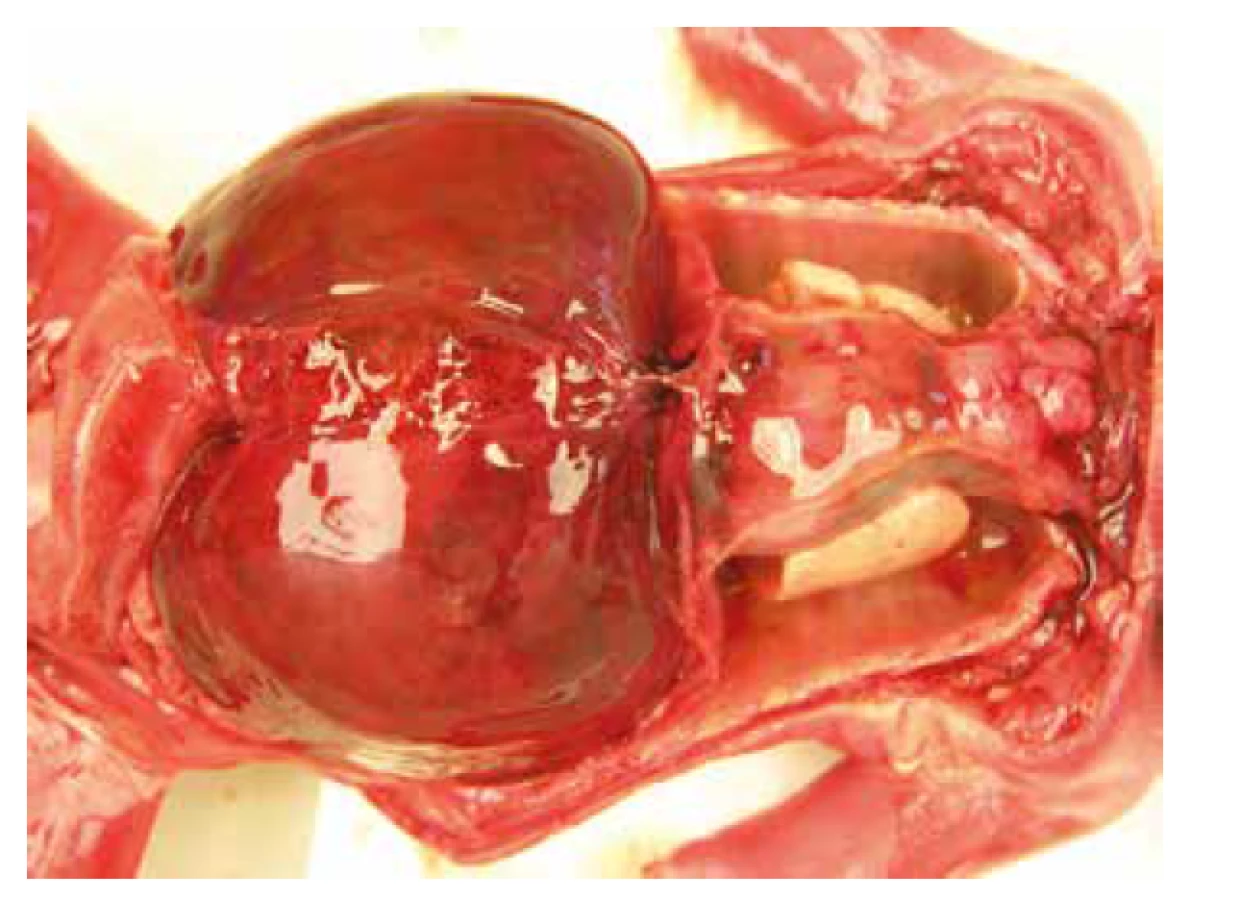
DNA analýza u plodu potvrdila mutáciu génu PTPN11 c.182A>G; p.Asp61Gly (c.[182A>G];[182A=]. Rodičia neboli nosičmi tejto mutácie, jednalo sa o mutáciu de novo.
Prípad č. 4
K nekroptickému vyšetreniu na ÚPA bol dodaný plod mužského pohlavia odpovedajúci biometrickými parametrami 19. týždňu gestačného veku. Matka 36 rokov, multigravida, podstúpila prenatálne ultrazvukové vyšetrenie v 13., 14., 15. a 17. gestačnom týždni, kvôli postupne sa zvyšujúcej hodnote nuchálneho prejasnenia. NT dosahovala hodnôt od 5–10 mm, súčasne bol zistený atypický tok v ductus venosus a výrazná trikuspidálna regurgitácia. Osobná anamnéza matky obsahovala informáciu o užívaní tetracyklinového antibiotika v perikoncepčnom období.
Nekroptické vyšetrenie potvrdilo výrazný nuchálny edém s náznakom cystického hygromu, výpotky v hrudných dutinách s tendenciou vývoja k celkovému hydropsu plodu. Bola zistená tzv. koňská noha a mierna stigmatizácia tváre s nižšie umiestnenými ušami (tab. 1). Venózny duktus nebol z dôvodu malej veľkosti plodu vyšetrený. Karyotyp plodu bol normálny mužský 46,XY, bez aneuploidií chromozómov 13, 18, 21, X a Y. Genealógia rodov obidvoch rodičov nepreukázala žiadnu genetickú záťaž. Vzhľadom k súboru prenatálnych znakov, patologických nálezov získaných pri pitve a normálnemu karyotypu, bola klinickým genetikom navrhnutá diagnóza NS. Materiál plodu bol zaslaný k DNA analýze. Výsledky genetickej analýzy rodičovskej a fetálnej DNA nie je možné zverejniť, pretože rodičia vyslovili nesúhlas s ich publikovaním v odborných článkoch.
DISKUSIA
V prezentovaných prípadoch bola diagnóza Noonanovej syndrómu stanovená na základe sumarizácie poznatkov získaných prenatálnym ultrazvukovým vyšetrením, nekroptickým vyšetrením a v neposlednom rade molekulárne genetickým vyšetrením príslušných génových mutácií. Diagnóza NS je diferenciálne diagnosticky dôležitá v prípadoch gravidít, kde prenatálné ultrazvukové vyšetrenie plodu v prvom trimestri tehotenstva odhalí zvýšené hodnoty nuchálneho prejasnenia, polyhydramnion, výpotky v pleurálnej, peritoneálnej dutine alebo celkový hydrops plodu. Nekropsie plodov s podozrením na NS častokrát preukážu pridružené vrodené vývojové vady srdca, skeletu, močového systému a dysmorfické zmeny tváre.
Charakteristické dysmorfické znaky tváre sa menia s vekom. V prenatálnom a rannom postnatálnom období pozorujeme vysoké a široké čelo, hypertelorizmus, epikanty (kožné riasy zakrývajúce vnútorný očný kútik), nízko umiestnené a abnormálne dorzálne rotované uši so zhrubnutou chrupavkou, vysoko vyklenuté podnebie a malé čeľuste. Krk je široký a krátky s nízko umiestnenou vlasovou hranicou (3).
Vrodené vývojové vady srdca sa vyskytujú v 60 % prípadov NS, najčastejšie zastúpené sú stenóza pľúcnice (27-65 %) a hypertrofická kardiomyopatia (9-25 %). K menej častým patria sieňové a komorové defekty septa, atrioventrikulárny kanál, Fallotova tetralógia alebo koarktácia aorty. Kombinácia vývojových vad srdca, predovšetkým stenóza pulmonálnej artérie, a prenatálne popisovaných cystických lymfatických vakov na krku, je silnou indikáciou k ďalšiemu došetreniu so zameraním sa na NS (4,5).
V troch zo štyroch prezentovaných prípadov bola nekroptickým vyšetrením potvrdená agenézia venózneho duktu. V poslednom prípade nebol duktus vyšetrovaný z dôvodu malej veľkosti plodu. Absencia venózneho duktu je všeobecne spojená so zlou prognózou pre plod, hlavne ak sa jedná o plody v prvom trimestri tehotenstva so zvýšenou hodnotou NT (6). Agenézia venózneho duktu s extrahepatálnou cestou pupočníkového žilového návratu u plodov zvyšuje riziko srdcového zlyhania. V prípadoch NS je popisovaná kombinácia so stenózou pulmonálnej artérie, ktorá je zároveň považovaná za najčastejšiu srdcovú vadu vrámci tohoto syndrómu. Prenatálna suspekcia na agenéziu duktu a následná nekroptická verifikácia by mali viesť ku genetickej analýze chromozomálnych i nechromozomálnych syndrómov (7).
K príznakom vyskytujúcim sa so zvýšenou incidenciou patria deformity hrudníka. Hrudný kôš je široký, vzdialenosť medzi prsnými bradavkami je zväčšená. V 70-95 % prípadov sa vyskytujú sternálne deformity typu vpáčeného a vtáčieho hrudníka (pectus carinatum, pectus excavatum). Je zvýšené riziko rozvoja skoliózy. Bežnými ortopedickými komplikáciami sú vbočený lakťový kĺb (cubitus valgus), tzv. vrodená koňská noha (talipes equinovarus), polydaktýlia alebo zkrátenie stehnovej kosti. Malformácie močového traktu sa vyskytujú v 10 % prípadov, jedná sa najčastejšie o dilatáciu alebo naopak stenózy v jeho v rôznych úrovniach. Kryptorchizmus postihuje až 80 % chlapcov s NS (3,5).
V postnatálnom období sa okrem už zmienených príznakov, ktoré sa s vekom stávajú diskrétnejšie, môžeme stretnúť s vyššou incidenciou porúch zrážanlivosti krvi a krvácavých stavov (u viac ako 55 % pacientov s NS). U niektorých pacientov je zvýšené riziko rozvoja akútnych leukémií a myeloproliferativných ochorení. Štúdie venujúce sa vzťahu NS a hematologických ochorení uvádzajú zvýšený výskyt juvenilnej myelomonocytárnej leukémie (JMML) (8). Bežnou komplikáciou sú poruchy lymfatického systému v zmysle dysplázie, hypoplázie alebo aplázie lymfatických ciev. Výsledkom týchto abnormalít sú vyššie popisované a prenatálnym ultrazvukom detekované, generalizované alebo lokálne lymfedémy, lymfangiektázie postihujúce hlavne pľúcny parenchým alebo tráviacu trubicu (9). Zatiaľ čo dysmorfické rysy tváre sú vo vyššom veku menej výrazné, iné vyššie spomínané vady môžu v dospelosti vyžadovať špeciálnu medicínsku starostlivosť. Jedná sa hlavne o poruchy fertility pri kryptorchizme, kardiálne či hematologické komplikácie.
V prezentovaných prípadoch, nekroptické vyšetrenia potvrdili a doplnili súbor prenatálne zistených príznakov. Súčasne bol preukázaný normálny karyotyp plodov a molekulárne genetické vyšetrenie fetálnej DNA preukázalo mutácie asociovaných génov.
NS je ochorenie s autozomálne dominantným typom dedičnosti. Môže sa vyskytovať de novo, poprípade môže byť zdedený od jedného z rodičov. Na základe recentných publikácií je identifikovaných približne 11 kauzálnych génov, ktorých mutácie sú zodpovedné za príznaky NS. Ako prvý bol identifikovaný gén PTPN11, následovaný génmi SOS1, RAF1, KRAS, BRAF, NRAS, MAP2K1, RIT1 a nedávno popísanými SOS2, LZTR1 a A2ML1. 52 % všetkých mutácií zodpovedných za NS tvoria mutácie génu PTPN11, druhým najčastějšie alterovaným génom je SOS1 v 16 % a mutácie RIT1 a RAF1 majú rovnakú prevalenciu 8 %. Tieto najčastejšie mutácie (PTPN11, SOS1, RIT1, RAF1) pokrývajú až 93 % mutácií u NS (10). Hlavný gén zodpovedný za NS je PTPN11, fenotypovým prejavom jeho mutácií je hypertrofická kardiomyopatia, pulmonálna stenóza a septálne srdcové defekty. S týmto typom mutácie sú najčastejšie asociované i rôzne typy neoplázií, ako vyššie zmienené leukémie, JMML, myeloproliferatívne ochorenia a solídne nádory, neuroblastóm a rabdomyosarkóm (11). Mutácie SOS1 génu sa prejavujú menším vzrastom a skeletálnymi defektami, mutácie v géne RAF1 vedú taktiež k hypertrofickej kardiomyopatii. Asociované neoplázie sú u týchto génových mutácií zriedkavejšie (12).
ZÁVER
Vzhľadom k širokému spektru nešpecifických fenotypových prejavov NS, je prenatálna a nekroptická diferenciálna diagnostika obtiažná. Na prvom mieste je odlíšenie od syndrómov s abnormálnym karyotypom. Jedná sa o Turnerov syndróm, u ktorého sa príznaky najviac prekrývajú a ďalšie chromozomálne aberácie ako Downov a Edwardsov syndróm. Druhú skupinu tvoria syndrómy a vady, u ktorých je preukázaný normálny karyotyp plodu, ale DNA analýza potvrdí mutáciu niektorého z génov spomínanej RAS/MAPK signálnej cesty. Sem zaraďujeme vzácné jednotky: faciokutaneoskeletálny syndróm (Costello sy.), kardiofaciokutánny syndróm (CFC sy.), neurofibromatózu I. typu (NF1), Noonan-like syndróm s mnohými kožnými hyperpigmentáciami skôr označovaný ako LEOPARD syndróm (z ang. Lentigines, EKG abnormalities, Ocular hypertelorism, Pulmonary stenosis, Abnormalities of genitalia, Retardation of growth, Deafness). Ďalšiu skupinu tvoria napr. Aarskogov syndróm s podobnými faciálnymi dysmorfiami, malým vzrastom a srdcovými vadami, ktorý je charakterizovaný dedičnosťou viazanou na X chromozóm a Williamsov syndróm patriaci do skupiny tzv. mikrodelečných syndrómov (3,13).
PREHLÁSENIE
Autor práce prehlasuje, že v súvislosti s témou, vznikom a publikáciou tohto článku nieje v konflikte záujmov a vznik ani publikácia článku neboli podporené žiadnou farmaceutickou firmou. Toto prehlásenie sa týka i všetkých spoluautorov.
Adresa pro korespondenci:
MUDr. Tatiana Stupková
Ústav patologie FN Brno
Jihlavská 20, 625 00 Brno
tel.: +420 532 23 3083
fax: 532 232 005
e-mail: gajdosova.tatiana@fnbrno.cz
Sources
1. Nisbet D, Griffin D, Chitty L. Prenatal features of Noonan syndrome. Prenatal Diagnosis 1999; 19(7): 642-647.
2. Croonen E, Nillesen W, Stuurman K, et al. Prenatal diagnostic testing of the Noonan syndrome genes in fetuses with abnormal ultrasound findings. European Journal of Human Genetics 2013; 21(9): 936-942.
3. Van der Burgt I. Noonan syndrome. Orphanet Journal of Rare Diseases 2007; 2(1): 4.
4. Moczulska H, Piotrowicz M, Janiak K, Jakubowski L, Respondek-Liberska M. Prenatal suspicion of Noonan syndrome on the basis of echocardiographic findings - a case report. PRENAT CARDIO 2013; 3(2): 26-30.
5. Romano A. Noonan syndrome - Clinical perspectives and growth issues. US Endocrinology 2008; 04(02): 93-96.
6. Staboulidou I, Pereira S, Cruz J, Syngelaki A, Nicolaides K. Prevalence and Outcome of Absence of Ductus Venosus at 11+0 to 13+6 Weeks. Fetal Diagnosis and Therapy 2011; 30(1): 35-40.
7. Demirci O, Yavuz T, Arisoy R, Pekin O, et al. Agenesis of the ductus venosus a case with Noonan syndrome. Genetic counseling 2015; 26(3): 373-376.
8. Derbent M, Oncel Y, Tokel K, Varan B, et al. Clinical and hematologic findings in Noonan syndrome patients with PTPN11 gene mutations. Am J Med Genet Part 2010; A 152 A: 2768-2774.
9. Sharland M, Burch M, McKenna WM, Paton MA. A clinical study of Noonan syndrome. Archives of Disease in Childhood 1992; 67(2): 178-183.
10. Bouchikhi I, Belhassan K, Moufid F, et al. Noonan syndrome-causing genes: molecular update and an assessment of the mutation rate. International Journal of Pediatrics and Adolescent Medicine 2016; 3(4): 133-142.
11. Aoki Y, Matsubara Y. Ras/MAPK syndromes and childhood hemato-oncological diseases. International Journal of Hematology 2013; 97(1): 30-36.
12. Ilenčíková D, Čižmárová M, Krajčiová A, Požgayová S, Rybárová A, Kovács L. Klinické dysmorfické syndrómy s tumorigenézou: Clinical dysmorphic syndromes with tumorigenesis. Klinická onkologie. Hereditární nádorová onemocnění III. 2012; 25 : 39-48.
13. Bakker M, Pajkrt E, Mathijssen I. B, Bilardo C. M. Targeted ultrasound examination and DNA testing for Noonan syndrome, in fetuses with increased nuchal translucency and normal karyotype. Prenat Diagn 2011; 31 : 833–840.
Labels
Anatomical pathology Forensic medical examiner ToxicologyArticle was published in
Czecho-Slovak Pathology

2019 Issue 1
Most read in this issue
- Aktuální otázky tenkojehlové aspirační biopsie štítné žlázy
- Aktuální pohled na močovou cytologii: Co přináší Pařížská klasifikace?
- Noonanovej syndróm z pohľadu fetopatológa
- Pneumologická cytodiagnostika – state of the art 2019