
Ložiskové léze kostí – diagnostické využití imunohistochemie a molekulární patologie
Bone lesions – diagnostic approach using immunohistochemistry and molecular pathology
Immunohistochemistry and molecular pathology play an essential role in the diagnosis of some focal bone lesions. These techniques may greatly help to distinguish primary bone tumors from metastatic diseases and allow a biologically important refinements in subclassification of round cell sarcomas.
Recently, the diagnostic accuracy of organ and tumor specific antibodies has improved significantly. Knowledge of new type of antibodies and their meaningful use enables an accurate classification of the most undifferentiated carcinomas of unknown primary. However, the interpretation of immunohistochemical stains and molecular genetic analysis can be difficult in bone biopsies due to previous decalcification.
This article summarizes the most important algorithmic approach to the diagnosis of bone tumors. It outlines the most frequently used tissue-specific antibodies. New advances in the understanding of bone tumorigenesis are also discussed.
Keywords:
decalcification – immunohistochemistry – Molecular pathology – round cell tumors – bone metastases – primary bone lymphomas
Authors:
Iva Staniczková Zambo 1; Tetiana Shatokhina 1
Authors‘ workplace:
I. ústav patologie FN u sv. Anny a LF MU, Brno
1
Published in:
Čes.-slov. Patol., 57, 2021, No. 1, p. 30-39
Category:
Reviews Article
Overview
Imunohistochemie a molekulární patologie hrají důležitou roli v diagnostice některých ložiskových lézí kostí. Tyto metody mohou značně prospět pro odlišení primárních nádorů kostí od metastáz a umožňují biologicky významnou subklasifikaci kulatobuněčných sarkomů.
V poslední době se výrazně zlepšila diagnostická přesnost orgánových a nádorově specifických protilátek. Znalost nových typů protilátek a jejich smysluplné použití dovoluje přesné zařazení většiny nediferencovaných karcinomů neznámého origa. V kostních biopsiích může být ale v důsledku přechozí dekalcifikace interpretace výsledků imunohistochemického barvení a molekulárně genetických analýz obtížná.
Tento článek shrnuje nejdůležitější algoritmy pro diagnostiku kostních nádorů. Zmíněny jsou nejčastěji užívané tkáňově specifické protilátky. Diskutovány jsou také nové pokroky v pochopení onkogeneze kostních nádorů.
Klíčová slova:
odvápnění – imunohistochemie – molekulární patologie – kulatobuněčné tumory – kostní metastázy – primární kostní lymfomy
Diagnostika a klasifikace ložiskových lézí kostí vykazuje řadu specifik. Navzdory nepopiratelným pokrokům v poznání histogeneze a molekulárně genetického pozadí mnoha primárních kostních tumorů zůstávají diagnostické algoritmy u většiny těchto nádorů bez zásadních změn. Pro stanovení správné diagnózy hraje klíčovou roli zhodnocení morfologie léze se zvláštním důrazem na interpretaci nálezu v kontextu s klinickými údaji a nálezem na zobrazovacích vyšetřeních. Posouzení vzhledu nádorových buněk, rozpoznání typu produkované extracelulární matrix a charakter růstu tumoru mnohdy umožňuje stanovit finální diagnózu již z hematoxylinu-eozinu. Případné pochybnosti je nutné konzultovat s ošetřujícím klinickým lékařem dobře orientovaným v problematice patologických kostních nálezů.
V indikovaných případech lze s výhodou využít imunohistochemické a/nebo molekulárně genetické vyšetření. Aplikace těchto metod je nezbytná u metastatických tumorů, a u pouze několika typů primárních kostních nádorů, mezi které patří zejména kulatobuněčné sarkomy, dále maligní cévní nádory, chordom, adamantinom, nediferencovaný pleomorfní sarkom kostí a hematoonkologické malignity. Imunohistochemie nachází uplatnění i při odlišení low-grade osteosarkomu od benigních ložiskových lézí (1,2).
Využití imunohistochemie a molekulárních technik v kostních biopsiích však může být limitováno charakterem materiálu (viz níže). Jednoznačný závěr tedy nelze vždy formulovat. Z hlediska léčebné strategie je ovšem důležité rozpoznat těchto osm kategorií ložiskových kostních lézí:
- metastáza (se snahou o co nejpřesnější typizaci; léčbu ovlivňuje rozsah onemocnění, primární lokalizace i histologický typ tumoru)
- lymfom (pokud není možno specifikovat imunofenotyp, lze většinou vyslovit suspekci na plazmocelulární myelom/plazmocytom nebo DLBCL, ostatní typy lymfomů jsou vzácné; léčeny na hematoonkologických klinikách)
- Ewingův sarkom, Ewing-like sarkomy, včetně kulatobuněčných sarkomů s rearanží genů BCOR či CIC, a mezenchymální chondrosarkom (léčba agresivní, multimodální v režimu indukční chemoterapie – resekce – adjuvantní chemoterapie, pro všechny tyto tumory se užívá protokol Euro Ewing 2012)
- high-grade osteosarkom (léčba agresivní, multimodální v režimu indukční chemoterapie – resekce – adjuvantní chemoterapie, protokol EURAMOS 1)
- konvenční chondrosarkom (většinou řešen resekcí; chemoterapie pouze u dediferencovaného chondrosarkomu či paliativně)
- ostatní high-grade sarkomy (léčba agresivní, multimodální: resekce + adjuvantní chemoterapie podobná léčbě high-grade osteosarkomu)
- lokálně agresivní tumory s nízkým metastatickým potenciálem a benigní tumory hrozící patologickou frakturou (řešeny resekcí, u obrovskobuněčného nádoru kosti v indikovaných případech také podáváním denosumabu)
- nenádorové ložisko (typu zánětu, nekrózy, fraktury apod.), benigní léze s tendencí k samovolné regresi a asymptomatické malé benigní nádory (většinou pouze sledování, případně resekce)(3).
OBECNÉ PRINCIPY IMUNOHISTOCHEMIE A MOLEKULÁRNÍ PATOLOGIE V KOSTNÍCH BIOPSIÍCH
Je nutno mít na paměti, že interpretace výsledků imunohistochemického a molekulárně genetického vyšetření může být u kostních biopsií svízelná. Mnohé „primární kostní“ imunohistochemické markery sice pomáhají identifikovat histogenetický původ buněčné populace (osteoblastická, chondroblastická diferenciace), nejsou však využitelné pro odlišení reaktivních lézí od benigních a maligních tumorů (2). Obezřetnost je tak např. nutná při hodnocení imunofenotypu při okraji léze, v místech reaktivní osteoplázie, kde se nenádorové osteoblasty (SATB2 pozitivní) mísí s vyšetřovaným tumorem. Reaktivní osteoblasty bývají nápadné, poměrně velké, s velkými jádry, obklopují trámce pletivové kosti i depozita osteoidu a hrozí tedy záměna s osteosarkomem.
V průběhu zpracování kostní tkáně je ve většině případů nezbytná dekalcifikace materiálu, která může zásadním způsobem negativně ovlivnit kvalitu tkáně a vést k falešně pozitivním i falešně negativním výsledkům. Nešetrné odvápnění likviduje epitopy antigenů a většina dekalcifikačních činidel degraduje nukleové kyseliny – dochází k hydrolýze a fragmentaci nukleotidů (4).
U málo mineralizovaných biopsií lze zvážit zpracování bez odvápnění, hrozí však riziko zhotovení nekvalitních řezů, a tedy obtížné hodnocení cytonukleárních detailů, navíc je tím výrazně ztíženo odlišení diskrétních depozit nádorového osteoidu od počínající hyalinizace extracelulární matrix. To vše může vést k mylné interpretaci histologického nálezu. V neodvápněných řezech autofluorescence přítomných kostních trámců částečně nebo zcela znemožňuje získat použitelné výsledky fluorescenční in situ hybridizace (FISH).
S ohledem na skutečnost, že zhotovení kvalitních preparátů je pro histopatologickou diagnostiku zásadní, bývá tedy dekalcifikace nutná. Její kvalita závisí na:
- množství mineralizované matrix (velikosti vzorku, zastoupení kortikální versus spongiózní kosti, množství sklerotické kosti)
- zvolené metodě dekalcifikace
- době trvání dekalcifikace
- dalších fyzikálních vlivech (teplotě, pH, použití ultrazvuku atp.)
V praxi se pro odvápnění nejčastěji používají roztoky anorganických a/nebo organických kyselin či chelační činidla. Optimální dekalcifikační metoda má být rychlá a ke tkáni šetrná. V současné době existuje celá řada komerčně vyráběných roztoků. Bez ohledu na pořizovací náklady však žádné činidlo bez výhrady nesplňuje kritéria ideálního dekalcifikačního roztoku. Anorganické kyseliny odvápňují rychle, ale mohou vést k výraznému poškození tkáně. Chelátory jsou velmi šetrné a umožňují využití imunohistochemie i molekulárních technik v plné šíři, odvápňují však velmi pomalu a v rutinní praxi jsou proto téměř nevyužitelné (prodlužují dobu dekalcifikace až na desetinásobek, u výrazně mineralizované tkáně může odvápnění trvat i více než 70 dní!). Dle literárních údajů i našich zkušeností jsou nejlepším kompromisem organické kyseliny nebo roztoky organických a anorganických kyselin. Ty většinou umožňují validní hodnocení imunohistochemie a použití samotné 5% kyseliny mravenčí by mělo být dostatečně šetrné pro zachování DNA a tedy umožňovat např. i FISH analýzu (5). Na našem pracovišti se čistá kyselina mravenčí nepoužívá. Průměrně dvojnásobně prodlužuje dobu dekalcifikace, přičemž u výrazněji mineralizované tkáně může být proces odvápnění ještě znatelně pomalejší. Průběh dekalcifikace je nutné kontrolovat a v případě potřeby odvápňovací roztok vyměnit. Po odstranění vápenatých solí další působení dekalcifikačního roztoku vede k rychlé devastaci tkáně. Odvápnění tedy musí být ukončeno včas (6).
I přes úzkostlivé dodržení všech doporučených postupů během dekalcifikace je nutno hodnotit výsledky speciálních vyšetření velmi uvážlivě. Obecně platí pravidlo, že na daném pracovišti zavedená dekalcifikační metoda má být v podmínkách tamějšího provozu validovaná na použití různých typů imunohistochemických markerů (s membránovou, cytoplazmatickou a jadernou expresí) a výsledek imunohistochemického i molekulárně genetického vyšetření musí být interpretován v kontextu s histologickým obrazem a klinicko-radiologickým nálezem.
VYUŽITÍ MOLEKULÁRNÍ PATOLOGIE V KOSTNÍCH BIOPSIÍCH
Naše znalosti molekulárních změn zodpovědných za vznik a progresi řady maligních nádorů rychle narůstají. Poslední dekáda je označována za postgenomickou éru, během které mnoho nádorových onemocnění začíná být chápáno v kontextu genomových změn, což je umožněno zejména využitím nových technologií (zejména NGS – next-generation sequencing) schopných současně detekovat celé spektrum genetických abnormalit (7).
Díky tomu mohly být rozpoznány nové mechanizmy onkogeneze. Jedním z nich je např. chromotripse. Dochází při ní k roztříštění jednoho nebo několika málo chromozomů na malé fragmenty (hovoří se také o „explozi chromozomu“) a jejich opětovnému spojení pomocí DNA reparačních procesů v náhodném pořadí, přičemž současně může docházet k deleci některých lokusů. Během jediné události tak vznikají desítky až stovky přestaveb. Při komplexních přestavbách chromozomů se uplatňuje odlišný mechanismus – vznikají opakované, mnohonásobné, a na sobě nezávislé zlomy a opravy, které se v postižených oblastech genomu v průběhu času hromadí. Na rozdíl od chromotripse se tedy jedná o proces mnohastupňový (8).
Vzrůstající tlak na rutinní užívání molekulárních technik u primárních kostních tumorů má především diagnostický, případně prognostický význam. V kostních biopsiích se jako smysluplné jeví zejména tam, kde pomáhá vyšetřovaný nádor zařadit do některé z terapeutických kategorií se specifickým typem léčby (např. pro přesné zařazení kulatobuněčného tumoru, nebo u metastáz karcinomu mammy lze při nedostupném primárním tumoru šetrně odvápněný materiál použít pro FISH HER2). V ostatních případech pro diagnostické účely v naprosté většině postačuje zhodnotit histologický obraz, eventuálně imunofenotyp.
V rámci výzkumu je detailní poznání molekulárních mechanizmů charakteristických pro konkrétní tumor více než žádoucí. Umožňuje identifikovat nové imunohistochemické diagnostické markery, které lze následně aplikovat v rámci rutinní diagnostické praxe. Výčet nových, takto identifikovaných markerů pro tumory různých orgánových systémů se rychle rozrůstá. Mezi dnes již běžně používané patří např. TTF-1, PAX8, GATA3, CDX2, Fli1, ERG, SOX10, MDM2, STAT6, DOG1, TLE1 a řada dalších. Navíc je pouze otázkou času, kdy poznatky z molekulární patologie povedou k nalezení nové cílené terapie aplikovatelné i u high-grade mezenchymálních tumorů (4,9). Například již nyní je zcela zřejmé, že alespoň u pacientů s kulatobuněčným sarkomem s CIC přestavbou je vysoce žádoucí změna terapeutického režimu. V současnosti jsou tito pacienti léčeni dle protokolu pro Ewingovy sarkomy, nicméně jejich léčebná odpověď je výrazně nižší, rychleji dochází k systémové progresi a pětileté přežití dosahuje pouze 43 % oproti 77 % u Ewingova sarkomu. Intenzifikace již tak vysoce toxické léčby není dost dobře možná. Řešením by mohla být terapie zacílená např. na inhibici transkripčních faktorů z rodiny PEA3 (polyoma enhancer activator 3)(10).
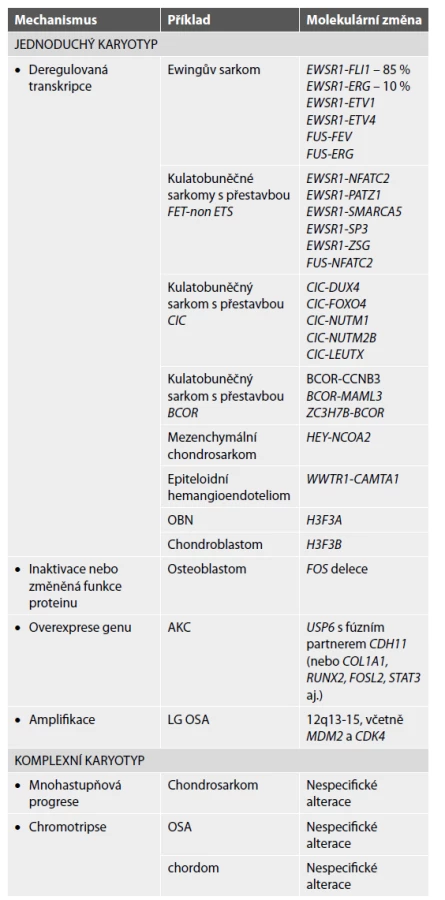
Z hlediska molekulárního pozadí lze tumory kostí rozdělit do dvou kategorií, na skupinu tumorů s jednoduchým karyotypem a na tumory s komplexním karyotypem. V první skupině jsou zahrnuty benigní i maligní kostní nádory se specifickými translokacemi, mutacemi nebo amplifikacemi. Chromozomální translokace obecně patří mezi časné události v tumorigenezi. Nádory s komplexním karyotypem mají nestabilní genom a jsou pro ně typické mnohočetné nespecifické molekulární alterace. Do této kategorie patří tři skupiny tumorů s typickými zástupci: chondrosarkom, osteosarkom a chordom. Při progresi enchondromu do sekundárního centrálního chondrosarkomu se uplatňuje model mnohastupňové progrese s postupnou akumulací početných genetických aberací. U chordomu a high-grade osteosarkomu bývá přítomna chromotripse (4,11). V tabulce 1 jsou uvedeny některé kostní tumory se simplexním i komplexním karyotypem.
KULATOBUNĚČNÉ TUMORY
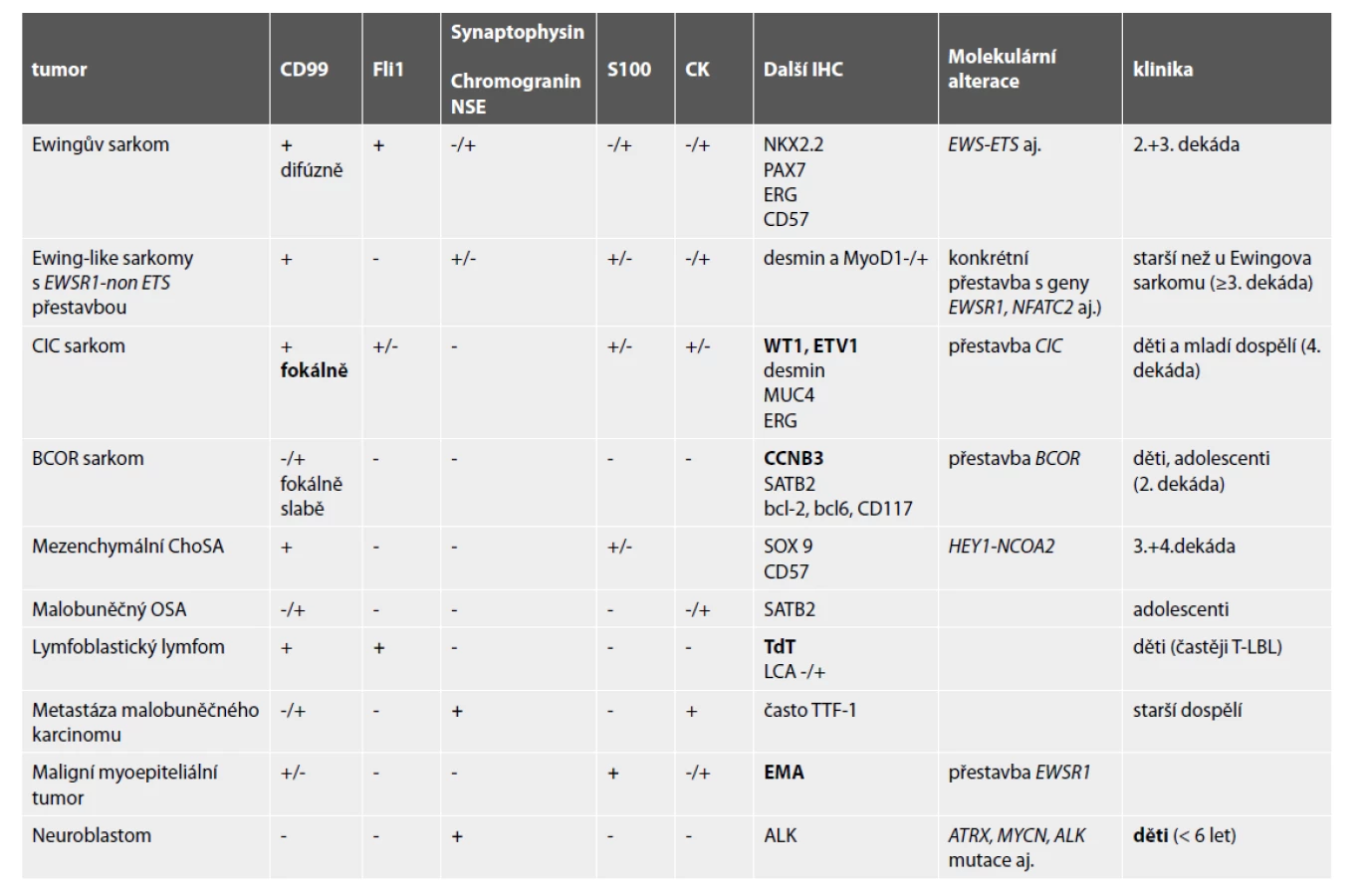
Diferenciální diagnostika kulatobuněčných tumorů zahrnuje širokou škálu primárních i metastatických nádorů s rozdílným typem léčby. Jejich vzájemné odlišení bez použití imunohistochemie a někdy i molekulární patologie není možné. V následujícím textu budou zmíněny nejdůležitější charakteristiky těchto malignit, Jejich hlavní diagnostické znaky jsou v Tabulce 2.
Ewingův sarkom
Ewingův sarkom typicky postihuje mladé jedince, v naprosté většině v 2. a 3. dekádě života a v žebříčku nejčastějších primárních mezenchymálních malignit kostí zaujímá po osteosarkomu a chondrosarkomu třetí místo (12).
Ewingův sarkom představuje prototyp primárního nehematoonkologického kulatobuněčného tumoru kosti. Diagnostickým je průkaz rearanže fúzních partnerů z rodiny FET a ETS genů, tedy mezi geny EWSR1 či FUS (t.j. rodinou FET genů) a geny FLI1, anebo ERG, ETV1, ETV4 či FEV (t.j. rodinou ETS genů; viz také tabulka 1). V přibližně 85 % případů je prokázána reciproční translokace t(11;22)(q24;q12), při které vzniká fúzní protein EWSR1-FLI1 s funkcí transkripčního faktoru. Druhou nejčastější aberací je translokace t(21;22)(q22;12)podmiňující vznik chimerického genu EWSR1-ERG. Je zastoupena v přibližně 10 % testovaných případů Ewingova sarkomu a má obdobnou roli v onkogenezi tohoto tumoru jako rearanže EWSR1-FLI1.Ve zbývajících případech lze identifikovat jiný typ translokace, a to buď s účastí genu EWSR1 nebo vzácně genu FUS. Metoda FISH při použití tzv. break apart sondy umožňuje detekovat zlom genu EWSR1. Pozitivní výsledek však musí být interpretován v kontextu s celkovým klinickým obrazem, histologickým nálezem a imunofenotypem. Gen EWSR1 totiž patří mezi geny „vysoce promiskuitní“, tzn., že je častým translokačním partnerem širokého spektra klinicky a patologicky odlišných tumorů a lze jej identifikovat např. v desmoplastickém nádoru z malých kulatých buněk, myxoidním liposarkomu, extraskeletálním myxoidním chondrosarkomu, angiomatoidním fibrózním histiocytomu, světlobuněčném sarkomu měkkých tkání, v myoepiteliálních tumorech (kůže, měkkých tkání i kostí), a vzácně i v jiných nádorech (4,13). Některé z těchto neoplázií mohou Ewingův sarkom připomínat i histologicky, mají ale odlišný průběh i klinický management. Při pochybnostech je tedy doporučováno výsledek FISH analýzy doplnit průkazem konkrétního fúzního transkriptu (např. metodou RT-PCR).
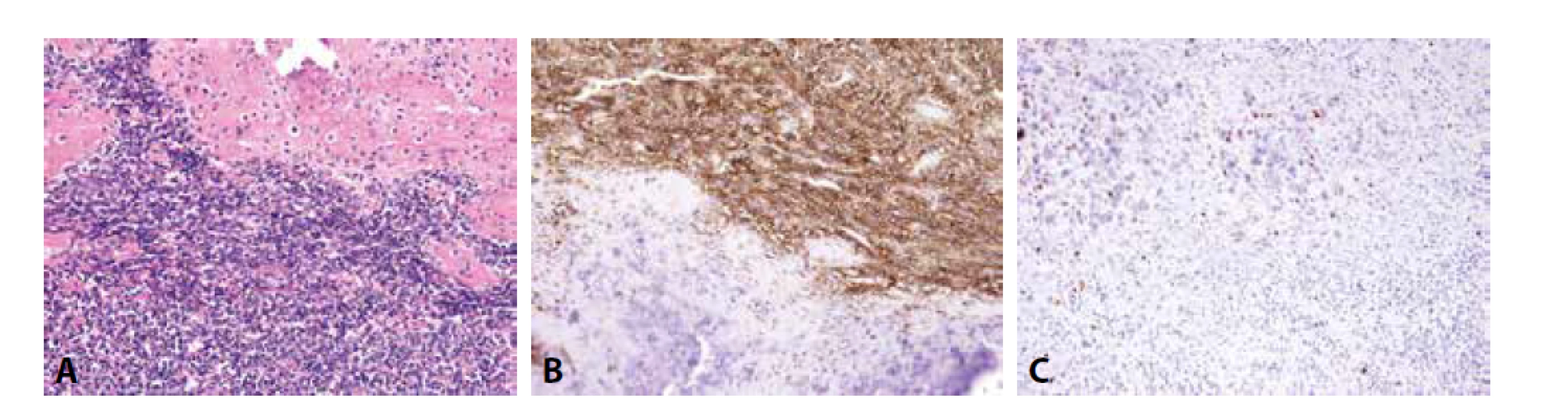
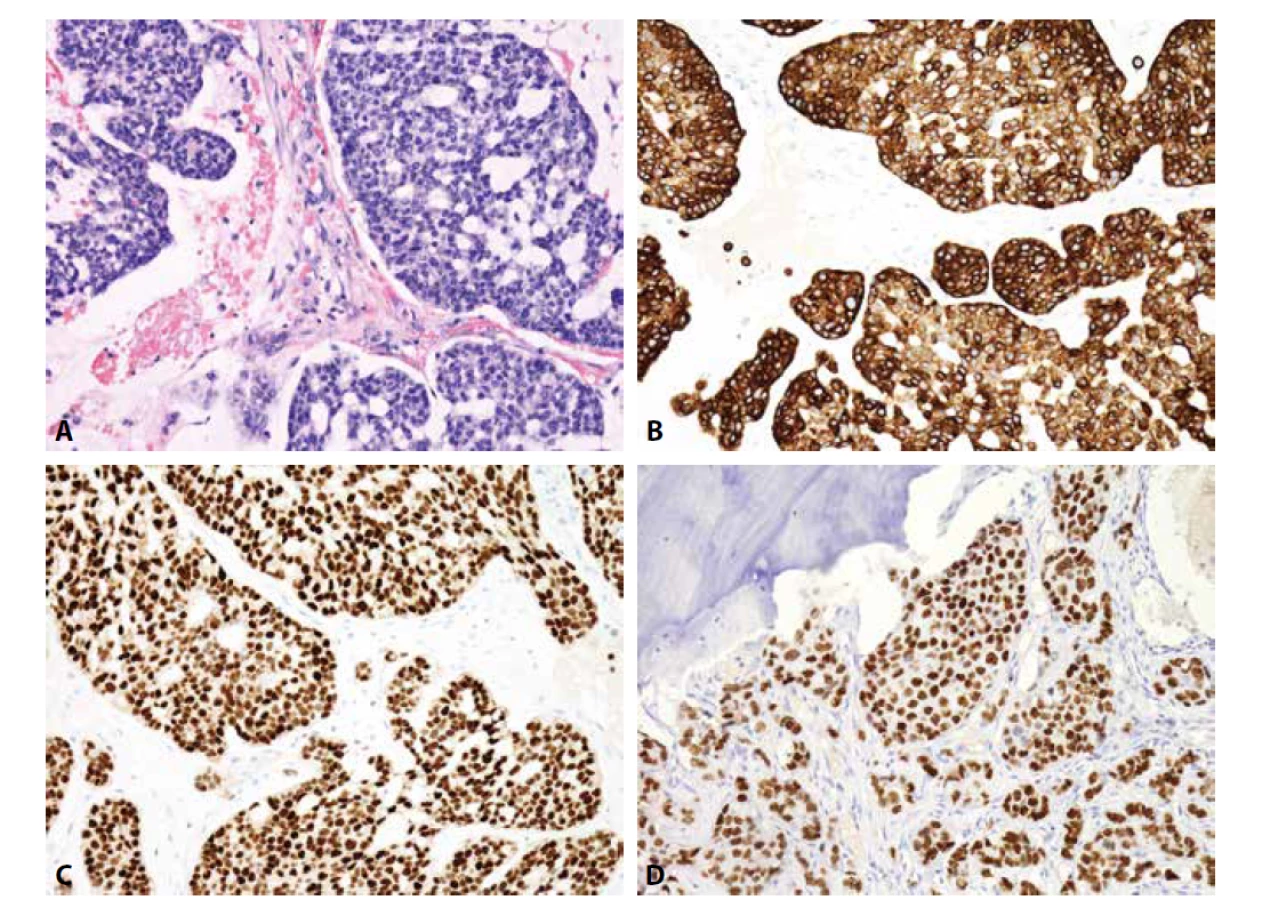
Imunofenotyp je pro stanovení finální diagnózy velmi nápomocný (obr. 1). Charakteristickou je difúzní silná membránová exprese CD99. Tato pozitivita je sice málo specifická, zato však vysoce senzitivní. Zatímco CD99 exprimuje celá řada kulatobuněčných i vřetenobuněčných tumorů, absence průkazu CD99 diagnózu Ewingova sarkomu vylučuje. Pro Ewingův sarkom je dále typickou jaderná pozitivita Fli1 (v cca 70 % případů (14)) či ERG, s variabilní expresí NSE, chromograninu a synapatophysinu. Více než v 90 % případů Ewingova sarkomu lze prokázat expresi NKX2.2 (15). Relativně vzácně může Ewingův sarkom exprimovat i cytokeratiny, což může při možné současné expresi neuroendokrinních markerů vést k záměně s metastázou neuroendokrinního karcinomu, včetně karcinomu z Merkelových buněk. Metastatický karcinom je však u mladých jedinců málo pravděpodobný, mívá na rozdíl od Ewingova sarkomu synaptophysin a/nebo chromogranin exprimován difúzně a neprokážeme u něj žádnou z diagnostických translokací typických pro Ewingův sarkom.

Bez použití molekulárních konfirmačních metod hrozí záměna za lymfoblastický lymfom, který se s Ewingovým sarkomem shoduje ve věkové predilekci postižených pacientů i v expresi CD99 a Fli1. Tento lymfom z malých kulatých buněk je navíc většinou LCA negativní. K jeho odlišení od Ewingova sarkomu pomůže exprese TdT a absence některé z diagnostických translokací typických pro Ewingův sarkom.
Diagnosticky obtížné, zejména v punkčních biopsiích, může být odlišení Ewingova sarkomu od mezenchymálního chondrosarkomu, zejména tehdy, pokud je v biopsii zachycena pouze kulatobuněčná komponenta tumoru. Mezenchymální chondrosarkom se vyskytuje u adolescentů a mívá nehomogenně exprimován CD99. Chybí však exprese Fli1 i přestavba genu EWSR1, diagnostickou je translokace HEY1-NCOA2. V současné době je ale mezenchymální chondrosarkom léčen podle stejného protokolu jako Ewingův sarkom. Stanovení mylné diagnózy tak zatím nemá dopad na typ onkologické péče.
Kulatobuněčné sarkomy „Ewing-like“
Do této skupiny patří tumory histologicky v různé míře připomínající Ewingův sarkom. V současné době lze dle rozpoznaného typu translokace kulatobuněčné „Ewing-like“ sarkomy rozdělit do tří podkategorií (16-18):
sarkomy s přestavbou genu CIC
sarkomy s přestavbou genu BCOR
sarkomy s translokací FET (nejčastěji EWSR1)-non-ETS (viz. také tabulka 1)
Vůbec nejčastějším subtypem „Ewing-like“ tumorů jsou sarkomy s přestavbou CIC genu. Vyrůstají v hlubokých měkkých tkáních končetin mladých dospělých (ve 4. dekádě) a vzhledem k rychle se vyvíjející chemorezistenci na léčiva užívaná při terapii Ewingova sarkomu a tedy rychlé progresi mají velmi špatnou prognózu. Morfologie nádorových buněk je znatelně pleomorfnější oproti Ewingově sarkomu. Jádra jsou velká, mají nápadná jadérka, cytoplazma je nezřetelná, mitózy početné, nekrózy často geografické. Extracelulární matrix bývá myxoidní. Exprese CD99 je spíše fokální než difúzní. Typickou je exprese ETV1 a WT1. Podobně jako Ewingův sarkom bývají Fli1 a ERG pozitivní. CIC sarkomy mohou být alespoň fokálně desmin a S100 pozitivní (18,19).
BCOR sarkomy se častěji vyskytují u mladých mužů (ve 2. dekádě) a rostou v dlouhých a plochých kostech nebo paraspinálně. Léčebná odpověď na neoadjuvantní i postoperační chemoterapii je srovnatelná s Ewingovým sarkomem, tedy i prognóza je obdobná (20). Nádorové buňky jsou drobnější, okrouhlé či naznačeně vřetenité, jádra prohnutá, s jemným chromatinem a nezřetelným jadérkem, cytoplazma světle eozinofilní, mitózy četné, často bývají přítomny drobné nekrózy. Extracelulární stroma může být myxoidní. Exprese CD99 je nehomogenní, většinou nevýrazná a jen fokální. Téměř vždy je tumor difúzně cyclin B3 pozitivní (CCNB3), často lze prokázat také SATB2, CD117, bcl2 a bcl6, někdy i TLE1 (21). Desmin ani cytokeratiny v BCOR sarkomech neprokazujeme.
Soubory pacientů s FET-non-ETS kulatobuněčným sarkomem jsou prozatím málo početné. Klinicky jde o velmi heterogenní skupinu nádorů postihující děti i o něco starší dospělé než u Ewingova sarkomu, a v porovnání s ním mají horší odpověď na chemoterapii (19). V této kategorii lze rozpoznat některé pomocné znaky u sarkomů s NFATC2 nebo PATZ1 přestavbou. Nádory EWSR1- či FUS-NFATC2 většinou postihují metafýzy nebo diafýzy dlouhých kostí, jsou tvořené kulatými nebo epiteloidními buňkami, často se zřetelnou anizonukleózou, nápadnými jadérky a světlou cytoplazmou (22). Marker CD99 mohou exprimovat difúzně. Sarkomy s PATZ1 rearanží častěji rostou v hlubokých měkkých tkáních hrudní stěny a břicha. Nádorové buňky jsou kulaté či mírně vřetenité a bývají obklopené denzní kolagenní matrix. Mitotická aktivita nebývá vysoká a nekrózy většinou chybí. Imunohistochemicky bývají CD99, S100, synaptophysin pozitivní a rovněž exprimují i svalové markery (16).
Další primární intraoseální kulatobuněčné tumory
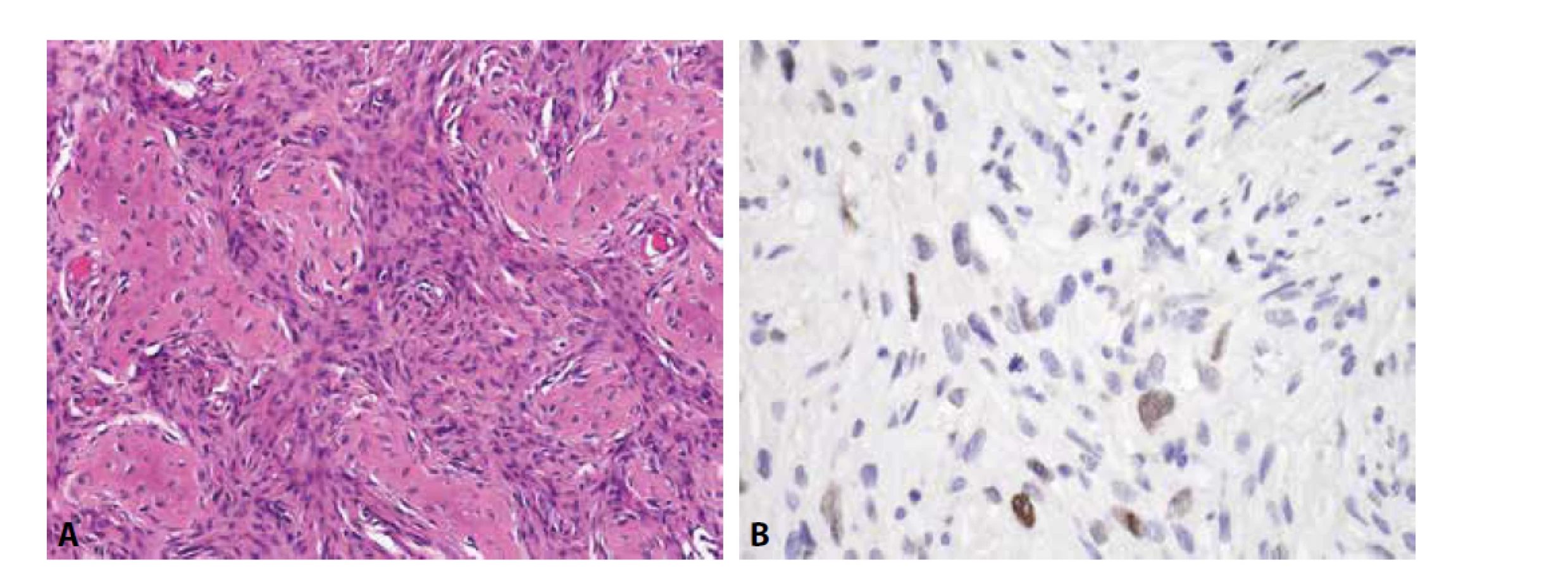
Mezenchymální chondrosarkom postihuje adolescenty, o něco častěji ženy, a typicky vyrůstá v kostech (v kraniofaciálním skeletu a kostech trupu), méně často v měkkých tkáních či parenchymových orgánech. Jedná se o vysoce agresivní tumor podmíněný translokací HEY-NCOA2, histologicky bifázické stavby. Různě rozsáhlé plochy okrouhlých či krátce vřetenitých buněk s hyperchromními jádry exprimují CD99, SOX9 a CD57. Fokálně jsou přítomny S100 protein pozitivní okrsky chrupavčité diferenciace (obr. 2). Zavádějící je difúzní exprese CD99 a NKX2-2, tedy markerů typických pro klasický Ewingův sarkom, za který může být mezenchymální chondrosarkom zaměněn. Molekulárně genetické vyšetření je v tomto případě rozhodující.
Kulatobuněčný osteosarkom je zastoupen necelými 2 % mezi všemi osteosarkomy. Je tvořen poměrně uniformní populací malých okrouhlých buněk a mnohdy málo výraznými depozity nádorového osteoidu. Většinou chybí exprese CD99. Důležitou, i když málo specifickou, je difúzní silná exprese SATB2. Hlavní diferenciálně diagnostickou jednotkou je tedy BCOR sarkom a Ewingův sarkom, není však přítomna přestavba genů BCOR ani EWSR1(23).
Lymfoblastický lymfom typicky postihuje děti a mladé dospělé, histologicky a částečně také imunohistochemicky napodobuje Ewingův sarkom. Bývá difúzně Fli1 pozitivní a exprimuje, i když nehomogenně, CD99. Navíc je velmi často LCA, CD3 a CD20 negativní. Důležitou je tedy exprese CD79a, CD43 a TdT. Lymfoblastický lymfom také bývá CD34 pozitivní. Určujícím je samozřejmě molekulárně genetické vyšetření (24).
OSTEOSARKOM, CHONDROSARKOM a jiné vybrané primární kostní nádory
Osteosarkom je nejčastějším primárním maligním tumorem kostí. V přibližně 80 % se jedná o konvenční high-grade osteosarkom, který vzniká intraoseálně a typicky postihuje metafýzy dlouhých kostí, nejčastěji oblast kolene. Pro stanovení diagnózy je nezbytná identifikace nádorového osteoidu, produkovaného maligními osteoblasty. Probatorní nebo punkční biopsie však nemusí osteoid zachytit. Pro osteosarkom zatím neexistuje žádný silný diagnostický marker. Jaderná exprese SATB2 neodliší reaktivní osteoblasty od nádorových (benigních ani maligních), a navíc exprese SATB2 není pro osteosarkom specifická (25). Je popisována u BCOR sarkomu, high-grade konvenčního chondrosarkomu, u dediferencovaných sarkomů (dediferencovaného chondrosarkomu, liposarkomu aj.), i v různých karcinomech (kolorektálním, světlobuněčném renálním, vzácněji v karcinomech Mülleriánské diferenciace aj.). Ani dříve preferované protilátky, osteocalcin a osteonectin, nebo novější protilátka ezrin nejsou z důvodů nízké specificity v rutinní praxi příliš využitelné (2). Zásadní tak nadále zůstává kombinace klinických údajů s rentgenologickým obrazem a patologickým nálezem. Pokud je zachycen high-grade sarkom s typickým nálezem na zobrazovacích vyšetřeních, je nutné osteosarkom zmínit v rámci diferenciálně diagnostické rozvahy. Onkolog dle konkrétní situace rozhodne o dalším postupu (rebiopsie, neoadjuvantní chemoterapie pro osteosarkomy, primární resekce).
U low-grade osteosarkomů (centrálních či povrchových) lze imunohistochemii s výhodou využít. Alespoň část oválných nádorových buněk je MDM2 a CDK4 pozitivní (obr. 3). Ve velmi šetrně odvápněném materiálu nebo v málo osifikované neodvápněné tkáni lze navíc např. metodou FISH prokázat amplifikaci genu MDM2, čímž je jednoznačně vyloučena záměna zejména za fibrózní dysplázii.
Konvenční chondrosarkom většinou nečiní diagnostické obtíže. Problematické může být odlišení high-grade chondrosarkomu od chondroblastického osteosarkomu, ve kterém nepomůže ani protilátka SATB2. Diagnostickou přesnost lze zvýšit mutační analýzou IDH1/2. U přibližně 60 % chondrosarkomů lze identifikovat některou z těchto mutací, častěji IDH1, přičemž osteosarkomy jsou IDH1/2 negativní (25). U IDH1/2 nemutovaných tumorů je tak opět nutno velmi pečlivě histologický obraz korelovat s klinickým a radiologickým nálezem. Navíc chondroblastický osteosarkom bývá značně histologicky heterogenní – na malé ploše se střídají chrupavčité okrsky výrazně pleomorfní s fokusy low-grade vzhledu.
U dediferencovaného chondrosarkomu je nutné zachytit přechod mezi chrupavčitou komponentou a high-grade sarkomem. Pokud máme k dispozici pouze hypercelulární komponentu, imunohistochemické vyšetření ke stanovení finální diagnózy příliš nápomocné není.
Mezenchymální chondrosarkom byl diskutován výše.
Chondroblastom patří mezi vzácné, lokálně agresivní tumory s chondrogenní diferenciací. Typicky roste v epifýzách dlouhých kostí u mladých jedinců. Histologicky může v malých biopsiích napodobovat obrovskobuněčný nádor kosti. V chondroblastomu by měla být patrná exprese S100, SOX9 a DOG1, většinou při absenci p63. Molekulárně geneticky lze prokázat mutaci genu H3F3B, díky které byla vyvinuta mutačně specifická protilátka proti histonu H3 rodiny 3B, konkrétně marker H3K36M. V chondroblastomech je tato protilátka exprimována difúzně jaderně (27).
Obrovskobuněčný nádor kosti (OBN) nejčastěji roste v metafýzách dlouhých kostí, mnohdy s přesahem do epifýz. Postihuje mladé dospělé. Mononukleární stromální buňky jsou většinou p63 pozitivní. Ve více než 90 % OBN lze prokázat mutaci v genu H3G34W, která podobně jako u chondroblastomu umožnila vývoj protilátky proti histonu H3, tentokrát rodiny 3A. Jedná se o nukleární marker H3G34W, který je v OBN difúzně pozitivní (28).
Aneurysmatická kostní cysta (AKC) v málo reprezentativním materiálu patří mezi diagnosticky obtížné jednotky. Může být zaměněna jak za benigní/lokálně agresivní tumory, většinou OBN, tak za vysoce agresivní sarkomy, zejména teleangiektatický osteosarkom. Klinický obraz může být také zavádějící. AKC nemá predilekci pro konkrétní část kosti, typicky postihuje mladé jedince (děti, dospívající a mladé dospělé) a v době diagnózy může být poměrně objemná. Imunohistochemie není přínosná. V necelé polovině případů AKC lze prokázat translokaci USP6 genu, naopak chybí mutace H3F3A/B(28).
Adamantinom patří mezi velmi vzácné kostní nádory pacientů v širokém věkovém rozpětí, nejčastěji dospělých ve 3. a 4. dekádě. Postihuje metafýzu nebo diafýzu tibie či fibuly. Ačkoli je řazen mezi low-grade maligní tumory, jedná se o velmi úporně recidivující nádor s metastatickým potenciálem (29). Histologicky se jedná o bifázický tumor s pruhy a klastry epitelií pozitivních CK5, CK14, CK19, EMA, p63 a D2-40. Epiteliální ostrůvky jsou lemovány stromatem z vřetenitých či oválných buněk s chudým imunoprofilem (vimentin, někdy fokálně i galectin-3 pozitivní). Diferenciálně diagnosticky je nutno pomýšlet na adamantinomu podobný Ewingův sarkom, který však lze odlišit imunohistochemicky i molekulárně geneticky. Obtížné může být odlišení od metastatického dlaždicobuněčného karcinomu s bazaloidní morfologií nebo uroteliálního karcinomu. Věk pacienta s adamantinomem bývá nižší, nápomocnou může být i exprese p16 u části dlaždicobuněčných karcinomů nebo GATA3 u uroteliálního karcinomu. Nejdůležitější je však korelace s klinickým nálezem, adamantinom se prezentuje jako solitární osteolytické ložisko v typické lokalizaci. Metastatický synoviální sarkom odliší detekce přestavby genu SS18 a imunohistochemie, při které lze s výhodou využít nové, vysoce specifické a senzitivní protilátky SSX a SS18 (30).
Chordom je maligní nádor s notochodrální diferenciací. Typicky roste na bázi lební, v obratlových tělech či sakrokokcygeální oblasti u dospělých jedinců v 5. - 7. dekádě. Imunohistochemický profil je určující. Chordomy exprimují EMA a cytokeratiny, zejména CK5, CK8/18, CK19, jsou však CK7 i CK20 negativní. Bývají HBME1 a, v přibližně 50 %, také S100 pozitivní. Jaderná exprese markeru brachyury je velmi užitečná a pomáhá odlišit většinu metastatických karcinomů či lokálně pokročilý chondrosarkom. Tato protilátka však není striktně exprimována pouze v chordomech, může být pozitivní také v části germinálních tumorů, malobuněčném karcinomu a vzácně i v některých sarkomech (31).
METASTÁZY SOLIDNÍCH TUMORŮ DO KOSTÍ
Kostní metastázy jsou vůbec nejčastější malignitou skeletu. Kostní tkáň je po plicním a jaterním parenchymu třetím nejčastějším terčem hematogenních metastáz karcinomů, a druhým u sarkomů (četnější je diseminace do plic).
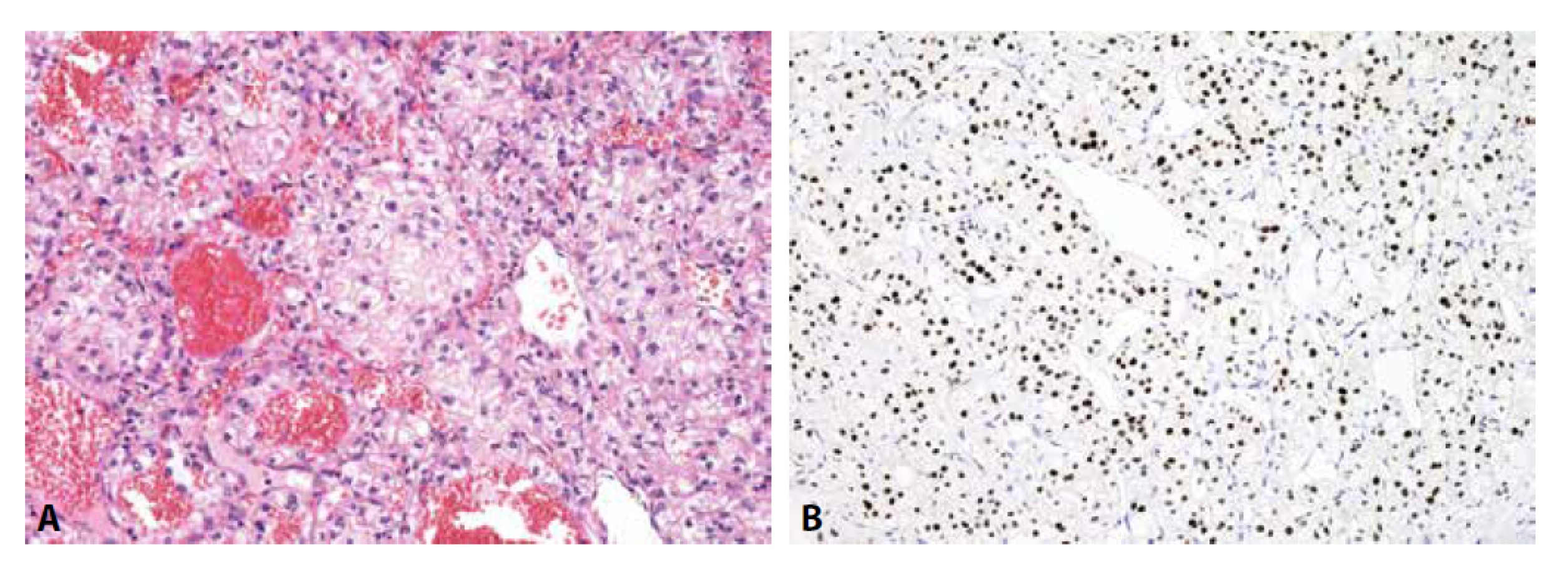
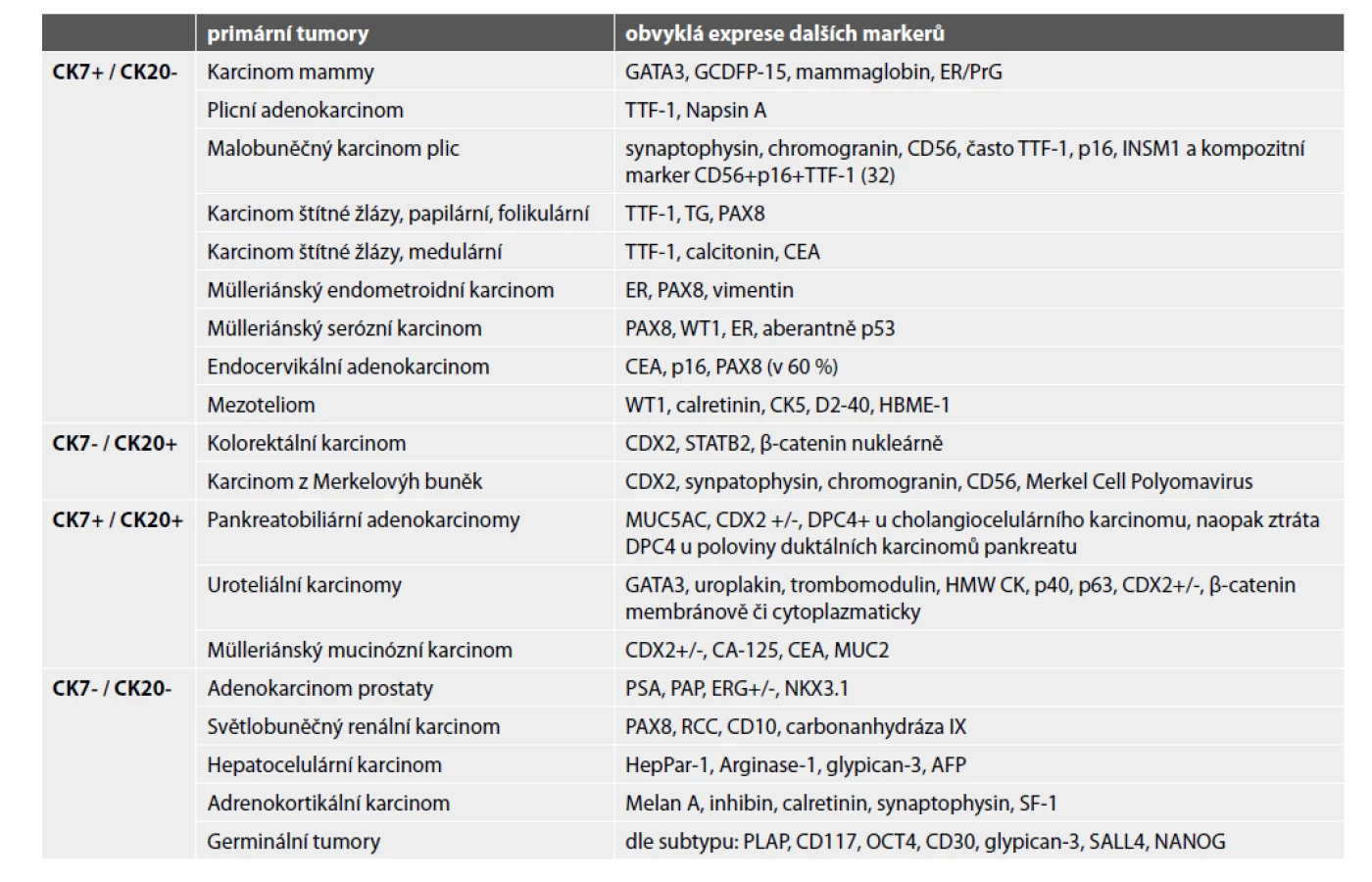
Určení histologického typu a odhalení primární lokalizace metastatického karcinomu zlepšuje prognózu pacienta. Do kostí může diseminovat prakticky kterýkoli karcinom, typicky karcinomy mammy, plic, ledvin, prostaty a kolorektální karcinom. Užití imunohistochemie je nezbytné (obr. 4-6). V prvním kole by neměly chybět cytokeratiny 7 a 20. Dle konkrétního nálezu lze již iniciálně nebo v dalším kroku vyšetření rozšířit o další vhodné protilátky (viz tabulka 3). Je důležité připomenout, že neexistuje žádný unikátní marker, který by byl výlučně specifický pro jeden typ tumoru, nicméně výčet nezbytných protilátek umožňujících identifikaci primárního origa může být poměrně krátký, a to zejména díky novým markerům identifikovaným pomocí molekulární patologie (1). Navzdory současným diagnostickým možnostem se v přibližně 5 % případů nepodaří primární lokalizaci identifikovat i s ohledem na fakt, že metastázy nízce diferencovaných/nediferencovaných tumorů často ztrácí expresi obvyklých markerů a naopak mohou vykazovat aberantní pozitivitu s „netypickými“ protilátkami (33).

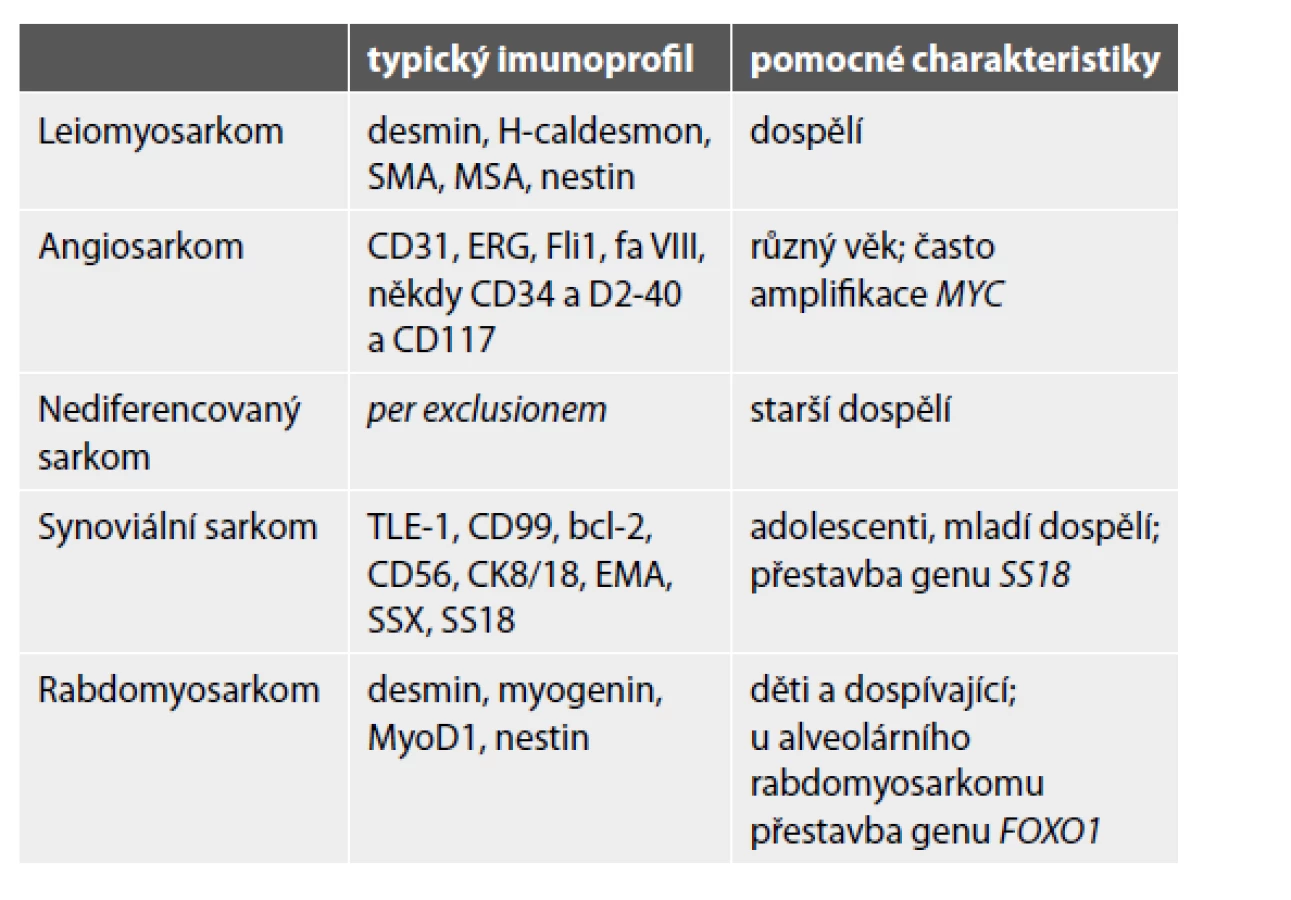
Do kostí mohou metastazovat také sarkomy. Kromě progrese primárních kostních sarkomů (zejména osteosarkomu a Ewingova sarkomu – viz výše) to mohou být i sarkomy měkkých tkání, dle klesající četnosti jmenovitě leiomyosarkom, angiosarkom, nediferencovaný sarkom, synoviální sarkom a rabdomyosarkom (34). Nejdůležitější patologická specifikace do kostí metastazujících sarkomů je uvedena v tabulce 4. Pozor na možnou expresi různého spektra cytokeratinů a/nebo EMA, která nevylučuje diagnózu sarkomu. Kromě synoviálního sarkomu či angiosarkomu mohou být epiteliální markery pozitivní také v high-grade sarkomech nejrůznějších diferenciací (v leiomyosarkomu, nediferencovaném sarkomu, Ewingově sarkomu a dokonce i osteosarkomu).
Kosti jsou také obvyklým místem metastáz maligního melanomu. Depozita typického pigmentu nemusí být zachycena a morfologie tumoru může být značně heterogenní, většinou však výrazně polymorfní. Imunofenotyp S100, SOX10, HMB-45, Melan A, MITF či tyrozináza pozitivní je spolu s histologickým obrazem diagnostický. Pokud to umožňuje kvalita materiálu, lze i v metastáze melanomu indikovat genetické vyšetření ke stanovení RAS, BRAF, KIT, PTEN mutace.
HEMATOONKOLOGICKÉ MALIGNITY V KOSTECH
Maligní lymfomy mohou kostní dřeň infiltrovat primárně nebo v rámci progrese základního onemocnění. Primární kostní lymfomy jsou vzácné a představují přibližně 7 % ze všech malignit kostí u dospělých pacientů (35). Většinou se jedná o non-Hodgkinské lymfomy, nejčastěji difúzní velkobuněčný B-lymfom (DLBCL) či plazmocytom. Zhodnocení histologického nálezu v kombinaci s obvyklým imunoprofilem je určující. U DLBCL prokazujeme LCA, CD79a, CD20, PAX5, a variabilně bcl6, bcl2, CD10 a MUM1. Pro plazmocytom je typická exprese CD138, CD79a a EMA s prokázanou restrikcí lehkého řetězce kappa nebo lambda. Exprese CD20 je slabá nebo chybí. Kost může být vzácněji postižena i jiným typem lymfomu, např. folikulárním lymfomem či T-buněčnými lymfomy. Problematika infiltrace lymfoblastickým lymfomem byla diskutována v rámci diferenciální diagnostiky malobuněčných kostních tumorů.
ZÁVĚR
Diagnostika ložiskových lézí kostí se opírá o histologický obraz, klinické údaje a radiologický nález. Doplňková laboratorní vyšetření lze užít zejména u kulatobuněčných tumorů, metastatických nádorů a hematoonkologických malignit. Interpretace výsledků imunohistochemického, případně molekulárně genetického vyšetření může být v kostních biopsiích obtížná zejména v důsledku dekalcifikace materiálu. Nešetrné odvápnění v různé míře poškozuje vyšetřovaný materiál, znemožňuje užití molekulárních metod a může vést k falešně pozitivním i falešně negativním imunohistochemickým výsledkům. Každé pracoviště patologie by mělo mít ověřeno fungování imunohistochemických protilátek s membránovou, cytoplazmatickou a jadernou expresí i na materiálu standardně dekalcifikovaném v daném provozu.
Expanze vědomostí v oblasti molekulární patologie zejména kulatobuněčných sarkomů umožnila významně zúžit kategorii nediferencovaných kulatobuněčných mezenchymálních tumorů. Odlišné genetické pozadí některých nově definovaných „Ewing-like“ sarkomů ukazuje na nutnost rozvoje cílené terapie. Odpověď na léčebné protokoly pro Ewingovy sarkomy je u těchto tumorů často nižší a zejména tzv. CIC sarkomy mají výrazně horší prognózu ve srovnání s Ewingovým sarkomem. V budoucnu lze tedy očekávat rozšíření portfolia maligních tumorů, u kterých se molekulárně genetické vyšetření stane nezbytnou součástí diagnostického algoritmu.
PROHLÁŠENÍ
Autor práce prohlašuje, že v souvislosti s tématem, vznikem a publikací tohoto článku není ve střetu zájmů a vznik ani publikace článku nebyly podpořeny žádnou farmaceutickou firmou. Toto prohlášení se týká i všech spoluautorů.
∗ Adresa pro korespondenci:
MUDr. Iva Staniczková Zambo, Ph.D.
I. ÚP FN u sv. Anny a LF MU
Pekařská 53, 656 91 Brno
tel. +420 543 183 220
e-mail: iva.zambo@fnusa.cz
Sources
1. Conner JR and Hornick JL. Metastatic carcinoma of unknown primary: diagnostic approach using immunohistochemistry. Adv Anat Pathol 2015; 22(3): 149-167.
2. Gao Z, Kahn LB. The application of immunohistochemistry in the diagnosis of bone tumors and tumor-like lesions. Skeletal Radiol 2005; 34(12): 755-770.
3. Adámková Krákorová D et al. Sarkomy. Praha: Mladá fronta 2019; 244-252.
4. Lam SW, van IJzendoorn DGP, Cleton-Jansen AM et al. Molecular pathology of bone tumors. Journal Mol Diagn 2019; 21(2): 171-182.
5. Brown RS, Edwards J, Bartlett JW et al. Routine acid decalcification of bone marrow samples can preserve DNA for FISH and CGH studies in metastatic prostate cancer. J Histochem Cytochem 2002; 50(1):113-115.
6. Singh VM, Salunga RC, Huang RJ et al. Analysis of the effect of various decalcification agents on the quantity and quality of nucleic acid (DNA and RNA) recovered from bone biopsies. Ann Diagn Pathol 2013; 17(4): 322-326.
7. Bignell GR, Greenman CD, Davies H et al. Signatures of mutation and selection in the cancer genome. Nature 2010; 463(7283): 893-898.
8. Maher CA, Wilson RK. Chromothripsis and human disease: piecing together the shattering process. Cell 2012; 148(1-2): 29-32.
9. Sugita S and Hasegawa T. Practical use and utility of fluorescence in situ hybridization in the pathological diagnosis of soft tissue and bone tumors. J Orthop Sci 2017; 22(4): 601-612.
10. Antonescu CR, Owosho AA, Zhang L et. Al. Sarcomas with CIC-rearrangements are a distinct pathologic entity with aggressive outcome: A clinicopathologic and molecular study of 115 cases. Surg pathol 2017; 41(7): 941-949.
11. Franceschini N, Lam SW, Cleton-Jansen AM, Boveé JVMG. What´s new in bone forming tumors of the skeleton? Virchows Arch 2020; 476(1): 147-157.
12. Fletcher CDM, Bridge JA, Hogendoorn PCW et al. WHO classification of tumours of soft tissue and bone (4th edn). Lyon: IARC; 2013.
13. Seningen JL, Inwards CY. Small round cell tumors of bone. Surg Pathol Clin 2012; 5(1): 231-256.
14. Folpe AL, Hill CE, PArham DM et al. Immunohistochemical detection of FLI-1 protein expression: a study of 132 round cell tumors with emphasis on CD99-positive mimics of Ewing’s sarcoma/primitive neuroectodermal tumor. Am J Surg Pathol 2000; 24(12): 1657-1662.
15. Hung YP, Fletcher CD, Hornick JL. Evaluation on NKX2-2 expression in round cell sarcomas and other tumors with EWSR1 rearrangement: imperfect specificity for Ewing sarcoma. Mod Pathol 2016; 29(4): 370-380.
16. Sbaraglia M, Righi A, Gambarotti M, Dei Tos AP. Ewing sarcoma and Ewing-like tumors. Virchows Arch 2019; 476(1): 109-119.
17. Renzi S, Anderson ND, Light N, Gupta A. Ewing-like sarcoma: An emerging family of round cell sarcomas. J Cell Physiol 2019; 234(6): 7999-8007.
18. Kinkor Z, Grossmann P, Dubová M, et al. Co nového v Ewing-like family aneb malobuněčné/kulatobuněčné sarkomy měkkých tkání a kostí s rearanží genů CIC a BCOR. Přehled problematiky a naše prvotní zkušenosti. Cesk Patol 2017; 53(4): 175‐180.
19. Le Loarer F, Pissaloux D, Coindre JM et al. Update on families of round cell sarcomas other than classical Ewing sarcomas. Surg Pathol Clin 2017; 10(3): 587-620.
20. Cohen-Gogo S, Cellier C, Coindre JM et al. Ewing-like sarcomas with BCOR-CCNB3 fusion transcript: A clinical, radiological and pathological retrospective study from the Societe Francaise des Cancers de L’Enfant. Pediatr Blood Cancer 2014; 61(12): 2191-2198.
21. Puls F, Niblett A, Marland G et al. BCOR-CCNB3 (Ewing-like) sarcoma: A clinicopathologic analysis of 10 cases, in comparison with conventional Ewing sarcoma. Am J Surg Pathol 2014; 38(10): 1307-1318.
22. Kinkor Z, Vaneček T, Švajdler M Jr, et al. Kde končí a začíná diagnóza Ewingova sarkomu - popis dvou neobvyklých kostních nádorů s translokací t(20;22)(EWSR1-NFATc2) Cesk Patol 2014; 50(2): 87‐91.
23. Righi A, Gambarotti M, Longo S et al. Small cell osteosarcoma: clinicopathologic, immunohistochemical, and molecular analysis of 36 cases. Am J Surg Pathol 2015; 39(5): 691-699.
24. Ozdemirli M, Fanburg-Smith JC, Hartmann DP et al. Differentiating lymphoblastic lymphoma and Ewing’s sarcoma: lymphocyte markers and gene rearrangement. Mod Pathol 2001; 14(11): 1175-1182.
25. Machado I, Navarro S, Picci P et al. The utility of SATB2 immunohistochemical expression in distinguishing between osteosarcomas and their malignant bone tumor mimickers, such as Ewing sarcomas and chondrosarcomas. Pathol Res Pract 2016; 212(9): 811-816.
26. Kerr DA, Lopez HU, Deshpande V et al. Molecular distinction of chondrosarcoma from chondroblastic osteosarcoma through IDH1/2 mutations. Am J Surg Pathol 2013; 37(6): 787-795.
27. Schaefer IM, Fletcher JA, Nielsen GP et al. Immunohistochemistry for histone H3G34W and H3K36M is highly specific for giant cell tumor of bone and chondroblastoma, respectively, in FNA and core needle biopsy. Cancer Cytopathol 2018; 126(8): 552-566.
28. Rehkämper J, Steinestel K, Jeiler B et al. Diagnostic tools in the differential diagnosis of giant cell-rich lesions of bone at biopsy. Oncotarget 2018; 9(53): 30106-30114.
29. Southam BR, Crawford AH, Billmire DA et al. Long-term follow-up of adamantinoma of the tibia complicated by metastases and second unrelated primary cancer: a case report and literature review. Case Rep Orthop 2018; 2018 : 5493750.
30. Baranov E, McBride MJ, Bellizzi AM, et al. A novel SS18-SSX fusion-specific antibody for the diagnosis of synovial sarcoma. Am J Surg Pathol. In press 2020.
31. Miettinen M, Wang Z, Laosta J et al. Nuclear brachyury expression in consistent in chordoma, common in germ cell tumors and small cell carcinomas and rare in other carcinomas and sarcomas. An immunohistochemical study of 5229 cases. Am J Surg Pathol 2015; 39(10): 1305-1312.
32. Švajdler M, Mezencev R, Šašková B, Ondič O, Mukenšnábl P, Michal M. Triple marker composed of p16, CD56, and TTF1 shows higher sensitivity than INSM1 for diagnosis of pulmonary small cell carcinoma: proposal for a rational immunohistochemical algorithm for diagnosis of small cell carcinoma in small biopsy and cytology specimens. HumPathol 2019; 85 : 58‐64.
33. Lin F, Liu H. Immunohistochemistry in undifferentiated neoplasm/tumor of uncertain origin. Arch Pathol Lab Med 2014; 138(12): 1583-1610.
34. Vincenzi B, Frezza AM, Schiavon G et al. Bone metastasis in soft tissue sarcomas: a survey of natural history, prognostic value and treatment options. Clin Sarcoma Res 2013; 3(1): 6.
35. Demircay E, Hornicek FJ, Mankon HJ and Degroot H. Malignant lymphoma of bone: a review of 119 patients. Clin Orthop Relat Res 2013; 471(8): 2684-2690.
Labels
Anatomical pathology Forensic medical examiner ToxicologyArticle was published in
Czecho-Slovak Pathology

2021 Issue 1
Most read in this issue
- A basic immunohistochemical panel for the diagnosis of soft tissue tumors
- Bone lesions – diagnostic approach using immunohistochemistry and molecular pathology
- Secondary pulmonary hypoplasia associated with calcified Meckel´s diverticulum with osseous metaplasia
-
Consensus recommendations from the Czech Head and Neck Cancer Cooperative Group (2019):
definition of surgical margins status, neck dissection reporting, and HPV/p16 status assessment