
Arytmogenní dysplazie/kardiomyopatie pravé srdeční komory jako příčina náhlé smrti
Arrhythmogenic Right Ventricular Dysplasia / Cardiomyopathy (ARVD/C) as a Cause of Sudden Death
Common cause of sudden death in children and teenagers are primary cardiomyopathies, mostly hypertrophic. The authors refer to arrhythmogenic right ventricular dysplasia / cardiomyopathy as a possible cause of sudden death. The disease is not limited to the right hearth only, the presence of fat and scar tissue in the in the hearth muscle is variable.
Key words:
sudden cardiac death – arrhythmogenic right ventricular dysplasia / cardiomyopathy, ARVD/C
Authors:
I. Bouška 1; P. Klír 2; J. Markvartová 1
Authors‘ workplace:
Ústav soudního lékařství 2. LFUK a FN Na Bulovce Praha
1; Institut postgraduálního vzdělávání ve zdravotnictví, Praha
2
Published in:
Soud Lék., 53, 2008, No. 2, p. 18-20
Overview
Častou příčinou náhlé smrti u dětí a mladistvých jsou primární kardiomyopatie, nejčastěji hypertrofické. Autoři upozorňují na možnou příčinu náhlého úmrtí též při arytmogenní kardiomyopatii/dysplazii pravé srdeční komory. Onemocnění není vázáno pouze na pravou stranu srdce, ale přítomnost tukové a vazivové složky ve svalovině je variabilní.
Klíčová slova:
náhlá kardiální smrt – arytmogenní dysplazie / kardiomyopatie pravé srdeční komory
Úvod
Náhlá smrt u starších dětí a mladistvých není sice častým jevem a při autoptické diagnostice je nutné věnovat pozornost i diskrétním nálezům. Významnou roli v této problematice představuje skupina vrozených onemocnění srdce – kardiomyopatií (CM), a to ve věkové kategorii do 35 let (4). Pro soudnělékařskou diagnostiku je znalost všech popsaných typů tohoto onemocnění nezbytná a navíc může být taková náhlá smrt předmětem i znaleckého posuzování (2). Srdce nemusí být vždy při tomto onemocnění zvětšené (tj. přesahující 4 promile váhy těla) a při celkově nejasném sekčním nálezu je vždy nutné srdce podrobně vyšetřit. Samostatnou nozologickou jednotkou CM, popsanou v 80. letech minulého století, je arytmogenní dysplazie/kardiomyopatie pravé srdeční komory ARVD/C – Arrythmogenic right ventricular dysplasia/cardiomyopathy, původně považována za jistou formu kardiomyopatie hypertrofické – HCM (6).
Toto onemocnění, postihující pravou srdeční komoru, je považováno za možnou příčinu komorové tachyarytmie a též náhlé srdeční smrti u mladistvých, zejména při sportu (8).
Jsou známy dvě formy onemocnění – s dominantním a též s recesivním typem dědičnosti (Naxos disease) a z hlediska kauzální patogeneze se předpokládá, že se jedná o důsledek mutace možných až 7 genů, zejména cytoskeletálního genu – plakoglobinu a receptoru pro cardiac ryanodine gen (10).
Klinická diagnostika je obtížná a je založena na podrobném laboratorním vyšetření srdce zobrazovacími a funkčními metodami (11).
Pro morfologický průkaz onemocnění je významná ložisková přítomnost tuku a též vazivové složky ve svalstvu pravé komory. Tento nález nelze v žádném případě spojovat s přítomností tuku pod osrdcem v pravé srdeční komoře, označovaný jako lipomatózní atrofie myokardu (5). Z hlediska formální patogeneze tohoto onemocnění se uvažuje o možné apoptóze srdečních buněk, zánětu, nebo jako o důsledku vrozené dysplazie či transformace srdečních buněk (14).
Japonští autoři doporučují zařadit toto onemocnění (ARVD/C) do širšího kontextu akutních chorobných stavů a navrhují označení – ABCDE syndrom – to je zkráceně Arrhytmogenicity-Brugada Syndrom-Competitive sports-Death (sudden)-Electric Disturbance (12).
Onemocnění bylo též diagnostikováno u psů – boxerů (1).
V tomto sdělení demonstrujeme nálezy při této kardiomyopatii, které ukazují rozmanitost v lokalizaci postižení srdce u jednotlivých úmrtí. Postižení pravé komory srdeční nebylo výlučné, změny jsme zjistili i v jiných úsecích srdce, přitom ale náhlý průběh onemocnění zůstal
Vlastní pozorování
1. 10letý chlapec umírá náhle doma v říjnu 2007 v nočních hodinách, při probíhající akutní respirační infekci.
V anamnestických údajích je zaznamenán v 8 měsících věku pád z postele s nálezem fisury na kosti klenby lební, známky postižení mozku při následném neurologickém vyšetření neprokázány.
Od dvou let věku se k horečnatým stavům přidružovaly křeče, opakovaně sledován na neurologii, ale nebyla jednoznačně stanovena diagnóza, veden jako symptomatická epilepsie.
Poslední neurologické vyšetření bylo tři měsíce před úmrtím, na EEG zjištěn jen lehce abnormální záznam. U chlapce se též postupně rozvíjel obraz chronického zánětu průdušek s asthmoidními projevy.
Pitva byla provedena druhý den po smrti – přítomny známky výrazného městnání krve v orgánech, na játrech s četnými nekrózami v jaterních lalůčcích. V mikroskopickém nálezu byl edém mozku s perivaskulárními hemoragiemi a na neuronech nespecifické hypoxické změny. Ložiskové změny na mozku nalezeny nebyly.
Srdce nebylo zvětšené, hmotnost 145 g, pravá komora silně dilatovaná a uspořádání srdečních oddílů i cévních odstupů bylo fyziologické. Na mitrální chlopni byly drobné bradavčité výběžky a endokard výtokové části levé komory byl zkalený. Drobnohledné vyšetření přineslo objasnění až při vyšetření pravé srdeční komory a zejména srdečního septa, kde jsme mohli zjistit ohraničená depozita adipocytů s doprovodnou vazivovou složkou. Nápadné zde bylo postižení vodivého systému srdeční přepážky s ložisky tuku přímo v Hisově svazku (obr. 1). Známky vystupňovaného oběhového selhání ve velkém krevním oběhu odpovídalo pravostrannému srdečnímu selhání při této kardiomyopatii.
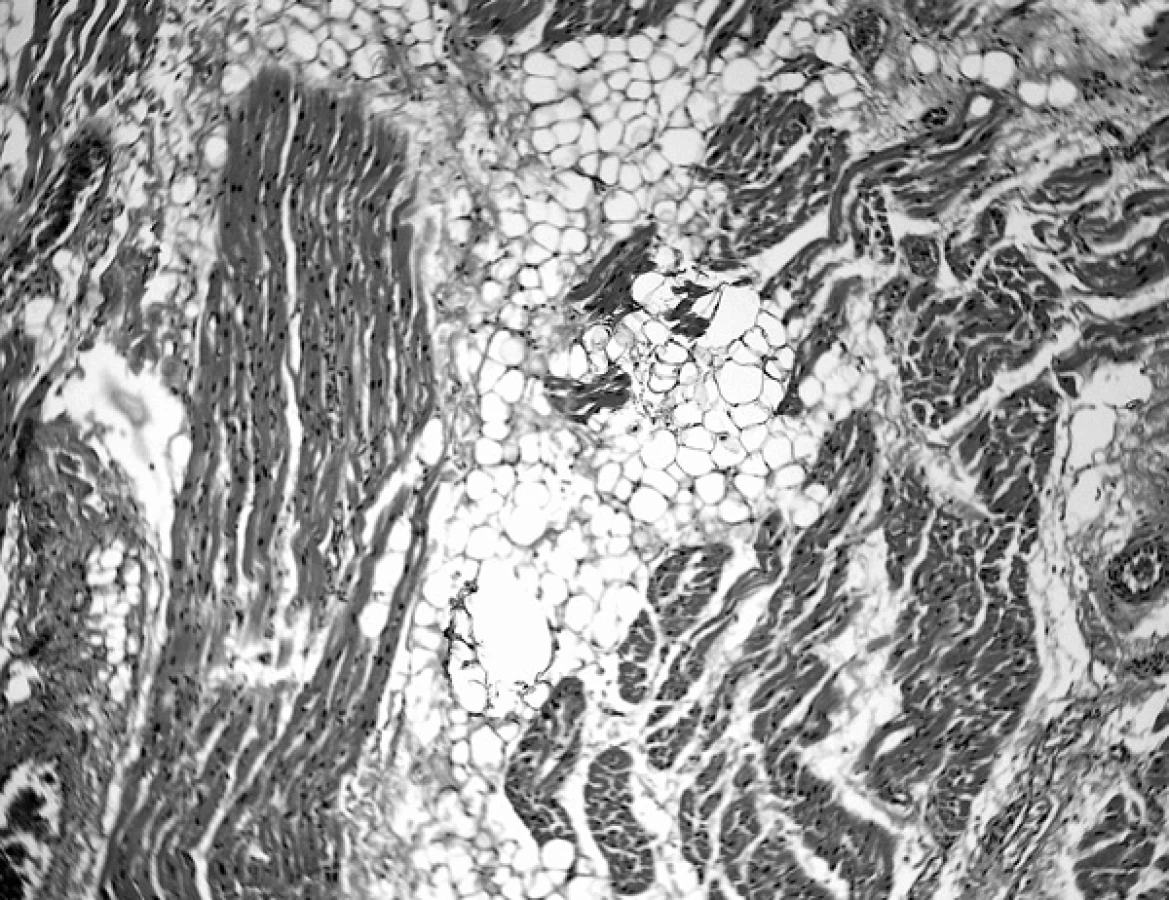
2. 9letý chlapec umírá v prosinci 2006 poté, co byla stanovena diagnóza smrti mozku po 15 dnech hlubokého bezvědomí.
Z anamnestických údajů lze zjistit, že graviditě předcházely spontánní aborty, předčasný porod byl ve 27. týdnu, porodní hmotnost 1610 g, délka 39 cm.
U dítěte byly v posledních letech diagnostikovány opakované stavy bezvědomí, podrobná opakovaná neurologická vyšetření neprokázala úchylky.
Poslední takový stav bezvědomí byl půl roku před smrtí a posléze dne 29. 11. 2006 obdobná porucha, kdy se ale po laické a do 5 minut zajištěné lékařské péči nezdařila úprava vědomí. Přetrvává fibrilace komor, echokardiografické vyšetření diagnostikuje sníženou svalovou činnost levé srdeční komory, ale při standardní šíři svaloviny (Dr. J. Gilík, Kardiocentrum FN Motol). Přes zavedenou elektrickou stimulaci a další léčbu se stav vědomí již nepodařilo obnovit a kříšení je ukončeno pro známky nezvratného poškození mozku.
Klinická diagnóza k sekci: Smrt mozku, maligní edém mozku, v. s. katecholaminergní komorová tachyarytmie.
Při pitvě byly zjištěny změny, které odpovídaly klinické diagnóze mozkové smrti, edém a posthypoxicko-ischemické změny. Významný byl nález na srdci. Zde koncentrická hypertrofie levé srdeční komory (váha 520 g), anatomické uspořádání bylo typické.
V srdeční svalovině levé komory byla patrna ložiska tukové tkáně a fibrózy, ale též drobná nekrotická ložiska (obr. 2). V imunohistochemickém nálezu tomu odpovídala ložiska výpadků myoglobinu a desminu, svědčící pro akutně probíhající ischemicko hypoxické poškození srdce. V pravé komoře jsme změny nezjistili.
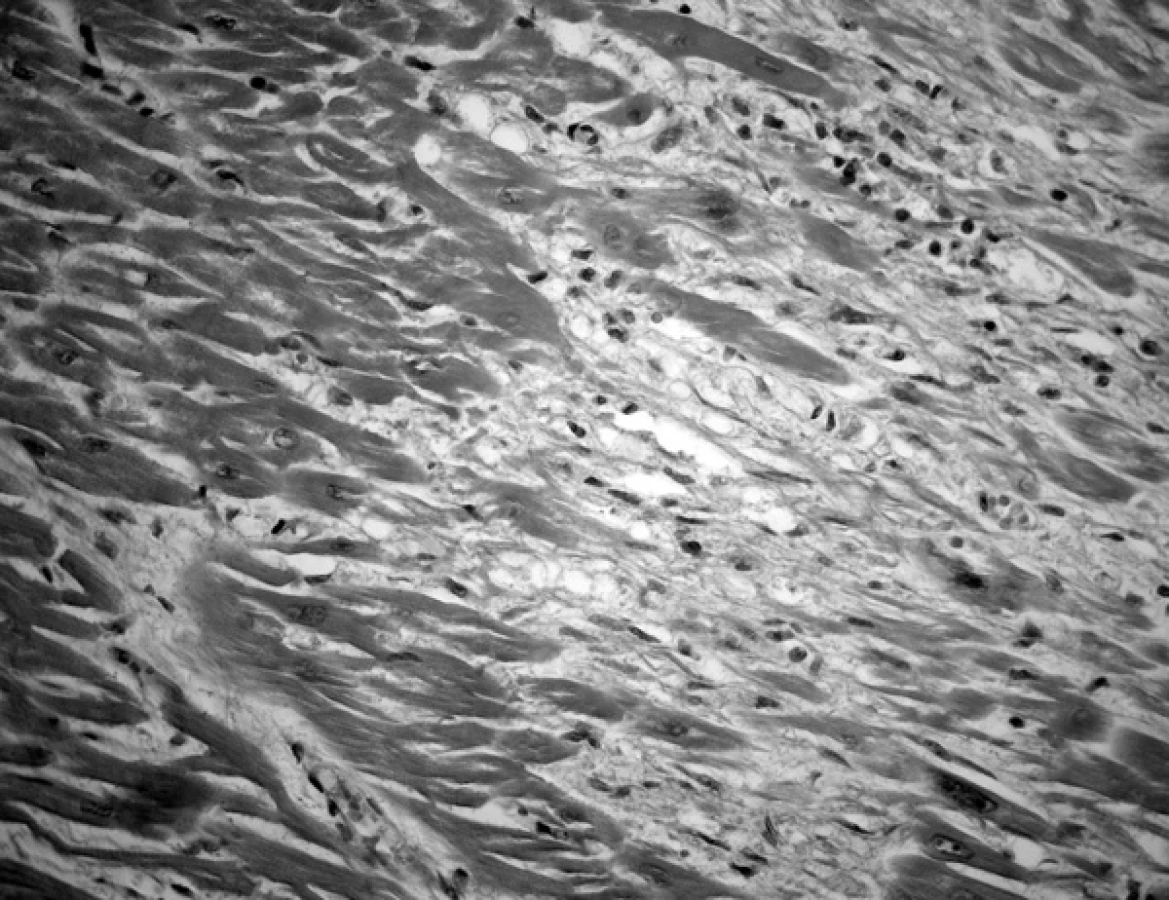
Diagnóza: arytmogenní dysplazie levé srdeční komory.
3. 29letý muž, náhle umírá v nočních hodinách. Anamnestické údaje byly sporé, údajně trpěl epileptickými záchvaty, jeho otec zemřel v mladém věku snad na infarkt myokardu a jeho bratr je veden v kardiologické poradně.
Při pitvě, provedené po dvou dnech od úmrtí, bylo zjištěno městnání krve ve velkém krevním oběhu, edém mozku a plic a též známky poranění sliznice jazyka. Na srdci zjištěna hypertrofie obou komor (520 g), známky arteriosklerózy věnčitých tepen až II. stupně, myofibróza a též ložiskové nekrózy myocytů. V pravé srdeční komoře kromě toho nalezena ložiska tukových buněk a též vaziva přímo ve tkáni a též pod epikardem. Ani hranice se svalovinou nebyla ostrá.
Nález sice nevylučoval epilepsii, ale na myokardu byl zřetelný obraz arytmogenní dysplazie pravé srdeční komory.
4. 26letá žena, normostenická, byla v roce 1995 léčena po prasknutí plicní buly pro spontánní pneumotorax vlevo. Pro přetrvávající nález buly v dolním laloku levé plíce a riziko opakovaného pneumotoraxu byla po běžném předoperačním vyšetření objednána k videotorakoskopické operaci. Předoperační vyšetření, kromě údajů o opakujících se bronchitidách, nepřineslo žádné neobvyklé nálezy. Pacientka byla již tři dny před výkonem hospitalizována a řádně premedikována.
V souvislosti s výkonem jí byl podán Thiopental, Fentanyl, Alloperin, Apaurin. Přesto krátce po zahájení výkonu došlo k zástavě srdce, kterou se nepodařilo nepřímou ani přímou masáží, ani kardiostimulací zvládnout. Po marných pokusech byla resuscitace za 2 a 1/2 hodiny ukončena.
Nález při pitvě svědčil pro oběhové selhání s obrazem akutní venostázy orgánů. Srdce o hmotnosti 336 g bylo dilatováno. Diskrétní subendokardiální fibróza v oblasti septa a mírná fibróza dvojcípé chlopně nevedly ke zjevnému hemodynamickému uplatnění. Dominantním nálezem však byla lipomatóza srdce, kde tuková tkáň masivně infiltrovala svalovinu nejen v oblasti pravé síně a pravé komory, ale též v enormním rozsahu svalovinu levé komory a mezikomorové přepážky.
Mnohé kardiomyocyty jevily zřetelné známka atrofie, v některých úsecích svaloviny obou komor vystupovala do popředí rovněž výrazná steatóza. Převodní systém zjevné odchylky nevykazoval, v jeho okolí však byla zmnožena tuková tkáň, obklopující i AV uzel. Imunohistochemicky zjištěny výpadky myoglobinu v kardiomyocytech.
Nález nasvědčuje arytmogenní displazii s postižením pravé i levé komory srdce.
Diskuse
Morfologická diagnóza arytmogenních kardiomyopatií nečiní potíže, pokud se na toto onemocnění pamatuje a srdce podrobně vyšetří. V klinické diagnostice i při použití zobrazovacích metod je její stanovení obtížné. Průběh onemocnění bývá asymptomatický, mohou předcházet funkční abnormality, charakterizované komorovou tachykardií a změnami na QRS komplexu a přítomností postexcitačních vln. Stav může být doprovázen poruchami vědomí a křečemi. Jako typické jsou popisovány změny v pravé srdeční komoře, charakterizované ložiskovou fibrózou a lipomatózou (fibrofatty infiltration).
Onemocnění není dosud jednoznačně z hlediska etiologie definováno, jedná se o geneticky heterogenní stavy. Ani z hlediska formální patogeneze není dosud zřejmé, proč k uvedenému nálezu v srdeční svalovině dochází.
Přesto, že bylo onemocnění popsáno jako postižení pravé srdeční komory, pozdější práce upozornily, že změny mohou být i na straně levé (7, 9, 13).
Změny na srdci, i u námi demonstrovaných úmrtí, lze charakterizovat jako významně variabilní. Přítomná tuková i vazivová tkáň byla zcela vyzrálá a uložená přímo v myokardu a bez souvislosti s epikardiálním tukem.
U 10letého chlapce jsme zjistili změny zejména v srdeční přepážce a též přímo v úseku převodního systému v AV junkci, u 9letého chlapce to bylo postižení levostranné, u 29letého muže byl nález pro toto onemocnění typický a u 26leté ženy, zmírající při úvodu do narkózy, byla lipomatóza myokardu na obou stranách srdce.
Mikroskopická vyšetření včetně imunohistochemických prokázala také čerstvé ischemicko-hypoxické změny na svalových buňkách, pro které nemáme jednoznačné vysvětlení. Byly ložiskové a jejich vznik mohl souviset též s letálně probíhající srdeční zástavou a následnou tkáňovou hypoxií (3).
Předneseno na 86. Sjezdu Německé společnosti pro soudní lékařství, 26. 9.–29. 9. 2007, Mohuč, Německo.
Prof. MUDr. Ivan Bouška, CSc.
FN Na Bulovce
Budínova 2
180 81 Praha 8
Sources
1. Basso, C., Fox, P. R., Meurs, K. M., Towbin, J. A., Spier, A. W., Calabre, F., Maron, B. J., Thiene, G.: Arrhythmogenie right ventricular cardiomyopathy causing sudden cardiac death in boxer dogs: a new animal model of human disease, Circulation, Mar. 2004, 9, 109 (9), 1180-1185.
2. Beran, M.: Lęexpertise en République Tchéque, Experts – Revue du technicien des conflicts, Mars 2000, 46, 6-7.
3. Bouška, I., Klír, P., Toupalík, P.: 1998, Imunohistochemické nálezy na převodním systému srdce, Soudní lékařství, 1998, 43, 55-57.
4. Bouška, I., Toupalík, P.: Diagnostika náhlé kardiální smrti v soudnělékařské praxi, Postgraduální medicína, 2007, 9, č. 6, 611-616.
5. Fornes, P., Ratel, S. and Lecomte, D.: Pathology of Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia – An Autopsy Study of 20 Forensic Cases, J. Forensic Sci., 1998, 43, 777-783.
6. Furlanello, F., Bertoldi, A., Dallago, M., Furlanello, C., Fernando, F., Inama, G., Pappone, C. and Chierchia S.: Cardiac arrest and sudden death in competitive athletes with arrhythmogenic right ventricular dysplasia, Pacing Clin. Electro-physiol., 1998,21, 331-335.
7. Gallo, P. DęAmati, J. and Pellicia, F.: Pathologic evidence of Extensive Left Ventricular Involvement in Arrhythmogenic Right Ventricular Cardiomyopathy, Hum. Pathol., 1992, 23, 948-952.
8. Marcus, F., Fontaine, G., Guiraudon, G., Frank, R., Laurenceau, J. L., Malergue, C. and Grosgogeat, Y.: Right ventricular dysplasia: a report of 24 adult cases. Circulation, 1982, 65, 384-398.
9. Michalodimitrakis, M., Papadomanolakis, A., Stiakakis, J., Kanaki, K.: Left Side Right Ventricular Cardiomyopaty, Med. Sci. Law, 2002, 42 (4), 313-314.
10. Paul, M., Schulze-Bahr, E., Breithardt, G., Wichter, T.: Genetics of arrhythmogenic right ventricular cardiomyopathy – status quo and future perspectives, Z – Kardiol, Feb. 2003, 92 (2), 128-136.
11. Peters, S., Peters, H. and Thierfelder, L.: Risk stratification of sudden cardiac death and malignant ventricular arrhythmias in right ventricular dysplasia-cardiomyopathy, Int. J. Cardiol, 1999, 71, 243-250.
12. Sekiguchi, M., Kinoshita, O.: From arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVD/ARVC) to a broader concept of ABCDE syndrome, Nippon-Rinsho., Jan. 2000, 58 (1), 108-116. (Summary).
13. Shrapnel, M., Gilbert, J. and Byard, R.: Arrhythmogenic left ventricular dysplasia and sudden death, Med. Sci. Law, 2001, 41, 159-162.
14. Vzernowska, E., Wlodarska, E. K., Zaleska, T.: Arytmogenna kardiomyopatia (dysplazia) prawej komory. Etiologia, objawy, diagnostyka i leczenie, Kardiol. – Pol., Jan. 2003, 58 (1), 58-63.
Labels
Anatomical pathology Forensic medical examiner ToxicologyArticle was published in
Forensic Medicine

2008 Issue 2
Most read in this issue
- Arytmogenní dysplazie/kardiomyopatie pravé srdeční komory jako příčina náhlé smrti
- Pitevní nález otravy etanolem a psychotropními látkami
- Technologický skríning pív, vyrábaných v Slovenskej republike, z pohľadu forenznej alkohológie