
Vybrané novinky v imunohistochemii a molekulární genetice nádorů měkkých tkání
An update on immunohistochemical and molecular genetic markers of selected soft tissue tumors
Recent years have brought an immense increase of knowledge regarding the molecular genetic background of mesenchymal tumors which in turn has significantly expanded the repertoire of molecular markers available for the routine diagnostic practice. This progress has also been followed by a rising number of available immunohistochemical markers useful for the diagnosis of soft tissue neoplasia. Both lineage specific and tumor-specific immunohistochemical antibodies have been discovered and subsequently tested in the surgical pathology practice. This article will review some of the immunohistochemical and molecular genetic markers useful in the diagnosis of vascular tumors, malignant peripheral nerve sheath tumors, low-grade fibromyxoid sarcomas/sclerosing epithelioid fibrosarcomas, solitary fibrous tumors, epithelioid sarcomas, rhabdomyosarcomas and other lesions showing skeletal muscle differentiation. The immunohistochemical and molecular genetic features of some recently characterized and clinically particularly important entities will be discussed as well.
Keywords:
soft tissue neoplasia – mesenchymal tumors – immunohistochemistry – Molecular genetics
Authors:
Michael Michal 1,2
Authors‘ workplace:
Bioptická laboratoř, s. r. o., Plzeň
1; Šiklův ústav patologie LF UK v Plzni a FN Plzeň
2
Published in:
Čes.-slov. Patol., 57, 2021, No. 1, p. 19-29
Category:
Reviews Article
Overview
Nebývalé prohloubení poznatků o molekulárně genetickém pozadí mezenchymálních tumorů v posledních letech přineslo i výrazné rozšíření palety dostupných molekulárních markerů pro rutinní diagnostickou praxi. Spolu s těmito pokroky a často právě díky nim, došlo také ke značnému rozvoji na poli imunohistochemie. Byly objeveny a následně praxí ověřeny nové protilátky jak pro detekci různých směrů diferenciace, tak i protilátky vyvinuté přímo k diagnostice konkrétních nádorových typů. V tomto textu budou shrnuty imunohistochemické a molekulárně genetických markery užitečné v diagnostice vaskulárních tumorů, maligních tumorů z pochev periferních nervů, low-grade fibromyxoidních sarkomů/sklerozujících epiteloidních fibrosarkomů, solitárních fibrózních tumorů, epiteloidních sarkomů, rhabdomyosarkomů a dalších lézí exprimujících markery diferenciace do příčně pruhované svaloviny. Přehledový článek se rovněž zabývá imunohistochemickými a molekulárně genetickými znaky pro diagnostiku některých nověji popsaných a klinicky obzvlášť významných lézí.
Klíčová slova:
nádory měkkých tkání – mezenchymální nádory – imunohistochemie – molekulární genetika
Nebývalé prohloubení poznatků o molekulárně genetickém pozadí mezenchymálních tumorů v posledních letech přineslo i výrazné rozšíření palety dostupných molekulárních markerů pro rutinní diagnostickou praxi. Spolu s těmito pokroky a často právě díky nim, došlo také ke značnému rozvoji na poli imunohistochemie (IHC). Byly objeveny a následně praxí ověřeny nové protilátky jak pro detekci různých směrů diferenciace (např. vaskulární markery, markery příčně pruhované diferenciace), tak i protilátky vyvinuté přímo k diagnostice konkrétních nádorových typů (např. MUC4, STAT6). V tomto textu budou shrnuty poznatky o imunohistochemických a molekulárně genetických metodách užitečných v diagnostice mezenchymálních tumorů častěji se vyskytujících, případně těch, jejichž správné rozpoznání je obzvláště klinicky významné; pochopitelně však nejde o přehled vyčerpávající. Toto review záměrně detailně nepojednává o rozsáhlé problematice kulatobuněčných sarkomů či některých neurogenních tumorů, neboť o nich informují poměrně nedávné přehledové články v tomto časopise (1,2). Mezenchymální tumory zažívacího traktu byly též probrány v samostatném článku v minulém čísle ČS patologie. Tabulkový seznam téměř všech v praxi využitelných imunohistochemických a molekulárně genetických metod byl publikován spolu s guidelines pro diagnostiku nádorů měkkých tkání a distribuován společně s časopisem Česko-slovenská patologie v průběhu loňského roku. Tyto guidelines je možné dohledat i na internetových stránkách České společnosti patologů (3).
Rhabdomyosarkomy (RMS) a léze s diferenciací do příčně pruhované svaloviny
Ačkoliv desmin je nejvíce senzitivní protilátka pro detekci rhabdomyosarkomové diferenciace (4), v případě podezření na RMS je samozřejmě vždy nutné nález potvrdit některým z více specifických imunohistochemických markerů. Za nejlepší metodu je momentálně považována detekce dvou transkripčních faktorů hrajících klíčovou roli ve vývoji příčně pruhované svaloviny, tj. MyoD1 a myogeninu. Podobně jako u ostatních protilátek detekujících proteiny transkripčních faktorů, i zde je nutné hledat pouze nukleární pozitivitu. Takováto exprese je velmi specifická pro rhabdomyosarkomovou diferenciaci a každá z protilátek má podle různých studií senzitivitu od 91 do 97 %. Ne vždy jsou však exprimovány současně (4,5), a proto se obvykle používají obě najednou. Obecně lze říci, že obě protilátky fungují o něco lépe v případech alveolárního RMS (5,6). Dalším transkripčním faktorem, který lze imunohistochemicky detekovat, je nedávno charakterizovaná protilátka PAX7. Její exprese je přítomna v celkem zhruba 70 % všech RMS, o něco slabší je pouze její senzitivita u alveolárních RMS. Naopak v případě embryonálního podtypu často reaguje i s případy, které jsou negativní jak s MyoD1, tak s myogeninem (7). Významná je rovněž exprese PAX7 v Ewingových sarkomech (7), která je natolik konzistentním nálezem, že ji lze pravděpodobně využít i diagnosticky (8). Z dosavadní zkušenosti na našem pracovišti se PAX7 skutečně jeví jako užitečný marker příčně pruhované svalové diferenciace. Setkali jsme se s několika případy s difúzní expresí PAX7 při současné negativitě či pouze fokální pozitivitě MyoD1 či myogeninu u jinak typických RMS či jiných tumorů s diferenciací do kosterní svaloviny (viz dále).
Genetické změny vyskytující se u embryonálních RMS jsou poměrně heterogenní, a protože jde navíc o změny poměrně často se u nádorů vyskytující (mutace v RAS signální dráze, mutace genu TP53, numerické aberace různých chromosomů (9)), je jejich detekce jen obtížně využitelná diagnosticky. Naopak naprostá většina alveolárních RMS obsahuje rekurentní fúze genu FOXO1 s partnery PAX3 a PAX7, jejichž detekce je snadno a relativně levně proveditelná například metodou FISH. Ačkoliv typický embryonální i alveolární RMS mají každý své charakteristické histologické znaky, bývá popisováno zhruba 25% případů morfologicky vypadajících spíše jako alveolární RMS, které však neobsahují výše popisované fúzní geny. Vzhledem k tomu, že i jejich klinické chování je podobné embryonálnímu RMS, většina z nich ve skutečnosti představuje nejspíše tento podtyp. Terapie embryonálního a alveolárního RMS se v některých situacích výrazně liší, zejména co se týče použití radioterapie či dávky chemoterapeutik, v posledních letech je proto vzhledem k výše zmíněnému nutné diagnózu alveolárního RMS vždy potvrdit či vyloučit molekulárně geneticky. Z klinického a terapeutického hlediska se pak tyto tumory jednoduše dělí do skupin „fusion positive“ a „fusion negative“ (10).
Zásadními změnami prošla kategorie vřetenobuněčných/sklerozujících RMS, o nichž se ještě nedávno uvažovalo jako o podtypu embryonálního RMS. Řada studií v posledních letech však potvrzuje, že nejenže se nejedná o podtyp embryonálního RMS, ale že i v rámci kategorie vřetenobuněčných/sklerozujících RMS se nachází několik dalších, biologicky zcela odlišných podskupin, dostatečně charakterizované jsou však zatím pouze dvě. První obsahuje tumory vyskytující se převážně u novorozenců a výhradně pak u dětí do 5 let věku a molekulárně geneticky se vyznačuje fúzemi genů VGLL2 a/nebo NCOA2. Druhá zahrnuje tumory obsahující většinou bodové mutace genu MyoD1, které postihují všechny věkové kategorie. Ačkoliv existují určité histologické rozdíly mezi nádory z obou skupin, pouze na základě morfologie je s jistotou rozlišit nelze. V obou případech se jedná o převážně vřetenobuněčné tumory s různou mírou hyalinizace stromatu a různě vyjádřenou rhabdomyosarkomovou diferenciací. Zatímco tumory z první skupiny jsou prognosticky velmi příznivé a nemetastazují, tumory s mutací MyoD1 jsou naopak značně agresivní a obvykle rychle vedou k úmrtí (11). Z důvodu zásadního klinického významu je tak k rozlišení nutné molekulárně genetické vyšetření.
Zcela nově popsanou kategorií jsou RMS s epiteloidní a vřetenobuněčnou morfologií a rearanžemi genů FUS-TFCP2, případně EWSR1-TFCP2. Klinicky jsou neobvyklé predilekcí pro kosti kraniofaciální oblasti s častým šířením do okolních měkkých tkání, malá část případů pak vzniká přímo v měkkých tkáních. Další zvláštností je konzistentní exprese cytokeratinů a protilátky ALK. Jedná se možná o vůbec nejagresivnější podtyp RMS, u něhož byl však zaznamenám určitý terapeutický efekt ALK inhibitorů (12,13).
V diferenciální diagnostice RMS je však třeba mít na paměti, že pozitivita s rhabdomyosarkomovými markery se nevyskytuje pouze u RMS či u nesrovnatelně vzácnějších rhabdomyomů. Lze se s ní setkat i ve formě heterologní rhabdomyosarkomové diferenciace v různých, převážně dediferencovaných formách měkkotkáňových tumorů (dediferencovavaný liposarkom, GIST, maligní tumor z pochev periferních nervů atd), a lze ji detekovat i u některých recentně popsaných lézí (9).
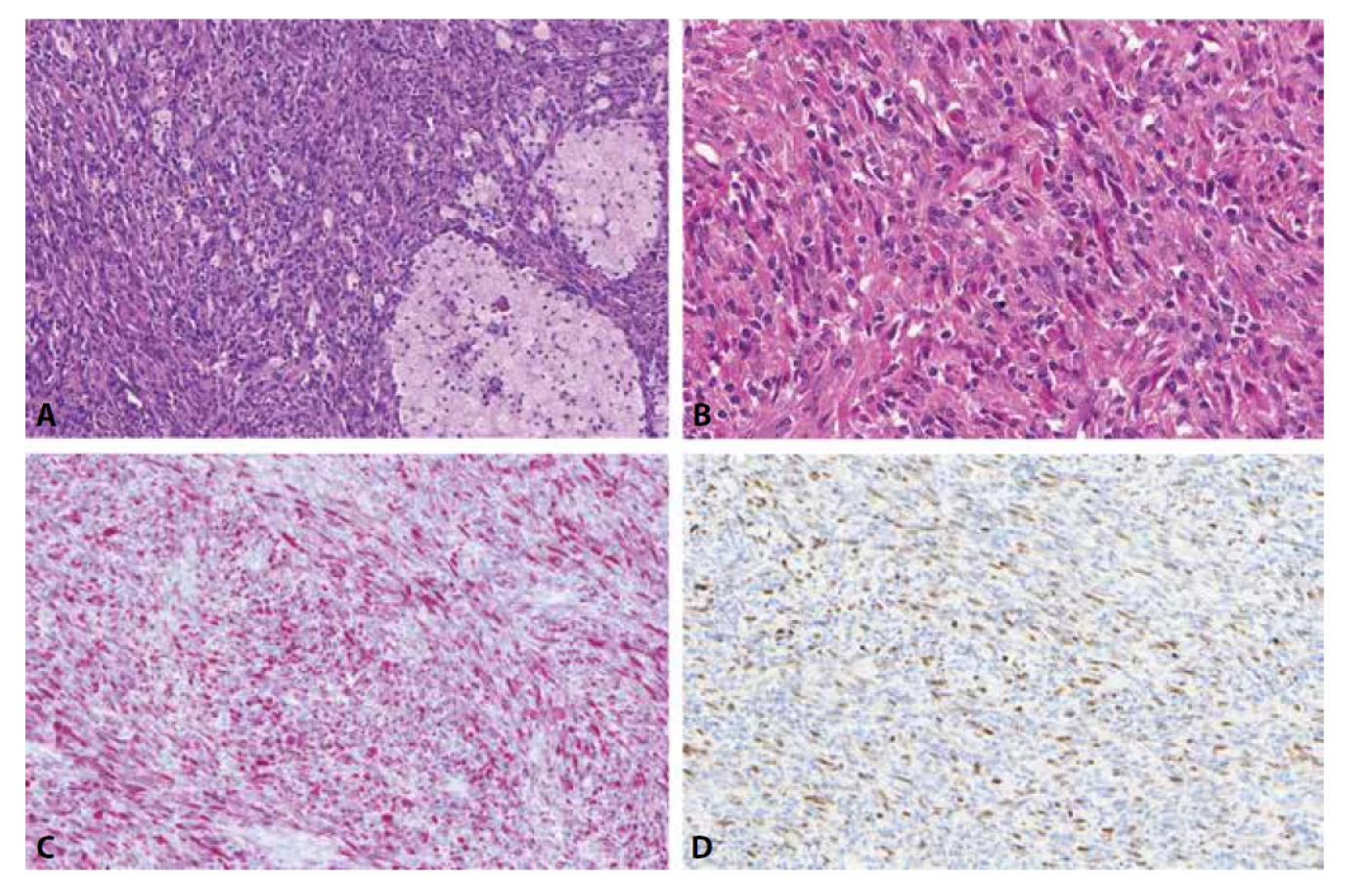
Low-grade inflamatorní myogenní tumor je naší skupinou nově charakterizovaná jednotka vyznačující se jak imunohistochemickými, tak většinou i morfologickými známkami diferenciace do kosterní svaloviny. Nejčastěji se jedná o ostře ohraničený a intramuskulárně uložený tumor na stehnech či zádech mladších mužů. Je složený z vřetenitých, místy epiteloidních až rhabdoidních buněk a je nápadný svou výraznou inflamatorní příměsí ve formě disperzně rozesetých lymfocytů a prominentních histiocytárních agregátů (obr. 1A, B)(14). Nedávno popsaný histiocyte-rich rhabdomyoblastic tumor dle našeho názoru představuje pouze fenotypickou variantu téže jednotky (15). Mezi tyto tumory spadají i dříve poněkud vágně charakterizované inflamatorní leiomyosarkomy (16). Bez ohledu na použitou terminologii je zásadní nezaměnit tyto neoplazie za některý z RMS či jiný maligní tumor, poněvadž biologické chování low-grade inflamatorních myogenních tumorů je podstatně příznivější, recidivy i metastázy jsou ojedinělými jevy. Protože mohou exprimovat jak desmin (obr. 1C), tak MyoD1 (obr. 1D), myogenin či PAX7, k rozlišení od vřetenobuněčného RMS poslouží zejména jejich velmi nízká mitotická a proliferační aktivita, přítomnost prominentní inflamatorní příměsi, obvykle dobré opouzdření/ohraničení tumoru. V nejobtížnějších případech pak lze provést molekulárně genetické vyšetření k vyloučení RMS (14).
Další novou jednotkou, která běžně exprimuje markery kosterní svaloviny, jsou sarkomy s EWSR1-PATZ1 fúzí (17,18). Podle 18 dosud publikovaných případů a dle naší zkušenosti s 9 případy se zdá, že ty to tumory postihují všechny věkové kategorie, mají zřetelnou anatomickou predilekci pro měkké tkáně v oblasti břišní stěny a zejména v oblasti hrudníku. Jedná se o fenotypicky extrémně variabilní tumory, jejichž morfologie zahrnuje jak čistě kulatobuněčné Ewing-like sarkomy (obr. 2A), tak low-grade vřetenobuněčné sarkomy, které připomínají spíše solitární fibrózní tumor či myoepitheliomy měkkých tkání (obr. 2D). Často je přítomné nápadné fibrózní stroma, které může spolu s kulatobuněčnou morfologií a překryvným imunoprofilem vést k záměně za desmoplastický kulatobuněčný tumor. Imunohistochemický profil, ač velmi pestrý, je poměrně konzistentní a zahrnuje především expresi markerů příčně pruhované svaloviny jako desmin (obr. 2C) a MyoD1 (obr. 2D) a dále expresi neurogenních markerů - S100 proteinu (obr. 2E) a GFAP (obr. 2F). Ačkoliv biologicky jde často o vysoce maligní tumory, v námi připravované sestavě měla část tumorů méně agresivní průběh než jaký udává dosud dostupná literatura (nepublikovaný nález). Rozlišení některých těchto případů od RMS na základě morfologie není spolehlivě možné. K indikaci molekulárně genetického vyšetření, které je pro určení diagnózy nezbytné, může navést mnohem pestřejší imunoprofil EWSR1-PATZ1 sarkomů.

Bifenotypický sinonazální sarkom je lokálně agresivní tumor sinonazální oblasti, který kromě S100 proteinu a hladkosvalového aktinu může v části případů fokálně exprimovat MyoD1 či myogenin (11). Dalším tumorem, u kterého lze občas nalézt expresi rhabdomyosarkomových markerů, je mezenchymální chondrosarkom (19).
Vaskulární tumory
V porovnání s ostatními nádory měkkých tkání hraje IHC v diagnostice většiny vaskulárních tumorů spíše omezenou roli. Její hlavní úloha spočívá v potvrzení vaskulárního původu u špatně diferencovaných lézí, výhodná může být též pro zvýraznění architektoniky vaskulární léze. Výjimku představují epiteloidní vaskulární tumory, pro které jsou dnes k dispozici dvě velmi specifické a diagnosticky užitečné protilátky FOSB a CAMTA1 (viz dále). Nesmírně nápomocná je též protilátka HHV8, jejíž nukleární exprese je, až na naprosté výjimky, specifická a senzitivní pro Kaposiho sarkom a velmi usnadnila stanovení této často obtížné diagnózy (20,21).
Nejlepšími vaskulárními markery jsou protilátky ERG a CD31. Alternativně je možné použít CD34, Von Willebrandův faktor (Faktor VIII) či jiný endoteliální marker, ve všech případech jde však již o markery méně specifické či senzitivní (22).
ERG je výhodný nejen pro svoji vysokou senzitivitu a v rámci vaskulárních lézí i vysokou specificitu (23) a vzhledem k nukleární expresi je i interpretace tohoto barvení většinou bezproblémová. U protilátky s afinitou k N-konci ERG proteinu je třeba pamatovat na její pozitivitu s velkou částí epiteloidních sarkomů (viz dále)(24). Stejný typ protilátky je diagnosticky užitečný i jako dobrý marker chondrogenní diferenciace (25,26). Mimo skupinu vaskulárních lézí reaguje rovněž s některými extramedulárními myeloidními tumory (27) a také s neoplaziemi, kde se vyskytuje fúzní gen ERG, tedy zhruba s 5 % Ewingových sarkomů (28) a polovinou prostatických adenokarcinomů (29). Užitečná může být exprese ERG v 90 % fosfaturických mezenchymálních tumorů (30) a ve všech EWSR1-SMAD3 rearanžovaných tumorech (31,32).
CD31 má obdobně vynikající senzitivitu i specificitu jako ERG. Hlavní nástraha tohoto barvení spočívá v jeho afinitě k histiocytům/makrofágům, což může vést k mylné diagnóze vaskulárního tumoru (obr. 3)(33). Obdobný chyták platí i pro některé histiocytární léze, např. histiocytární sarkom (33), histiocytózu z Langerhansových buněk (34) či juvenilní xantogranulom (35), které mohou být též CD31 pozitivní. U poslední jmenované léze jde zároveň o užitečný diagnostický znak (obr. 4A). Způsob exprese protilátky CD31 v histiocytech/makrofázích se však od pozitivity ve vaskulárních lézích liší. Zatímco v prvém případě jde o slabší granulární membranózní pozitivitu (obr. 4A), u vaskulárních lézí se vždy jedná o silnou a lineární membranózní pozitivitu spolu s cytoplazmatickou pozitivitou (obr. 4B)(33). Pokud jsou přítomny, k porovnání lze využít nenádorové endotelie v okolí


Všechny zmiňované markery reagují s nenádorovými endoteliemi jak krevních, tak lymfatických cév. Existuje však několik protilátek, které jsou v tomto kontextu specifické pro lymfatické cévy a nenádorové krevní endotelie nebarví. Nejpoužívanější z nich jsou podoplanin (D2-40) a prox-1. Zatímco podoplanin se barví na cytoplazmatické membráně a/nebo v cytoplazmě, exprese prox-1 je nukleární. Ani jeden z těchto markerů ovšem není specifický pouze pro lymfatické cévy a reaguje v různé míře nejen s většinou vaskulárních tumorů (22), ale i s řadou dalších mezenchymálních a epiteliálních tumorů (36,37). Za zmínku stojí reaktivita podoplaninu v mnoha karcinomech (zejména v kožních adnexálních), seminomech a také v mezoteliích a mezoteliomech. To může být využito v diferenciální diagnóze s některými adenokarcinomy, zejména plicními, kde je podoplanin většinou negativní (36,38).
Epiteloidní hemangiom je benigní vaskulární tumor, který může v některých případech být obtížně odlišitelný od reaktivních vaskulárních lézí či jiných vaskulárních tumorů včetně těch maligních. Proto přijde vhod, že přibližně polovina epiteloidních hemangiomů – částečně ve shodě se svým genetickým pozadím charakterizovaným výskytem FOSB či FOS genových fúzí – vykazuje nukleární imunoreaktivitu s protilátkou FOSB, přičemž jde o poměrně specifický nález. Z morfologického hlediska lze epiteloidní hemangiomy rozdělit na tři podtypy, z nichž každý reaguje s FOSB s jinou frekvencí: konvenční podtyp - 75 % případů je pozitivních (obr. 5 A,B), celulární - 10 % případů je pozitivních, a podtyp zvaný angiolymfoidní hyperplazie s eosinofilií, kde byla reaktivita pozorována ve všech případech (39). Jediný další známý konzistentně FOSB-pozitivní tumor je pseudomyogenní hemangioendoteliom, který rovněž patří k epiteloidním vaskulárním neoplaziím. Vzhledem ke své morfologii byl poprvé popsaný pod názvem epithelioid sarcoma-like hemangioendothelioma (40). V jeho patogenezi hrají hlavní roli rearanže genů FOSB (SERPINE1-FOSB či ACTB1-FOSB)(41), a proto takřka všechny případy exprimují FOSB protein v nádorových jádrech (39). To je v praxi velmi cenné, protože diferenciální diagnostika pseudomyogenního hemangioendoteliomu je obzvláště obtížná. Tento tumor exprimuje vaskulární markery (CD31, ERG) i cytokeratiny a morfologicky i imunohistochemicky se překrývá jak s epiteloidním sarkomem, tak s epiteloidním angiosarkomem a epiteloidním hemangioendoteliomem. Rozlišení má velký klinický význam, neboť pseudomyogenní hemangioendoteliom je převážně lokálně agresivní tumor a metastazuje extrémně vzácně, v protikladu k ostatním zmíněným jednotkám.

Kromě protilátky FOSB může ve zmíněné diferenciální diagnostice velmi pomoci protilátka CAMTA1, jejíž nukleární exprese je téměř 100% specifická a zároveň velmi senzitivní pro epiteloidní hemangioendoteliom vznikající na podkladě genové fúze WWTR1-CAMTA1(42,43). Zhruba 5-10 % tumorů, které se dnes řadí mezi epiteloidní hemangioendoteliomy, vzniká fúzí genů YAP1-TFE3, tyto jsou negativní s CAMTA1 protilátkou a naopak vykazují difúzní nukleární expresi TFE3 (44). Bohužel protilátka TFE3 se ukazuje jako velmi nespecifická, a tudíž nepříliš užitečná (45). Je možné, že YAP1-TFE3 rearanžované epiteloidní hemangioendoteliomy budou v budoucnu vyčleněny jako svébytná jednotka, neboť oproti klasickým epiteloidním hemangioendoteliomům mají určité klinickopatologické odlišnosti (46). Je u nich pozorována predilekce pro mladší pacienty (průměr okolo 30 let), morfologicky je zde mnohem více vyjádřená vazoformativní aktivita a výrazná myxochondroidní matrix typická pro klasickou variantu téměř vždy chybí (44,46).
V problematice vaskulárních lézí dětského věku má své důležité místo protilátka GLUT1. Ta je velmi užitečná jako marker infantilních hemangiomů, jejichž léčba se podstatně liší oproti léčbě vaskulárních malformací a ostatních typů hemangiomů. Obě tyto dvě skupiny jsou přitom GLUT1 negativní, stejně jako některé kongenitální hemangiomy (47, 48). Mimo specializované mikrovaskulární struktury (cévy placenty, retiny či hematoencefalické bariéry) jsou normální cévy též GLUT1 negativní (48).
Epiteloidní sarkom (ES)
Reaktivita tohoto tumoru s nízko i vysokomolekulárními cytokeratiny, případně s protilátkou EMA, je notoricky známá a v praxi značně užitečná vlastnost. Některé případy ES je však obtížné odlišit od dalších cytokeratin pozitivních epiteloidních tumorů jako např. metastatického karcinomu, epiteloidního angiosarkomu či např. pseudomyogenního hemangioendoteliomu (dříve též nazývaného epithelioid sarcoma-like hemangioendothelioma, viz výše (40)). Pro některé tyto situace se jako velmi praktická ukazuje protilátka SMARCB1 (INI1). Delece či epigenetické změny genu SMARCB1 stojí za většinou ES a v 90 % případů mají za následek ztrátu tvorby tohoto proteinu v nádorových buňkách (49). Protože všechny ostatní buňky v těle tento protein tvoří a exprimují, je ztráta nukleární exprese v rámci měkkých tkání poměrně specifická pro ES. Pouze zhruba 70 % epiteloidních maligních tumorů z pochev periferních nervů, 40 % epiteloidních schwannomů a několik dalších vzácných sarkomů je také SMARCB1 deficientních (49-52). Rovněž rozlišení od SMARCB1 deficientních karcinomů (52) touto metodou samozřejmě možné není. K tomu se naopak může hodit protilátka CD34, která je pozitivní až v 60 % ES, zatímco u karcinomů (nejen SMARB1 deficientních) zcela výjimečně. V diferenciální diagnostice s epiteloidními vaskulárními lézemi je třeba pamatovat na reaktivitu ES s některými typy protilátek ERG, a to až v 70 % případů (24). Zde je jako vaskulární marker vhodnější použití protilátky CD31, jejíž exprese se u ES nevyskytuje.
USP6-rearanžované léze
Mezi diagnosticky nejvýznamnější novinky v patologii mezenchymálních tumorů patří bezpochyby objev rearanže genu USP6 v několika relativně běžných a často velmi obtížně diagnostikovatelných lézích. Mezi ně patří především nodulární fasciitida, myositis ossificans, fibrooseózní pseudotumor prstu, aneuryzmatická kostní cysta a několik dalších tumorů, které však lze převážně považovat za anatomickou obdobu jmenovaných (53). Tyto benigní tumory navíc představují jedny z nejčastěji mylně diagnostikovaných lézí s potenciálně velmi vážnými klinickými důsledky, neboť jejich diferenciální diagnóza zahrnuje různé typy sarkomů. Translokace genu USP6 se nacházejí ve většině případů těchto lézí, u nodulární fasciitidy, v závislosti na studii, v70-90 % případů (54). Jejich detekce je snadno proveditelná např. metodou FISH.
Low-grade fibromyxoidní sarkom a sklerozující epiteloidní fibrosarkom
Další relativně recentně objevenou protilátkou je MUC4, která nebývalým způsobem usnadnila dříve velmi svízelnou diagnostiku jak sklerozujícího epiteloidního fibrosarkomu (SEF), tak zejména low-grade fibromyxoidního sarkomu (LFMS). Ačkoliv obě léze nejspíše představují morfologickou variantu jedné jednotky (obr. 6), díky výrazně odlišnému vzhledu se jejich diferenciální diagnostika značně liší. Morfologie SEF je poměrně charakteristická (obr. 6A) a mezi hlavní úkoly patologa patří odlišit tento sarkom od metastatického karcinomu, a to zejména lobulárního prsního, který je MUC4 negativní (55). K tomu nicméně poslouží i běžné širokospektré cytokeratiny, které jsou v SEF exprimovány zcela výjimečně (56). Možnost potvrzení diagnózy pomocí detekce exprese MUC4 je však i přesto velmi užitečná. Exprese MUC4 je přítomna ve zhruba 80% případů SEF (obr. 6B)(57) a dle velmi nedávno publikovaných studií představuje většina zbylých případů morfologicky prakticky identickou, avšak imunohistochemicky a molekulárně geneticky odlišnou lézi vyznačující se nejčastěji rekurentními rearanžemi genů KMT2A a YAP1(58,59).

Diagnosticky daleko problematičtější jsou tumory ze spektra LFMS, jejichž často velmi blandní vzhled (obr. 6A) snadno vede k záměně za některý z benigních vřetenobuněčných tumorů, zejména pak za perineuriom, nodulární fasciitidu či fibromatózu. Exprese EMA ve zhruba 90 % LFMS diagnózu ještě více komplikuje. Svému jménu navzdory je LFMS, podobně jako SEF, high-grade sarkom, který může recidivovat či metastazovat i několik dekád po prvotní diagnóze, a na který může nakonec zemřít až polovina pacientů (60). Z tohoto důvodu je správná diagnóza zásadní, jelikož kompletní široká excize a doživotní dispenzarizace výrazně zlepšuje prognózu pacientů (61). Exprese MUC4 je přítomna ve velké většině LFMS (obr. 6B)(62), vzácné případy mohou být MUC4 negativní a takové budou opět nejspíše představovat výše zmíněné sarkomy s KMT2A a YAP1 rearanžemi (58,59). Difúzní silná MUC4 pozitivita je velmi specifická pro LFMS a SEF. Výjimku (v rámci měkkých tkání) představuje pouze malý zlomek angiomatoidních fibrózních histiocytomů (63) a především extrakraniální meningiom, kde je difúzní a silná exprese MUC4 natolik konstantním nálezem, že bude nejspíše i užitečnou diagnostickou pomůckou (64). Z jiných lézích byla pouze fokální exprese nalezena u zhruba třetiny synoviálních sarkomů a osifikujících fibromyxoidních tumorů (57,62).
Mezenchymální tumory s kinázovými fúzemi (např. NTRK, BRAF, RET)
Se zavedením metod masivního paralelního sekvenování (next generation sequencing; NGS) do diagnostické praxe se v posledních několika letech čím dál častěji popisují tumory měkkých tkání s translokacemi kinázových genů jako např. NTRK, BRAF, RAF1, RET a dalších (65-69). Tyto tumory častěji postihují dětskou část populace, ale zcela identické léze se mohou vyskytovat i u dospělých. Některé z nich představují morfologicky blandní a biologicky převážně indolentní neoplazie s velmi pestrou a často necharakteristickou morfologií. Zbytek pak tvoří sarkomy s mikroskopickým obrazem spadajícím do kategorie infantilního/adultního fibrosarkomu, přičemž minimálně část z těchto sarkomů vzniká neoplastickou progresí tumorů z první skupiny (65,70). Ačkoliv některé z těchto lézí mají necharakteristický imunoprofil, relativně často se u nich vyskytuje exprese CD34 a/nebo S100 proteinu (70). Imunomorfologicky charakteristickou podskupinou jsou pak benigní fibroblastické tumory s nápadnou stromální a perivaskulární hyalinizací (obr. 7A, B), konzistentní CD34 (obr. 7C) a S100 koexpresí (obr. 7D) a translokacemi genů NTRK, RAF1, BRAF či RET (67,69). U této podskupiny je sarkomatózní transformace dobře zdokumentována a pravděpodobně bude poměrně častým jevem (obr. 7E), zejména u dlouhotrvajících a recidivujících lézí (67). Správná diagnostika je důležitá jak vzhledem k nutnosti kompletní excize, jež spolehlivě zabraňuje recidivám, tak ke zvyšující se dostupnosti cílené léčby pro pokročilé či metastatické tumory (65).

Maligní tumor z pochev periferních nervů (MPNST)
Odlišení MPNST od morfologicky podobných tumorů je často velmi obtížné. Před několika lety bylo zjištěno, že vlivem některých genetických změn dochází u velké části MPNST ke ztrátě trimetylace lyzinu 27 histonu H3 (H3K27me3), což lze imunohistochemicky detekovat absencí exprese tohoto proteinu (71). Později bylo bohužel opakovaně prokázáno, že interpretace tohoto barvení je nezřídka problematická vzhledem k častému výskytu pouze parciální ztráty exprese, která je pro MPNST daleko méně specifická. Především se však ukázalo, že i specificita kompletní ztráty exprese H3K27me3 je pro MPNST nižší, než ukazovaly prvotní studie (72,73) a protilátka tak na mnoha pracovištích pozbyla využití. Velmi recentně bylo zjištěno, že pokud se namísto trimethylované formy H3K27 detekuje forma dimethylovaná (H3K27me2), interpretace barvení je mnohem snazší a dosažené výsledky jsou pro MPNST podstatně specifičtější (74). Na základě našich úvodních zkušeností s protilátkou H3K27me2 lze konstatovat, že minimálně „odečítání“ tohoto barvení je skutečně jednoznačnější než u H3K27me3. K ověření specificity budou potřeba další nezávislé studie. V tomto časopise bylo o této problematice podrobněji pojednáno v rubrice monitor v čísle 2/2020.
Superficiální CD34 pozitivní fibroblastický tumor a PRDM10-rearanžovaný tumor
Tento teprve v roce 2014 charakterizovaný tumor se vyskytuje obvykle u adolescentů či mladších dospělých a má predilekci pro kůži a podkoží proximálnějších oblastí končetin, zejména stehen. Je složený z relativně buněčných snopců vřetenitých až epiteloidních buněk (obr. 8A). Morfologicky nejnápadnějšími znaky je většinou hojná, výrazně eozinofilní, granulární až sklovitá cytoplazma a výrazný jaderný pleomorfismus s častými nukleárními pseudoinkluzemi (75). V nápadném protikladu k výše zmíněnému je rovněž obvykle velmi nízká mitotická aktivita (obr. 8B). Mimo difúzní exprese CD34 (obr. 8C) může být velmi užitečná častá a typicky pouze fokální exprese cytokeratinů (obr. 8D). Znalost tohoto tumoru je významná, protože přes svůj na první pohled maligní vzhled – hlavní diferenciální diagnóza je nediferencovaný pleomorfní sarkom – se jedná o zcela indolentní tumor (75,76).

Ještě recentněji byly publikovány tumory s velmi podobnou morfologií, imunofenotypem i klinickými vlastnostmi, u kterých však byla molekulárně geneticky detekována rearanže genůCITED2 či MED12 s genem PRDM10. Jediným rozdílem se zdá být o něco hlubší anatomická lokalizace a vyšší mitotická aktivita u PRDM10-rearanžovaných tumorů. Autoři této práce měli možnost testovat i tumory dříve diagnostikované jako superficiální CD34 pozitivní fibroblastický tumor a 3/7 případů skutečně obsahovalo PRDM10 translokaci, zbylé 4 případy byly negativní. Vše tedy nasvědčuje tomu, že oba zde popisované tumory patří do jedné skupiny, u části z nich je však genetické pozadí zatím neznámé (77).
Solitární fibrózní tumor (SFT)
Do této nádorové entity je dnes zahrnován jak tradičně popisovaný SFT, tak dříve odděleně klasifikovaný hemangiopericytom, který, jak se ukázalo, představuje pouze celulární variantu SFT. Definitivní potvrzení, že se jedná o jednu a tutéž jednotku přinesl objev rekurentního fúzního genu NAB2-STAT6 ve všech těchto tumorech (78,79). Tato genetická změna vede k nukleární overexpresi proteinu STAT6 prakticky ve všech případech klasického a maligního SFT, a navíc se jedná o nález velmi specifický. Z morfologicky zaměnitelných lézí vykazuje pouze zhruba 10 % dediferencovaných liposarkomů převážně slabou nukleární expresi STAT6 (80,81). Vysoká specificita je o to cennější, že SFT je jednou z nejvíce morfologicky variabilních neoplazií měkkých tkání, zejména v případě maligní či dediferencované varianty. Dediferencovaný SFT se většinou nachází v těsném sousedství klasického SFT a může nabývat podoby kulatobuněčného (obr. 9A) či pleomorfního sarkomu, zde může ovšem exprese STAT6 (obr. 9B) nezřídka chybět. (82).

ZÁVĚR
Cílem tohoto přehledového článku bylo zprostředkovat některé z nejvýznamnějších novinek v diagnostice nádorů měkkých tkání. Přes neobyčejný pokrok v této části patologie se však stále běžně setkáváme s nádory, které na základě morfologie a imunohistochemie nelze nejen jednoznačně diagnostikovat, ale ani s dostatečnou mírou pravděpodobnosti směřovat k molekulárnímu genetickému potvrzení diagnózy metodami jako jsou FISH či RT-PCR, tedy metodami schopnými analyzovat jeden konkrétní gen. Ačkoliv je jistě pravda, že v řadě případů pro následný klinický management postačí diagnózy jako „neklasifikovatelný benigní mezenchymální tumor“ případně „nediferencovaný sarkom“, má podobná aproximativní klasifikace mnoho nedostatků. V první radě, zdaleka ne všechny blandně vyhlížející tumory jsou skutečně benigní, viz např. LGFMS. To samé platí i naopak, viz např. nodulární fasciitida či myositis ossificans. Navíc, i mezi vysoce maligními sarkomy je rozdíl v prognóze a velmi často i ve zvolené terapii. Správná klasifikace je klíčová i pro správný odhad maligního potenciálu. Kupříkladu celulární a mitoticky aktivní myofibrom je stále benigním tumorem, zatímco při záměně za leiomyomatózní tumor by nález mitotické aktivity vedl k mylné diagnóze leiomyosarkomu. V neposlední řadě je pak správná klasifikace nezbytná pro další medicínský výzkum, detekci vrozených nádorových syndromů a čím dál častěji i pro indikaci cílené terapie či zařazení do klinických studií.

Poslední možností, jak klasickými metodami nezařaditelné tumory správně klasifikovat, jsou metody masivního paralelního sekvenování (next generation sequencing; NGS). Nejčastěji se pomocí této technologie aplikují tzv. genové panely, a to buď komerčně dostupné či na míru vyrobené, které obsahují předdefinovaný soubor genů, o kterých je známo, že jejich translokace či mutace způsobují nádorové bujení. Pomocí těchto panelů pak lze vyšetřit několik desítek až stovek genů najednou a je tedy nasnadě, že podobná analýza výrazně zvyšuje pravděpodobnost detekce specifické aberace a následně možnost správné klasifikace. Pro ilustraci je uveden genový panel, který na našem pracovišti nejčastěji používáme k diagnostice sarkomů. Jde o na míru vytvořený genový panel (ArcherDx), schopný detekovat takřka veškeré známé a diagnosticky využitelné fúzní geny a některé mutace vyskytující se v mezenchymálních nádorech nejen měkkých tkání. Do budoucna lze očekávat, že tato metoda dále zlevní a do velké míry nahradí dnes široce používané, avšak méně „výkonné“ metody jako FISH či RT-PCR.
PROHLÁŠENÍ
Autor práce prohlašuje, že v souvislosti s tématem, vznikem a publikací tohoto článku není ve střetu zájmů a vznik ani publikace článku nebyly podpořeny žádnou farmaceutickou firmou. Tato studie byla podpořena Národním programem udržitelnosti I (NPU I) č. LO1503 a grantem SVV–2020 No. 260 391 poskytnutého Ministerstvem sportu, mládeže a tělovýchovy České republiky.
∗ Adresa pro korespondenci:
MUDr. Michael Michal, Ph.D.
Šiklův ústav patologie LF UK v Plzni a FN Plzeň,
Alej Svobody 80, 304 60 Plzeň,
tel.: +420 603 792 671
e-mail: michael.michal@medima.cz
Sources
1. Kinkor Z, Grossmann P, Dubová M, et al. Co nového v Ewing-like family aneb malobuněčné/kulatobuněčné sarkomy měkkých tkání a kostí s rearanží genu CIC a BCOR. Přehled problematiky a naše prvotní zkušenosti. Cesk Patol; 53(4): 175-180.
2. Michal M, Kazakov DV, Michal M. Hybrid peripheral nerve sheath tumors: a review. Cesk Patol; 53(2): 81-88.
3. Švajdler M, Zambo I, Michal M, Kinkor Z, Michal M. Nádory měkkých tkání. Doporučený postup pro bioptické vyšetření. Společnost českých patologů, ČSL JEP, 2019.http://www.patologie.info/soubory/all/2018-4_Guideline-web-v1.pdf
4. Wang NP, Marx J, McNutt MA, Rutledge JC, Gown AM. Expression of myogenic regulatory proteins (myogenin and MyoD1) in small blue round cell tumors of childhood. Am J Pathol 1995; 147(6): 1799-1810.
5. Morotti RA, Nicol KK, Parham DM, et al. An immunohistochemical algorithm to facilitate diagnosis and subtyping of rhabdomyosarcoma: the Children’s Oncology Group experience. Am J Surg Pathol 2006; 30(8): 962-968.
6. Dias P, Chen B, Dilday B, et al. Strong immunostaining for myogenin in rhabdomyosarcoma is significantly associated with tumors of the alveolar subclass. Am J Pathol 2000; 156(2): 399-408.
7. Charville GW, Varma S, Forgo E, et al. PAX7 expression in rhabdomyosarcoma, related soft tissue tumors, and small round blue cell neoplasms. Am J Surg Pathol 2016; 40(10): 1305-1315.
8. Charville GW, Wang WL, Ingram DR, et al. EWSR1 fusion proteins mediate PAX7 expression in Ewing sarcoma. Mod Pathol 2017; 30(9): 1312-1320.
9. Leiner J, Le Loarer F. The current landscape of rhabdomyosarcomas: an update. Virchows Arch 2020; 476(1): 97-108.
10. Borinstein SC, Steppan D, Hayashi M, et al. Consensus and controversies regarding the treatment of rhabdomyosarcoma. Pediatr Blood Cancer 2018; 65(2): 10.1002/pbc.26809.
11. Alaggio R, Zhang L, Sung YS, et al. A molecular study of pediatric spindle and sclerosing rhabdomyosarcoma: identification of novel and recurrent VGLL2-related fusions in infantile cases. Am J Surg Pathol 2016; 40(2): 224-235.
12. Le Loarer F, Cleven AHG, Bouvier C, et al. A subset of epithelioid and spindle cell rhabdomyosarcomas is associated with TFCP2 fusions and common ALK upregulation. Mod Pathol2020; 33(3): 404-419.
13. Brunac AC, Laprie A, Castex MP, et al. The combination of radiotherapy and ALK inhibitors is effective in the treatment of intraosseous rhabdomyosarcoma with FUS-TFCP2 fusion transcript. Pediatr Blood Cancer 2020: e28185.
14. Michal M, Rubin BP, Kazakov DV, et al. Inflammatory leiomyosarcoma shows frequent co-expression of smooth and skeletal muscle markers supporting a primitive myogenic phenotype: a report of 9 cases with a proposal for reclassification as low-grade inflammatory myogenic tumor. Virchows Arch. In press 2020.
15. Martinez AP, Fritchie KJ, Weiss SW, et al. Histiocyte-rich rhabdomyoblastic tumor: rhabdomyosarcoma, rhabdomyoma, or rhabdomyoblastic tumor of uncertain malignant potential? A histologically distinctive rhabdomyoblastic tumor in search of a place in the classification of skeletal muscle neoplasms. Mod Pathol 2019; 32(3): 446-457.
16. Merchant W, Calonje E, Fletcher CD. Inflammatory leiomyosarcoma: a morphological subgroup within the heterogeneous family of so-called inflammatory malignant fibrous histiocytoma. Histopathology 1995; 27(6): 525-532.
17. Bridge JA, Sumegi J, Druta M, et al. Clinical, pathological, and genomic features of EWSR1-PATZ1 fusion sarcoma. Mod Pathol 2019; 32(11): 1593-1604.
18. Chougule A, Taylor MS, Nardi V, et al. Spindle and round cell sarcoma with EWSR1-PATZ1 gene fusion: asarcoma with polyphenotypic differentiation. Am J Surg Pathol 2019; 43(2): 220-228.
19. Folpe AL, Graham RP, Martinez A, Schembri-Wismayer D, Boland J, Fritchie KJ. Mesenchymal chondrosarcomas showing immunohistochemical evidence of rhabdomyoblastic differentiation: a potential diagnostic pitfall. Hum Pathol 2018; 77 : 28-34.
20. Patel RM, Goldblum JR, Hsi ED. Immunohistochemical detection of human herpes virus-8 latent nuclear antigen-1 is useful in the diagnosis of Kaposi sarcoma. Mod Pathol 2004; 17(4): 456-460.
21. Hammock L, Reisenauer A, Wang W, Cohen C, Birdsong G, Folpe AL. Latency-associated nuclear antigen expression and human herpesvirus-8 polymerase chain reaction in the evaluation of Kaposi sarcoma and other vascular tumors in HIV-positive patients. Mod Pathol 2005; 18(4): 463-468.
22. Ordonez NG. Immunohistochemical endothelial markers: a review. Adv Anat Pathol 2012; 19(5): 281-295.
23. Miettinen M, Wang ZF, Paetau A, et al. ERG transcription factor as an immunohistochemical marker for vascular endothelial tumors and prostatic carcinoma. Am J Surg Pathol 2011; 35(3): 432-441.
24. Stockman DL, Hornick JL, Deavers MT, Lev DC, Lazar AJ, Wang WL. ERG and FLI1 protein expression in epithelioid sarcoma. Mod Pathol 2014; 27(4): 496-501.
25. Shon W, Folpe AL, Fritchie KJ. ERG expression in chondrogenic bone and soft tissue tumours. J Clin Pathol 2015; 68(2): 125-129.
26. Creytens D. ERG expression in chondrogenic bone and soft tissue tumours: importance of antibody clone. Comment on Shon et al (2015). J Clin Pathol 2015; 68(12): 1043.
27. Xu B, Naughton D, Busam K, Pulitzer M. ERG is a useful immunohistochemical marker to distinguish leukemia cutis from nonneoplastic leukocytic infiltrates in the skin. Am J Dermatopathol 2016; 38(9): 672-677.
28. Wang WL, Patel NR, Caragea M, et al. Expression of ERG, an Ets family transcription factor, identifies ERG-rearranged Ewing sarcoma. Mod Pathol 2012; 25(10): 1378-1383.
29. Chaux A, Albadine R, Toubaji A, et al. Immunohistochemistry for ERG expression as a surrogate for TMPRSS2-ERG fusion detection in prostatic adenocarcinomas. Am J Surg Pathol 2011; 35(7): 1014-1020.
30. Agaimy A, Michal M, Chiosea S, et al. Phosphaturic mesenchymal tumors: clinicopathologic, immunohistochemical and molecular analysis of 22 cases expanding their morphologic and immunophenotypic spectrum. Am J Surg Pathol 2017; 41(10): 1371-1380.
31. Kao YC, Flucke U, Eijkelenboom A, et al. Novel EWSR1-SMAD3 Gene Fusions in a Group of Acral Fibroblastic Spindle Cell Neoplasms. Am J Surg Pathol 2018; 42 : 522-528.
32. Michal M, Berry RS, Rubin BP, et al. EWSR1-SMAD3-rearranged fibroblastic tumor: an emerging entity in an increasingly more complex group of fibroblastic/myofibroblastic neoplasms. Am J Surg Pathol 2018; 42(4): 1325-1333.
33. McKenney JK, Weiss SW, Folpe AL. CD31 expression in intratumoral macrophages: a potential diagnostic pitfall. Am J Surg Pathol 2001; 25(9): 1167-1173.
34. Slone SP, Fleming DR, Buchino JJ. Sinus histiocytosis with massive lymphadenopathy and Langerhans cell histiocytosis express the cellular adhesion molecule CD31. Arch Pathol Lab Med 2003; 127(3): 341-344.
35. Vanchinathan V, Mirzamani N, Kantipudi R, Schwartz EJ, Sundram UN. The vascular marker CD31 also highlights histiocytes and histiocyte-like cells within cutaneous tumors. Am J Clin Pathol 2015; 143(2): 177-185; quiz 305.
36. Xu Y, Ogose A, Kawashima H, et al. High-level expression of podoplanin in benign and malignant soft tissue tumors: immunohistochemical and quantitative real-time RT-PCR analysis. Oncol Rep 2011; 25(3): 599-607.
37. Miettinen M, Wang ZF. Prox1 transcription factor as a marker for vascular tumors-evaluation of 314 vascular endothelial and 1086 nonvascular tumors. Am J Surg Pathol 2012; 36(3): 351-359.
38. Husain AN, Colby TV, Ordonez NG, et al. Guidelines for pathologic diagnosis of malignant mesothelioma 2017 update of the consensus statement from the international mesothelioma interest group. Arch Pathol Lab Med 2018; 142(1): 89-108.
39. Hung YP, Fletcher CD, Hornick JL. FOSB is a useful diagnostic marker for pseudomyogenic hemangioendothelioma. Am J Surg Pathol 2017; 41(5): 596-606.
40. Billings SD, Folpe AL, Weiss SW. Epithelioid sarcoma-like hemangioendothelioma. Am J Surg Pathol 2003; 27(1): 48-57.
41. Agaram NP, Zhang L, Cotzia P, Antonescu CR. Expanding the spectrum of genetic alterations in pseudomyogenic hemangioendothelioma with recurrent novel ACTB-FOSB gene fusions. Am J Surg Pathol 2018; 42(12): 1653-1661.
42. Tanas MR, Sboner A, Oliveira AM, et al. Identification of a disease-defining gene fusion in epithelioid hemangioendothelioma. Sci Transl Med 2011; 3(98): 98ra82.
43. Shibuya R, Matsuyama A, Shiba E, Harada H, Yabuki K, Hisaoka M. CAMTA1 is a useful immunohistochemical marker for diagnosing epithelioid haemangioendothelioma. Histopathology 2015; 67(6): 827-835.
44. Antonescu CR, Le Loarer F, Mosquera JM, et al. Novel YAP1-TFE3 fusion defines a distinct subset of epithelioid hemangioendothelioma. Genes Chromosomes Cancer 2013; 52(8): 775-784.
45. Sharain RF, Gown AM, Greipp PT, Folpe AL. Immunohistochemistry for TFE3 lacks specificity and sensitivity in the diagnosis of TFE3-rearranged neoplasms: a comparative, 2-laboratory study. Hum Pathol 2019; 87 : 65-74.
46. Habeeb O, Rubin BP. The molecular diagnostics of vascular neoplasms. Surg Pathol Clin 2019; 12(1): 35-49.
47. North PE, Waner M, James CA, Mizeracki A, Frieden IJ, Mihm MC, Jr. Congenital nonprogressive hemangioma: a distinct clinicopathologic entity unlike infantile hemangioma. Arch Dermatol 2001; 137(12): 1607-1620.
48. North PE, Waner M, Mizeracki A, Mihm MC, Jr. GLUT1: a newly discovered immunohistochemical marker for juvenile hemangiomas. Hum Pathol 2000; 31(1): 11-22.
49. Hornick JL, Dal Cin P, Fletcher CD. Loss of INI1 expression is characteristic of both conventional and proximal-type epithelioid sarcoma. Am J Surg Pathol 2009; 33(4): 542-550.
50. Schaefer IM, Dong F, Garcia EP, Fletcher CDM, Jo VY. Recurrent SMARCB1 inactivation in epithelioid malignant peripheral nerve sheath tumors. Am J Surg Pathol 2019; 43(6): 835-843.
51. Jo VY, Fletcher CDM. SMARCB1/INI1 loss in epithelioid schwannoma: aclinicopathologic and immunohistochemical study of 65 Cases. Am J Surg Pathol 2017; 41(8): 1013-1022.
52. Agaimy A. The expanding family of SMARCB1(INI1)-deficient neoplasia: implications of phenotypic, biological, and molecular heterogeneity. Adv Anat Pathol 2014; 21(6): 394-410.
53. Švajdler M, Michal M, Martínek P, et al. Fibro-osseous pseudotumor of digits and myositis ossificans show consistent COL1A1-USP6 rearrangement: a clinicopathological and genetic study of 27 cases. Hum Pathol 2019; 88 : 39-47.
54. Oliveira AM, Chou MM. USP6-induced neoplasms: the biologic spectrum of aneurysmal bone cyst and nodular fasciitis. Hum Pathol 2014; 45(1): 1-11.
55. Kasashima S, Kawashima A, Zen Y, et al. Expression of aberrant mucins in lobular carcinoma with histiocytoid feature of the breast. Virchows Arch 2007; 450(4): 397-403.
56. Meis-Kindblom JM, Kindblom LG, Enzinger FM. Sclerosing epithelioid fibrosarcoma. A variant of fibrosarcoma simulating carcinoma. Am J Surg Pathol 1995; 19(9): 979-993.
57. Doyle LA, Wang WL, Dal Cin P, et al. MUC4 is a sensitive and extremely useful marker for sclerosing epithelioid fibrosarcoma: association with FUS gene rearrangement. Am J Surg Pathol 2012; 36(10): 1444-1451.
58. Kao YC, Lee JC, Zhang L, et al. Recurrent YAP1 and KMT2A gene rearrangements in a subset of MUC4-negative sclerosing epithelioid fibrosarcoma. Am J Surg Pathol 2020; 44(3): 368-377.
59. Puls F, Agaimy A, Flucke U, et al. Recurrent fusions between YAP1 and KMT2A in morphologically distinct neoplasms within the spectrum of low-grade fibromyxoid sarcoma and sclerosing epithelioid fibrosarcoma. Am J Surg Pathol. In press 2020.
60. Evans HL. Low-grade fibromyxoid sarcoma: a clinicopathologic study of 33 cases with long-term follow-up. Am J Surg Pathol 2011; 35(10): 1450-1462.
61. Folpe AL, Lane KL, Paull G, Weiss SW. Low-grade fibromyxoid sarcoma and hyalinizing spindle cell tumor with giant rosettes: a clinicopathologic study of 73 cases supporting their identity and assessing the impact of high-grade areas. Am J Surg Pathol 2000; 24(10): 1353-1360.
62. Doyle LA, Moller E, Dal Cin P, Fletcher CD, Mertens F, Hornick JL. MUC4 is a highly sensitive and specific marker for low-grade fibromyxoid sarcoma. Am J Surg Pathol 2011; 35(5): 733-741.
63. Abrahao-Machado LF, Bacchi LM, Fernandes IL, Costa FD, Bacchi CE. MUC4 expression in angiomatoid fibrous histiocytoma. Appl Immunohistochem Mol Morphol. In press 2019.
64. Matsuyama A, Jotatsu M, Uchihashi K, et al. MUC4 expression in meningiomas: under-recognized immunophenotype particularly in meningothelial and angiomatous subtypes. Histopathology 2019; 74(2): 276-283.
65. Davis JL, Lockwood CM, Stohr B, et al. Expanding the spectrum of pediatric NTRK-rearranged mesenchymal tumors. Am J Surg Pathol 2019; 43(4): 435-445.
66. Kao YC, Fletcher CDM, Alaggio R, et al. Recurrent BRAF gene fusions in a subset of pediatric spindle cell sarcomas: expanding the genetic spectrum of tumors with overlapping features with infantile fibrosarcoma. Am J Surg Pathol 2018; 42(1): 28-38.
67. Suurmeijer AJH, Dickson BC, Swanson D, et al. A novel group of spindle cell tumors defined by S100 and CD34 co-expression shows recurrent fusions involving RAF1, BRAF, and NTRK1/2 genes. Genes Chromosomes Cancer 2018; 57(12): 611-621.
68. Antonescu CR, Dickson BC, Swanson D, et al. Spindle cell tumors with RET gene fusions exhibit a morphologic spectrum akin to tumors with NTRK gene fusions. Am J Surg Pathol 2019; 43(10): 1384-1391.
69. Michal M, Ptáková N, Martínek P, et al. S100 and CD34 positive spindle cell tumor with prominent perivascular hyalinization and a novel NCOA4-RET fusion. Genes Chromosomes Cancer 2019; 58(9): 680-685.
70. Agaram NP, Zhang L, Sung YS, et al. Recurrent NTRK1 gene fusions define a novel subset of locally aggressive lipofibromatosis-like neural tumors. Am J Surg Pathol 2016; 40(10): 1407-1416.
71. Schaefer IM, Fletcher CD, Hornick JL. Loss of H3K27 trimethylation distinguishes malignant peripheral nerve sheath tumors from histologic mimics. Mod Pathol 2016; 29(1): 4-13.
72. Cleven AH, Sannaa GA, Briaire-de Bruijn I, et al. Loss of H3K27 tri-methylation is a diagnostic marker for malignant peripheral nerve sheath tumors and an indicator for an inferior survival. Mod Pathol 2016; 29(6): 582-590.
73. Pekmezci M, Reuss DE, Hirbe AC, et al. Morphologic and immunohistochemical features of malignant peripheral nerve sheath tumors and cellular schwannomas. Mod Pathol 2015; 28(2): 187-200.
74. Marchione DM, Lisby A, Viaene AN, et al. Histone H3K27 dimethyl loss is highly specific for malignant peripheral nerve sheath tumor and distinguishes true PRC2 loss from isolated H3K27 trimethyl loss. Mod Pathol 2019; 32(10): 1434-1446.
75. Carter JM, Weiss SW, Linos K, DiCaudo DJ, Folpe AL. Superficial CD34-positive fibroblastic tumor: report of 18 cases of a distinctive low-grade mesenchymal neoplasm of intermediate (borderline) malignancy. Mod Pathol 2014; 27(2): 294-302.
76. Lao IW, Yu L, Wang J. Superficial CD34-positive fibroblastic tumour: a clinicopathological and immunohistochemical study of an additional series. Histopathology 2017; 70(3): 394-401.
77. Puls F, Pillay N, Fagman H, et al. PRDM10-rearranged soft tissue tumor: aclinicopathologic study of 9 cases. Am J Surg Pathol 2019; 43(4): 504-513.
78. Robinson DR, Wu YM, Kalyana-Sundaram S, et al. Identification of recurrent NAB2-STAT6 gene fusions in solitary fibrous tumor by integrative sequencing. Nat Genet 2013; 45(2): 180-185.
79. Chmielecki J, Crago AM, Rosenberg M, et al. Whole-exome sequencing identifies a recurrent NAB2-STAT6 fusion in solitary fibrous tumors. Nat Genet 2013; 45(2): 131-132.
80. Demicco EG, Harms PW, Patel RM, et al. Extensive survey of STAT6 expression in a large series of mesenchymal tumors. Am J Clin Pathol 2015; 143(5): 672-682.
81. Doyle LA, Vivero M, Fletcher CD, Mertens F, Hornick JL. Nuclear expression of STAT6 distinguishes solitary fibrous tumor from histologic mimics. Mod Pathol 2014; 27(3): 390-395.
82. Dagrada GP, Spagnuolo RD, Mauro V, et al. Solitary fibrous tumors: loss of chimeric protein expression and genomic instability mark dedifferentiation. Mod Pathol 2015; 28(8): 1074-1083.
Labels
Anatomical pathology Forensic medical examiner ToxicologyArticle was published in
Czecho-Slovak Pathology

2021 Issue 1
Most read in this issue
- A basic immunohistochemical panel for the diagnosis of soft tissue tumors
- Bone lesions – diagnostic approach using immunohistochemistry and molecular pathology
- Secondary pulmonary hypoplasia associated with calcified Meckel´s diverticulum with osseous metaplasia
-
Consensus recommendations from the Czech Head and Neck Cancer Cooperative Group (2019):
definition of surgical margins status, neck dissection reporting, and HPV/p16 status assessment