
Metabolické choroby a oko v detskom veku
Metabolic diseases and the eye in childhood
The authors demonstrate different metabolic diseases with ocular symptomatology from their own clinical materials. They emphasize that the patients with these rare diseases frequently come to an ophthalmologist with primarily ocular symptomatology. The metabolic basis is only proven after a child has been referred to other medical specialists, which requires a good knowledge of the ophthalmologist about these disorders. The correct diagnosis and often a speedy treatment are a big help in stabilizing the clinical condition of some metabolic diseases. The management of this class of general diseases requires adequate interdisciplinary cooperation.
Key words:
metabolic diseases, neurodegeneration, lysosomal disorders, peroxisomal disorders
Received:
1. 1. 2017
Accepted:
15. 2. 2017
Authors:
Anton Gerinec 1; Dana Tomčíková 1; Vladimír Krásnik 2
Authors‘ workplace:
Klinika detskej oftalmológie LF UK a DFNsP, Bratislava
1; Klinika oftalmológie LF UK a UNB, Nemocnica Ružinov, Bratislava
2
Published in:
Forum Diab 2017; 6(1): 6-10
Category:
Topic
Overview
Autori demonštrujú z vlastného klinického materiálu viaceré metabolické choroby s očnou symptomatológiou. Zdôrazňujú, že pacienti patriaci k týmto zriedkavým chorobám, prichádzajú nezriedka primárne s očnou symptomatológiou k oftalmológovi. Metabolický podklad sa dokazuje až odoslaním dieťaťa k iným medicínskym špecialistom, čo je náročné na znalosti oftalmológa o týchto chorobách. Správna diagnostika a často urýchlená liečba totiž veľmi pomôžu k stabilizácii klinického stavu niektorých metabolických ochorení. Práca s týmto druhom celkových ochorení si vyžaduje adekvátnu interdisciplinárnu spoluprácu.
Kľúčové slová:
lyzozómové poruchy, metabolické choroby, neurodegenerácie, peroxisómové poruchy
Úvod
Humánna medicína zahrňuje asi do 2 500 diagnostických jednotiek celkových ochorení, ktoré majú rôznorodú oftalmologickú symptomatológiu. Tieto choroby sú predmetom záujmu prevažne iných medicínskych disciplín, avšak ich očné prejavy diagnostikuje a lieči i oftalmológ, ktorý musí mať isté základné znalosti o ich klinickom obraze.
Asi 1 000 celkových ochorení má metabolický pôvod a často primárne prejavy na oku, čo akcentuje ich význam z hľadiska včasnej diagnostiky systémovej choroby a kladie zvýšené nároky na znalosti všeobecnej symptomatológie oftalmológom a naopak vedomosť o možných očných prejavoch musí mať i lekár neoftalmológ.
Na Klinike detskej oftalmológie LF UK a DFNsP v Bratislave sme počas 25-ročnej existencie mali viacero pacientov s primárnou očnou symptomatológiou, kde bolo podozrenie na celkovú chorobu a spoluprácou s ostatnými disciplínami sa dospelo k správnej diagnóze metabolického ochorenia.
Demonštrácia vybraných pacientov
Zo širokého spektra metabolických ochorení uvádzame niektoré, ktoré mali prvotnú očnú symptomatológiu a definitívnu diagnózu sme potvrdili spoluprácou s ostatnými medicínskymi disciplínami.
Galaktosémia
Galaktosémia je vážna metabolická choroba spôsobená deficienciou galaktokinázy a galaktózo-1-fosfát-uridyltransferázy alebo uridíndifosfát-galaktózo epimerázy.
Etiologicky je u galaktosémie prvého typu identifikovaný gén na 9p13 [4].
Prvé príznaky celkovej choroby sú očné, a to v prvých týždňoch katarakta olejovej kvapky, (jemná vakuola v zadnom kortexe šošovky), ktorá môže progredovať do lamelárnej až totálnej katarakty unilaterálne alebo bilaterálne. Systémové príznaky zahŕňajú poruchy CNS, pečene a obličiek s hepatomegaliou, žltačkou, hnačkou a letargiou. Vylúčenie mlieka zo stravy sa prejaví hneď v zlepšení celkového stavu a vymiznutím katarákt. Dôkaz je vo vyšetrení moču na redukujúce látky 2 hodiny po mliečnej strave. Najťažšia forma je pri defekte uridiltransferázy. Ostatné 2 sú miernejšie a prejavujú sa neskôr v detstve.
Pri forme z deficitu galaktokinázy (náš pacient obr. 1, obr. 2) je možný a v niektorých štátoch i zavedený neonatálny skríning.
Homocystinúria
Homocystinúria je vážna autosómovo recesívne dedičná porucha metabolizmu methionínu. Etiologicky je gén cystation beta syntázy mapovaný na 21q22.3 a v ňom je veľký počet mutácií pri homocystinúrii odlišný v rozličných krajinách [5].
Diagnostika vyžaduje nitroprusidový test dôkazu látok v moči. Enzymatický dôkaz v krvi sa vyžaduje pri negativite močového testu a prítomnosti iných vrátane očných príznakov homocystinúrie.
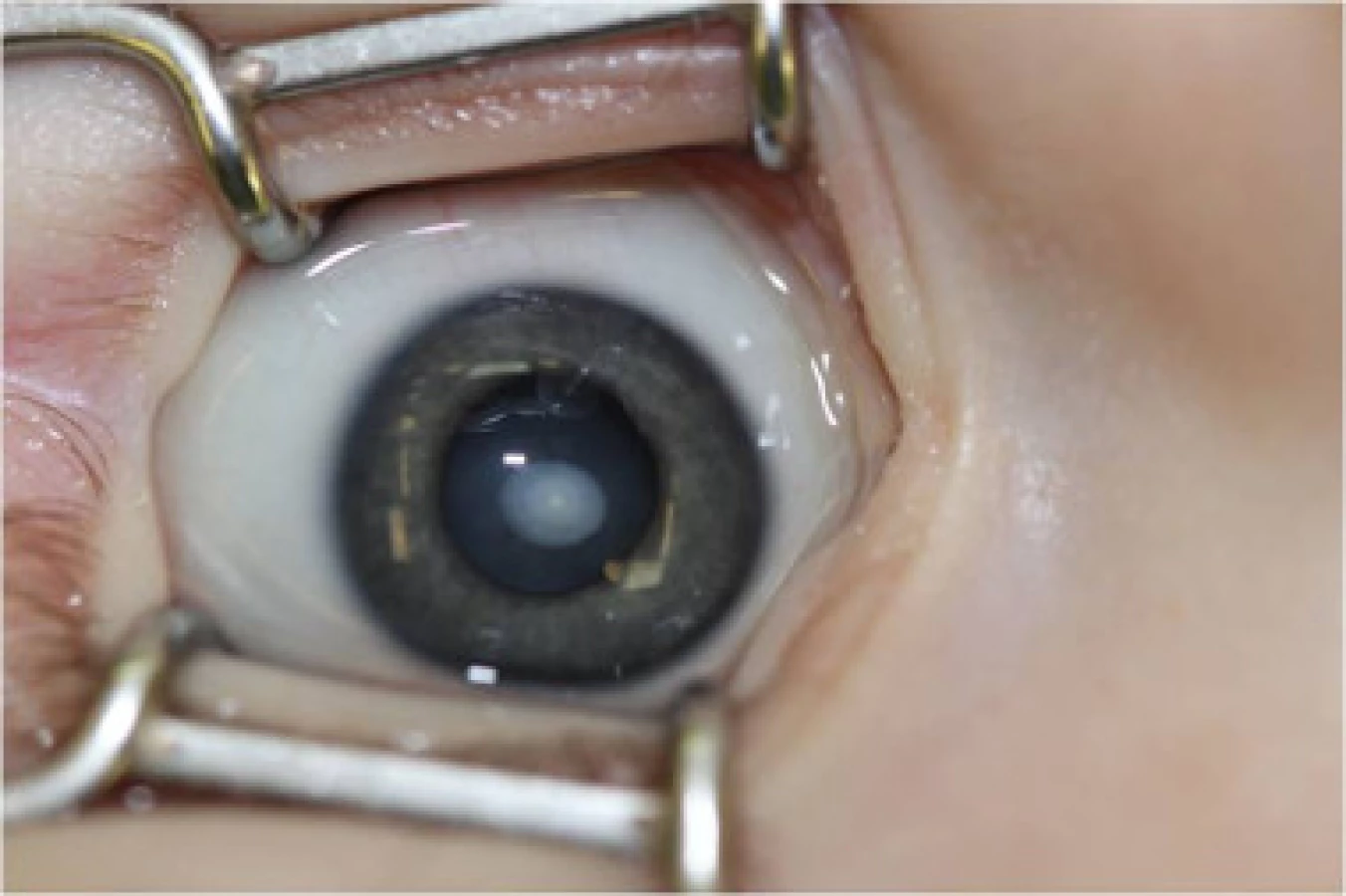
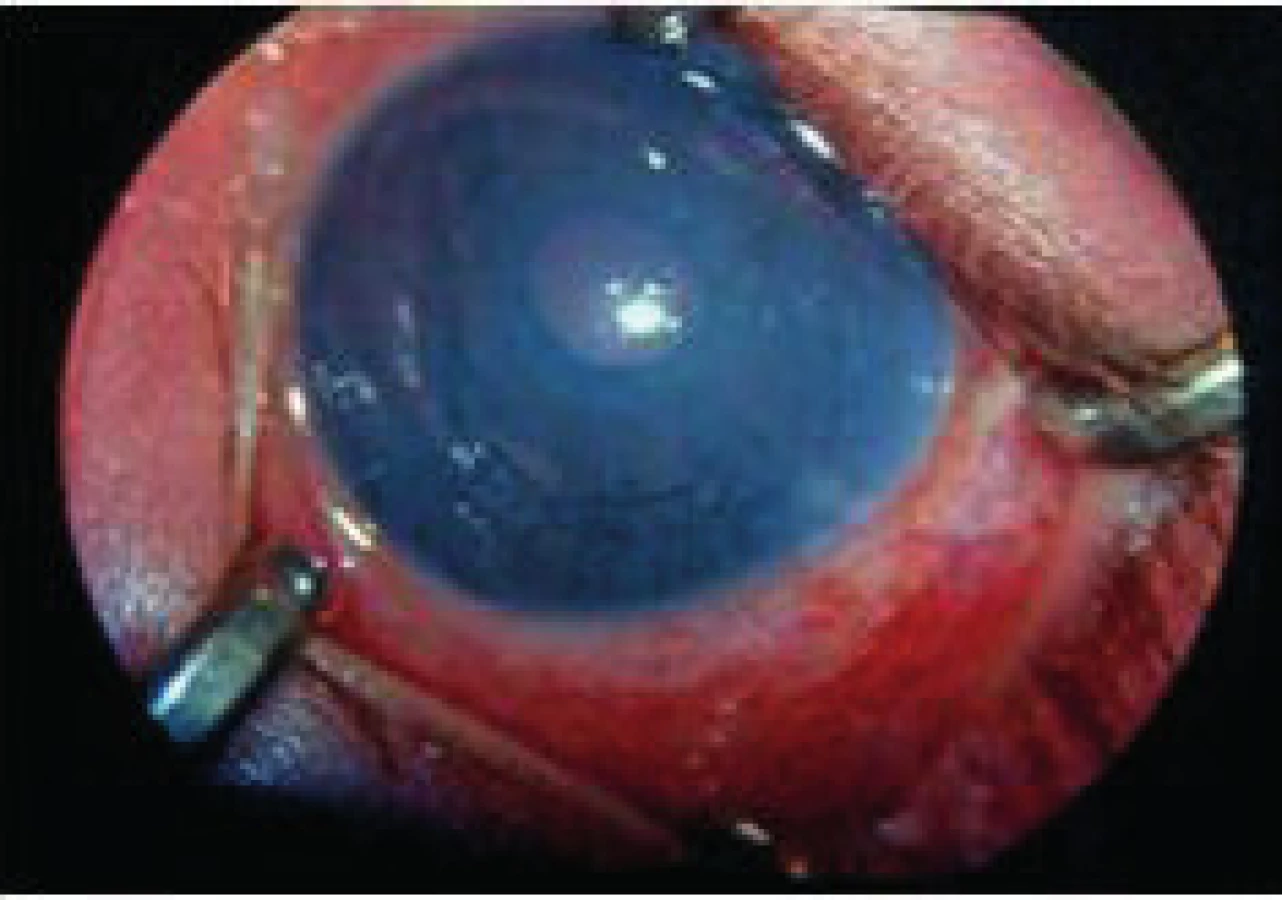
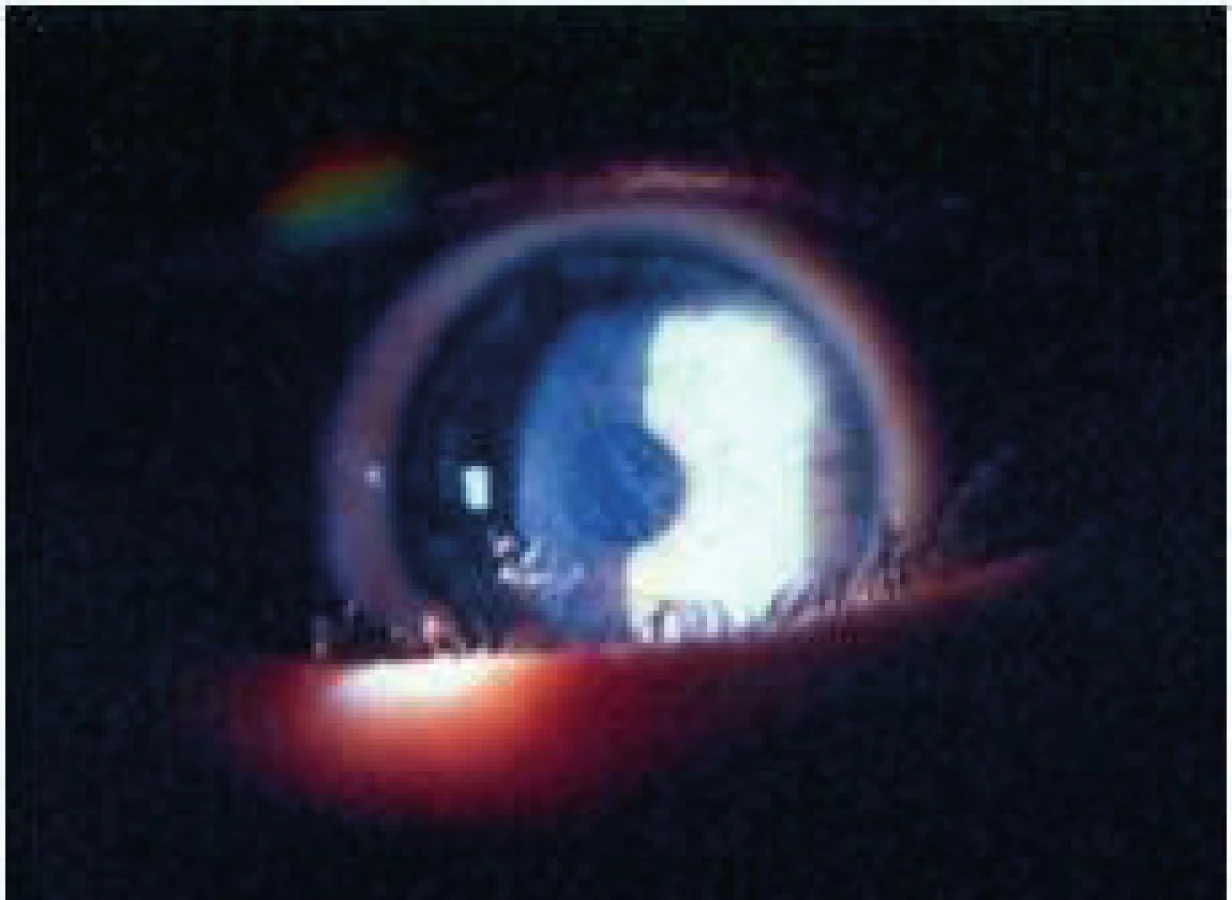
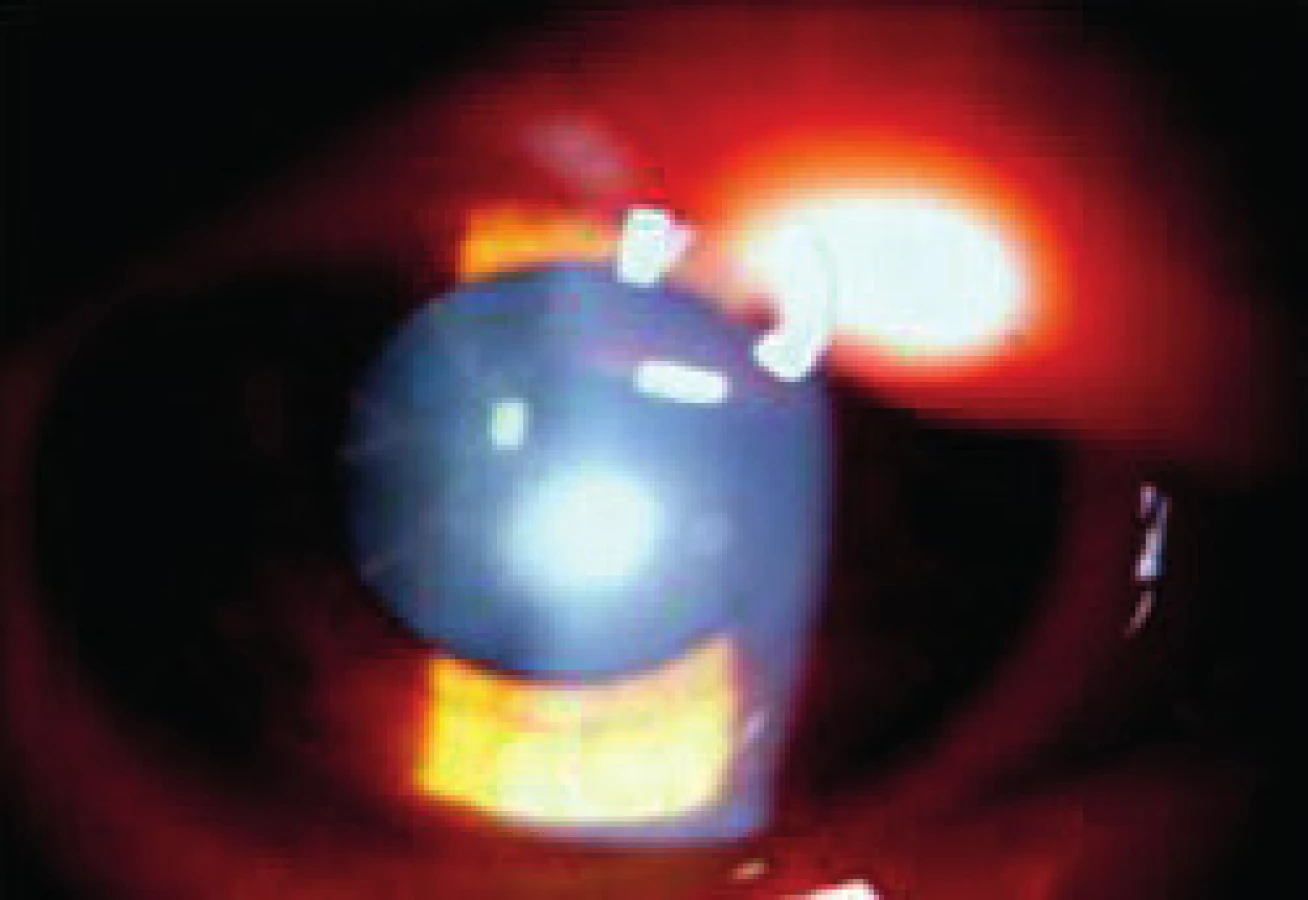
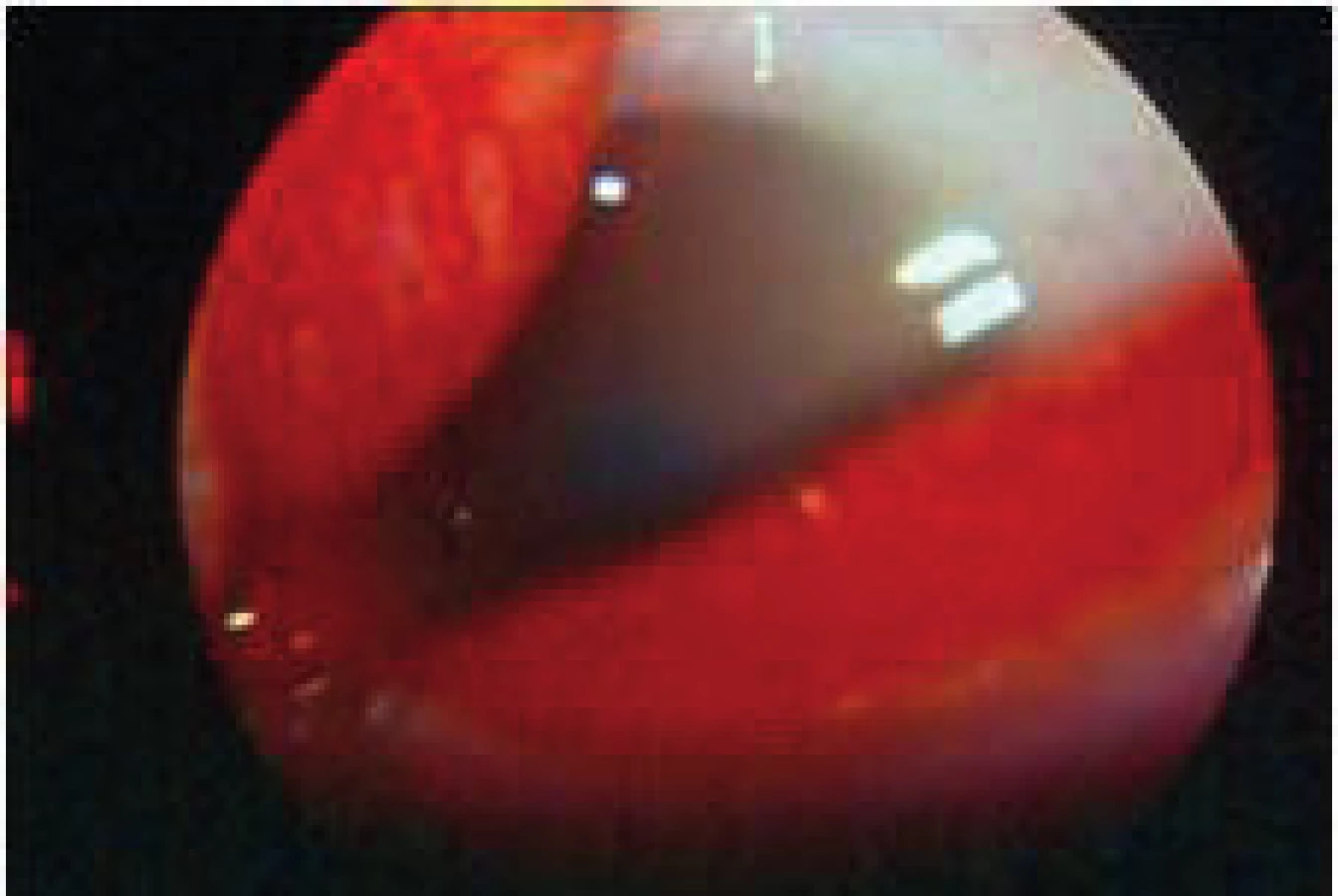
Postihnutí jedinci sú klinicky zdraví pri pôrode, avšak počas ďalšieho vývoja sa začína prejavovať mentálna retardácia. Okrem celkových príznakov dominuje v očnom náleze ektopia šošovky (obr. 3, obr. 4).

Liečba je podobná u všetkých typov ektopií šošovky. Veľmi dôležité však je pri operačných indikáciách zvážiť riziko tromboembolických príhod s letálnym koncom. Pacienti musia byť metabolicky kompenzovaní a až potom operovaní. Pri metabolicky nekompenzovanom pacientovi progreduje i dislokácia šošovky a zhoršuje sa celkový stav, čo správny liečebný režim stabilizuje. Do diéty sa pridáva cystín a vitamín B6 a eliminuje homocystín.
Loweho syndróm
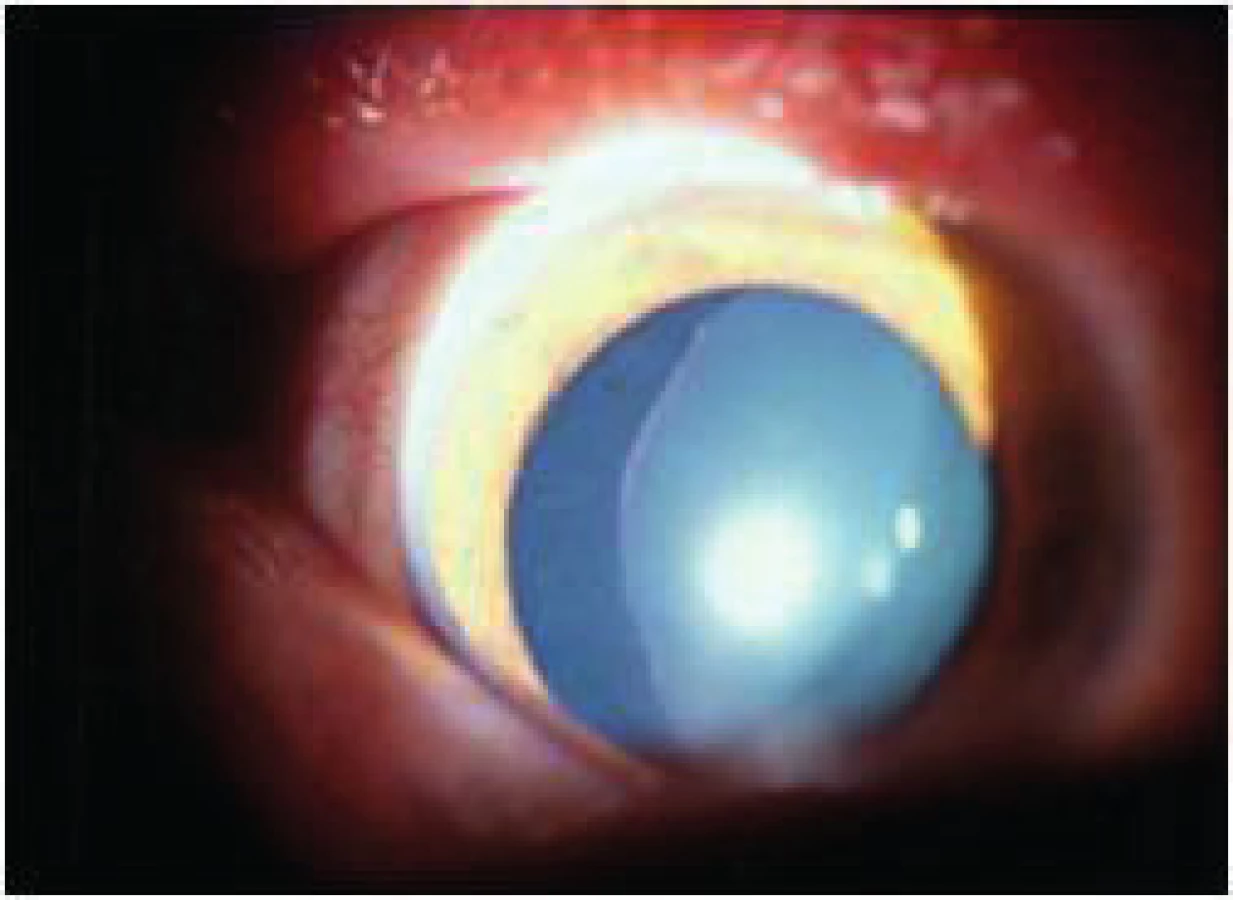
Loweho syndróm je gonosomálne recesívne viazané ochorenie postihujúce mozog, obličky a oči. Deti majú charakteristické vyklenutie čela s bucľatými lícami (obr. 5 a obr. 6), v ľadvinách je tubulárna dysfunkcia vedúca k metabolickej acidóze, sekundárnemu hyperparathyreoidizmu, demineralizácii kostí a hypotónii. V CNS sú ložiská demyelinizácie. Diagnóza sa potvrdí nálezom aminokyselín v moči.
Katarakty sú bilaterálne centrálne diskoidné, ktoré nemajú ohraničenie medzi kôrou a jadrom. Pravidelným nálezom je i kongenitálny glaukóm a primerane väčšie bulby. Ženskí konduktori majú zadnú kortikálnu kataraktu rôzneho rozsahu, ktorá mierne narúša zrakové funkcie. Ochorenie je často letálne v druhom decéniu. Stav si z očného hľadiska vyžaduje operáciu katarakty a glaukómu, čím sa videnie čiastočne zlepší [2].
Albinizmus
Albinizmus je vrodená a dedičná porucha aminokyselín v procese melanogenézy prejavujúca sa iba na koži, iba na očiach alebo na oboch orgánoch rôznym stupňom absencie pigmentu. Predstavuje stacionárny stav. Etiopatogeneticky je albinizmus heterogénny problém s identifikáciou početných mutácií najmä v TYR a OCA génoch [6].
Očná symptomatológia je typická tým, že obsahuje veľmi variabilný stupeň defektu rozličných štruktúr oka, ktoré obsahujú pigment: dúhovka, vráskovec, choroidea, pigmentový epitel sietnice (obr. 7, obr. 8).
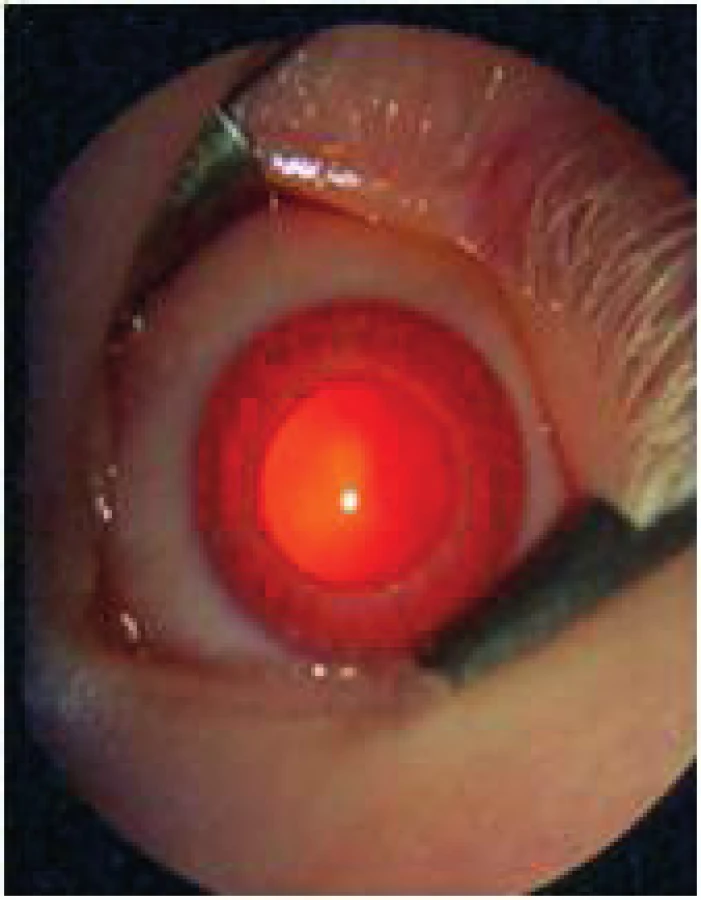
Príznakové komplexy zahŕňajú nystagmus, strabizmus, makulárnu hypopláziu, totálnu absenciu pigmentu v pigmentovom epiteli sietnice a uvey s transilumináciou dúhovky a vysoký astigmatizmus a myopiu. Oči majú typickú červenú farbu dúhovky danú vaskulatúrou dúhovky a odrazom červeného reflexu cez transiluminujúcu dúhovku. Zraková ostrosť kolíše medzi 0,2–0,5. Tyčinky sú lokalizované vo fovei a čapíky sú vzdialenejšie od fovey. U pacientov nie je žiaden pigment v koži ani vo vlasoch.
V liečbe všetkých foriem albinizmu musia byť deti dokonale korigované pre prevenciu amblyopie. Strabizmus je v prípade potreby indikovaný na chirurgické riešenie. Deti nikdy nedosiahnu binokulárne videnie. Musia byť trvale sledované a nosia fotochromatické ochranné okuliarové sklá. Pri ostatných ochoreniach v rámci systémového postihnutia je liečba v rukách iných disciplín.
Lyzosómové poruchy
Sú spôsobené dysfunkciou bunečných organel – lyzosómov – obsahujúcich rozličné enzýmy na metabolizmus cukrov a lipidov. Dôsledkom tejto poruchy je intracelulárna akumulácia rôznych metabolických produktov, ktoré sa akumulujú najmä v nervových bunkách CNS, sietnice, zrakového nervu a rohovky a obsahujú:
- sfingolipidy
- mukopolysacharidy
- mukolipidy
- oligosacharidy
- myelín (leukodystrofie)
Metachromatická leukodystrofia
Metachromatická leukodystrofia je taktiež autosomálne recesívne ochorenie s defektom arylsulfatázy-A a akumuláciou cerebrozid sulfátu v CNS, periférnych nervoch, viscerách a moči, čo spôsobuje demyelinizáciu.
Ochorenie má formy infantilnú, juvenilnú a adultnú, ktoré sú všetky letálne. Choroba má viacero génových mutácií arylsulfatázy.
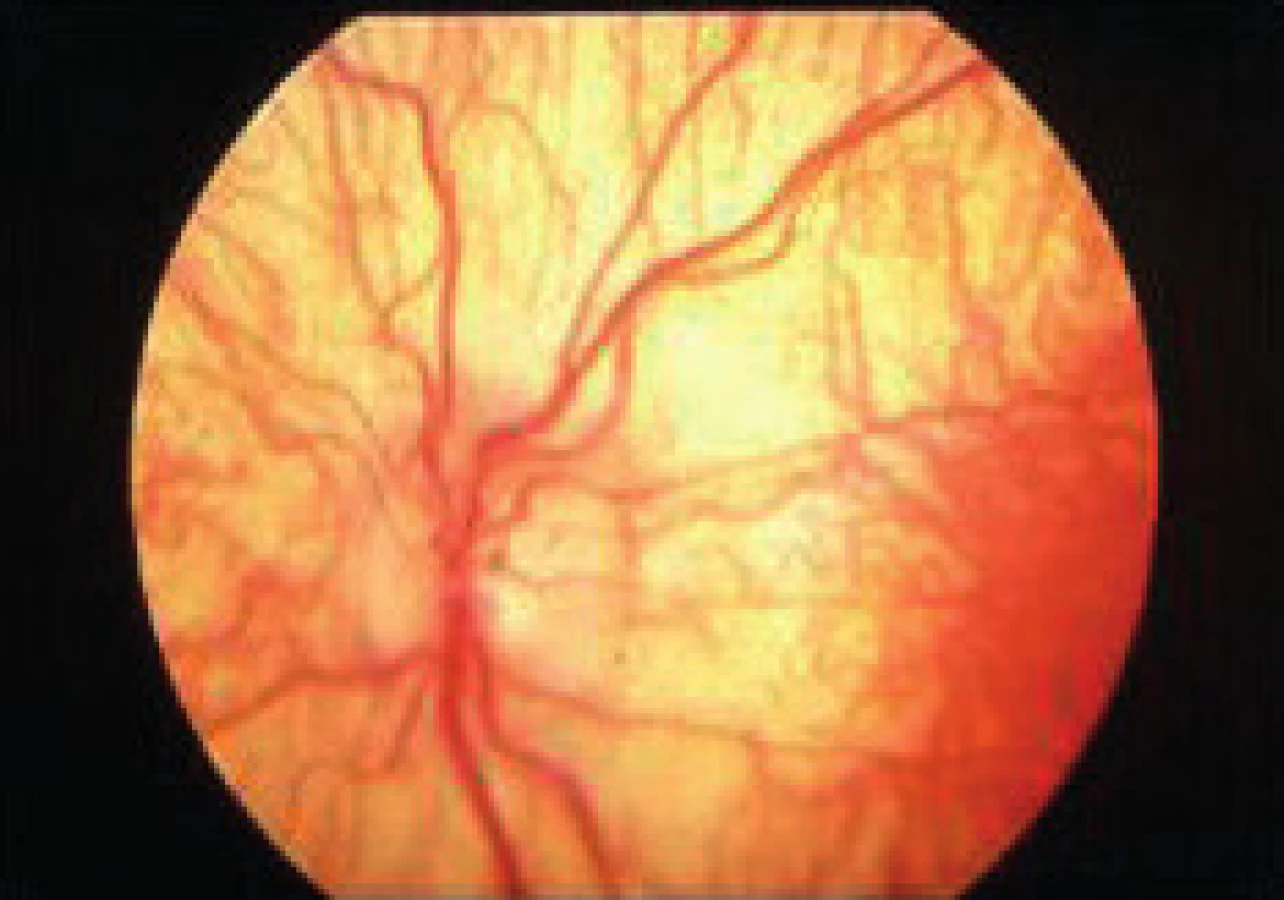
Symptomatológia – okolo tretieho roka začína infantilná forma ako progesívna slabosť, spasticita, demencia, ataxia a atrofia nervi optici (obr. 9). Do 16 rokov sa objavuje juvenilná a po tomto období dospelá forma miernejšou symptomatológiou.
Diagnostika sa opiera o symptomatológiu a enzýmový dôkaz arylsulfatázy A. Prenatálne je dôkaz z choriových klkov.
Liečba – pokusy s transplantáciou kostnej drene.
Neuronálna ceroidná lipofuscinóza (Battenova choroba)
Neuronálna ceroidná lipofuscinóza je skupina autosómovo recesívnych (chromosóm 1 a 16) ochorení s akumuláciou autofluorescentného lipopigmentu ceroidu v neuronálnych lyzosómoch. Enzymatický defekt nie je doteraz známy. Je najčastejšou neurodegeneráciou u detí. Jej klinické formy sa rozlišujú podľa veku nástupu:
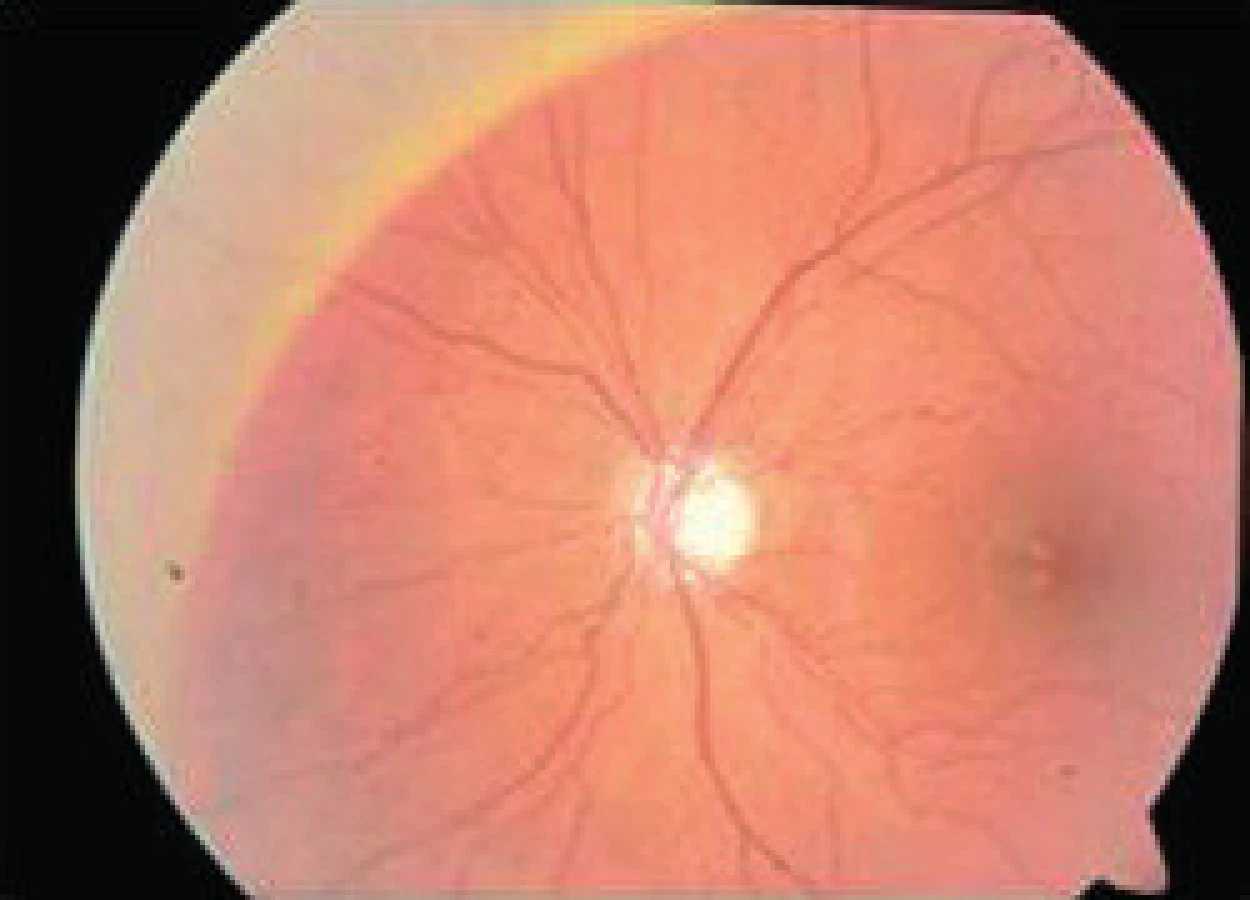
- infantilná (m. Santavuori-Haltia) v dvoch rokoch – má prejavy psychomotorickej retardácie, choreoatetózy a záchvatov, na očiach je pigmentózna dystrofia a atrofia terča zrakového nervu
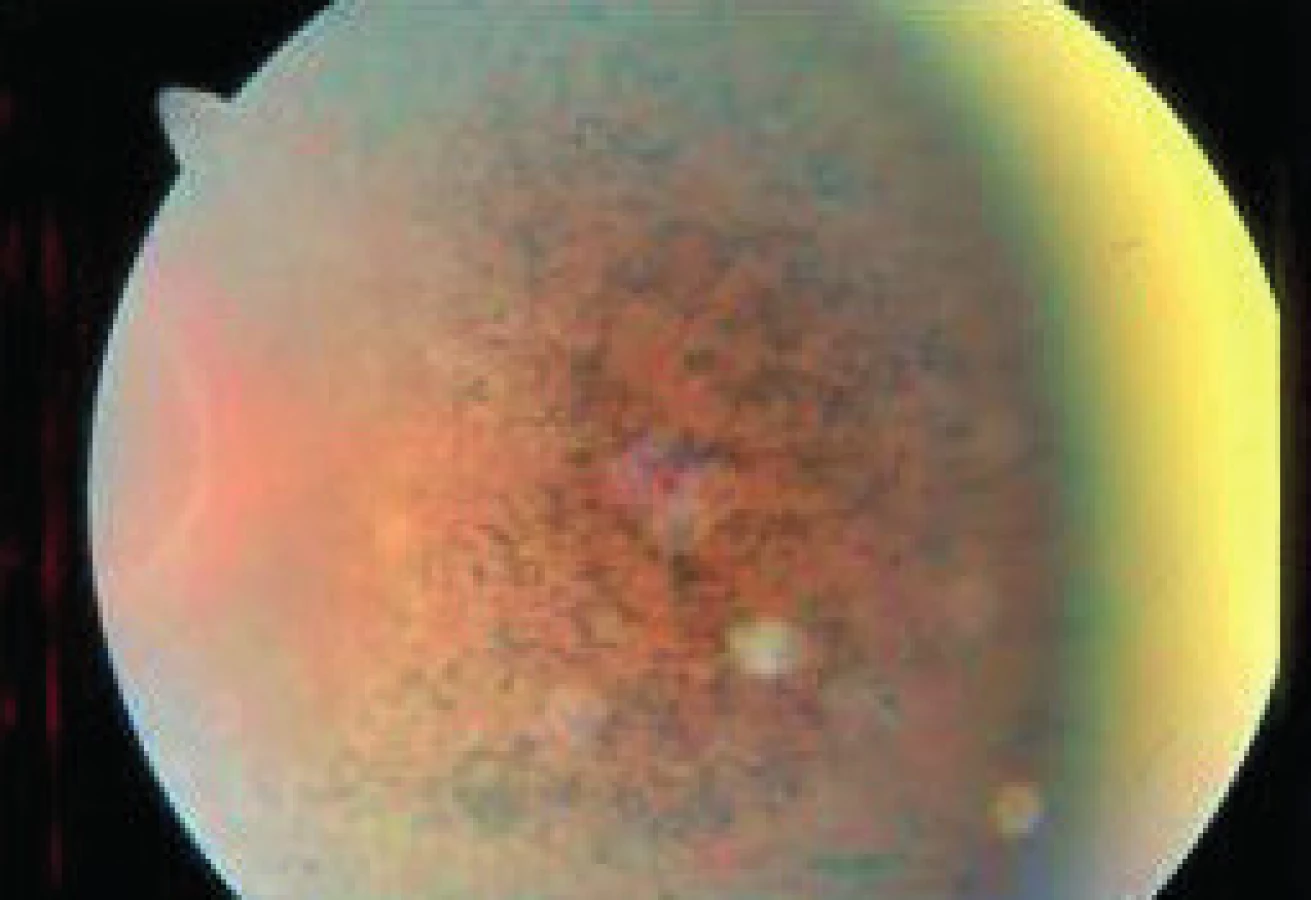
- neskoro infantilná (m. Jansky-Bielschovsky) medzi 2–4 rokmi – ataxia, záchvaty, strata reči a spasticita, na očiach je atrofia zrakového nervu a pigmentózna dystrofia (obr. 10)
- juvenilná (m. Spielmeyer-Vogt) medzi 5–10 rokmi má mentálnu retardáciu a pokles kognitívnych funkcií, záchvaty a spasticita, na očiach je atypická pigmentózna dystrofia
- adultná (m. Kufs) po 20 rokoch – degradácia intelektu od dospelosti, ataxia, na očiach iba výnimočne hemeralopia
Všetky formy sú letálne, tým skôr, čím skôr sa manifestuje choroba. Najčastejšie sú infantilná a juvenilná forma zahrňujúca až 85 % pacientov.
Symptomatológia – hypotónia, ataxia, záchvaty, psychomotorická retardácia. Slepota má pôvod v atrofii zrakového nervu a u juvenilnej formy i v retinálnej dystrofii atypického typu bez osteoblastov s obrazom granulovanej deštrukcie pigmentového listu. Spoločným nálezom je i tzv. bull’s eye makulopatia.
Diagnostika – symptomatológia a intracelulárne inklúzie ceroidu v neuronálnych bunkách, lymfocytoch a v spojovkovej biopsie [3]. Enzým sa zatiaľ nedá stanoviť, iba produkty rozpadu – dlhoreťazcové alkoholy (dolichol) sú zvýšené v moči.
Liečba nie je možná, i keď sú pokusy s transplantáciou kostnej drene a génovou terapiou.
Gangliozidózy
Gangliozidózy sú lyzosomálne sfingolipidózy autosómovo recesívne dedičné, ktoré majú viacero klinických foriem:
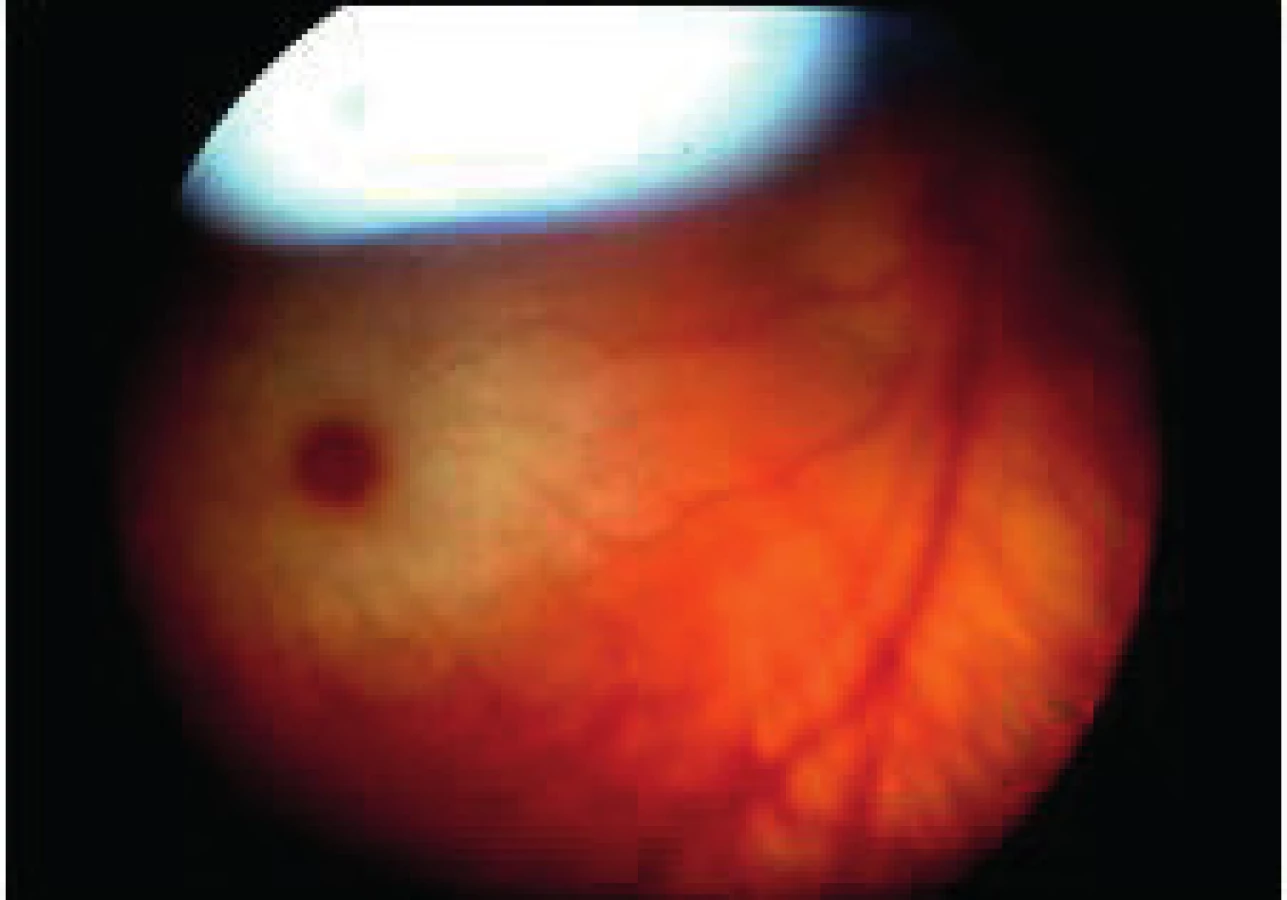
e.4/GM2 – typ 1 Tay-Sachs – je najčastejšou klinickou formou. Etiologicky je autosómovo recesíny defekt hexosaminidázy A s viacerými mutáciami jeho génu, čo sa prejaví krátko po narodení retardáciou vývoja, hypotóniou, záchvatmi a progresívnou slepotou.
Na očiach je konštatntne prítomná višňová škvrna v makule (obr. 11) s depozitmi gangliozidov. Nakoľko gangliová vrstva je najhrubšia juxtafoveálne, depozity sú najlepšie viditeľné okolo fovey. Rasové charakteristiky menia farbu vrstvy bez gangliových buniek vo fovei, ktorá je rôzne červená. Postupná atrofia terča a atenuácia retinálnej vaskulatúry pokračuje. Ak sa sietnica vyšetruje hneď po narodení alebo po niekoľkých rokoch môže višňová škvrna chýbať, nakoľko gangliové bunky postupnou atrofiou vymiznú asi do 2 rokov [1]. Deti exitujú zvyčajne do 3 rokov, ale na základe znalosti mutácie je možná prenatálna diagnostika.
Fabryho choroba
Fabryho choroba je gonosómovo recesívne ochorenie s deficienciou alfa-galaktozidázy. Ukladá sa pri nej abnormálny glykosfingolipid do CNS, periférnych nervov, obličiek, myokardu, cievneho endotelu a oka.
Symptómy začínajú okolo 10. roku bolesťami končatín a nevysvetliteľnou únavou a slabosťou. Vyvíjajú sa červené lézie kože anagiokeratoma corporis diffusum, pribúdajú hypertenzia, cerebrálne aneuryzmy, kardiomyopatie a vo veku 30–40 rokov vzniká renálna insuficiencia.
Očný nález je patognomický, v zadnom kortexe šošovky vzniká lúčovitá katarakta (spoke like) (obr. 13), spojovková a retinálna tortuozita ciev a typická cornea verticillata (obr. 12) granulárne jemné depozity glykosfingolipidov v rohovkovom epiteli a pod ním usporiadané radiálne v tvare šrobovnice [7]. U žien je iba rohovkový nález alebo ľahká forma choroby.
Mukopolysacharidózy
Je široká skupina tezaurizmóz, lyzosomálnych porúch postihujúcich viacero systémov a najmä rohovku, kde hrajú v metabolizme úlohu práve mukopolysacharidy (MPS).
MPS-I zahŕňa ťažkú Hurleovu formu a ľahšiu Scheieho formu, zriedkavá je i kombinovaná forma MPS-I-HS pri deficiencii alfa-L-iduronidázy. Gén pre deficienciu iduronidázy bol identifikovaný a taktiež i počiatky substitučnej enzymatickej liečby sa ukazujú ako veľmi nádejné.
Hurleov syndróm sa manifestuje hneď po narodení mentálnou retardáciou, dysostosis multiplex, organomegaliou (hepatosplenomegaliou) a tvárovou dysmorfiou. Očné príznaky sú až od 6 mesiacov pod obrazom stromálneho zákalu rohovky (obr. 14). V oblasti očí je synophrys, hypertelorizmus a zhrubnutie mihalníc. Neskôr sa pridružuje atypická pigmentózna dystrofia sietnice a atrofia terča zrakového nervu so zdôraznením ťažkého zrakového defektu. Atrofii predchádza často edém terča pravdepodobne z akumulácie mukopolysacharidov v ňom a kompresiou v sklerálnom kanáli. Z edému rohovky a trabekula akumuláciou glykosaminoglykanov sa vyskytuje i sekundárny glaukóm. Terapia je v poslednej dobe génová cestou transplantácie kostnej drene. Staršia metóda je keratoplastika.
Peroxisómové choroby
Peroxisómy sú malé cytoplazmatické subcelulárne organely ohraničené membránou. Obsahujú enzýmy zodpovedné za beta oxidáciu dlhoreťazcových mastných kyselín. Tento proces konzumuje molekulárny kyslík a generuje peroxid vodíka. Kontinuálna beta oxidácia krátkoreťazcových mastných kyselín sa totiž odohráva v mitochondriách. Okrem toho majú za úlohu čiastočnú syntézu plazmalogénu a žlčových kyselín. Význam a úloha peroxizómov v patogenéze mnohých neurodegenerácií sa vysvetlila v poslednej dobe. Zaujímavosťou je, že prakticky všetky nasledovné peroxizómové choroby majú oftalmologickú symptomatológiu:
- adrenoleukodystrofia
- Zellwegerov syndróm
- Refsumov syndróm
Adrenoleukodystrofia
Adrenoleukodystrofia je daná deficienciou lignoceroyl CoA ligázy v peroxisómoch, ktorý aktivuje metabolizmus dlhoreťazcových mastných kyselín [3]. V plazme sa zvyšuje hladina kyseliny pipekolovej, produktu metabolizmu lyzínu ako i žlčových kyselín.
Juvenilná manifestácia, ktorá je gonosomálne recesívne dedičná, sa vyznačuje prejavmi bronzovej kože sekundárne k insuficiencii nadobličky. Pokračuje tiež mentálna degradácia, dysfágia, dysartria a kvadruparéza. Manifestácia začína zvyčajne v neskoršom detstve.
Očné príznaky: kortikálna strata zraku pokračuje postupne i s prejavmi ezotropie a atrofie terča zrakového nervu. Na CT a NMR je masívna strata bielej hmoty mozgu.
Neonatálna manifestácia, ktorá je odlišná od juvenilnej, sa vyskytuje u oboch pohlaví. Obdobné príznaky nastupujú veľmi skoro a deti exitujú po 3 rokoch v respiračnej insuficiencii.
Očné príznaky majú stratu zraku spôsobenú degeneráciou fotoreceptorov. V sietnici sa nachádzajú cytoplazmatické inklúzie. V liečbe je nutný diétny režim redukcie mastných kyselín a transplantácia kostnej drene. Obe metódy sú málo účinné.
Refsumov syndróm
V roku 1946 popísal Refsum v prvej dekáde života hereditárne recesívne ochorenie s ataxiou a svalovou slabosťou a progresívnou hemeralopiou, senzorickou hluchotou a spinálnou degeneráciou. Boli popísané 2 formy:
- infantilný typ má deficit enzýmu alfa-hydroxylázy kyseliny fytánovej, ktorá spracováva kyselinu fytánovú, ktorá môže byť dodaná iba z rastlinnej potravy, pacienti s uvedenou symptomatológiou zomierajú na kardiálnu arytmiu
- juvenilný typ sa manifestuje hepatomegaliou, anosmiou, hluchotou, pigmentovou dystrofiou sietnice (obr. 15), ako príčina sa našla absencia peroxisómov spracovávajúcich kyselinu fytánovú
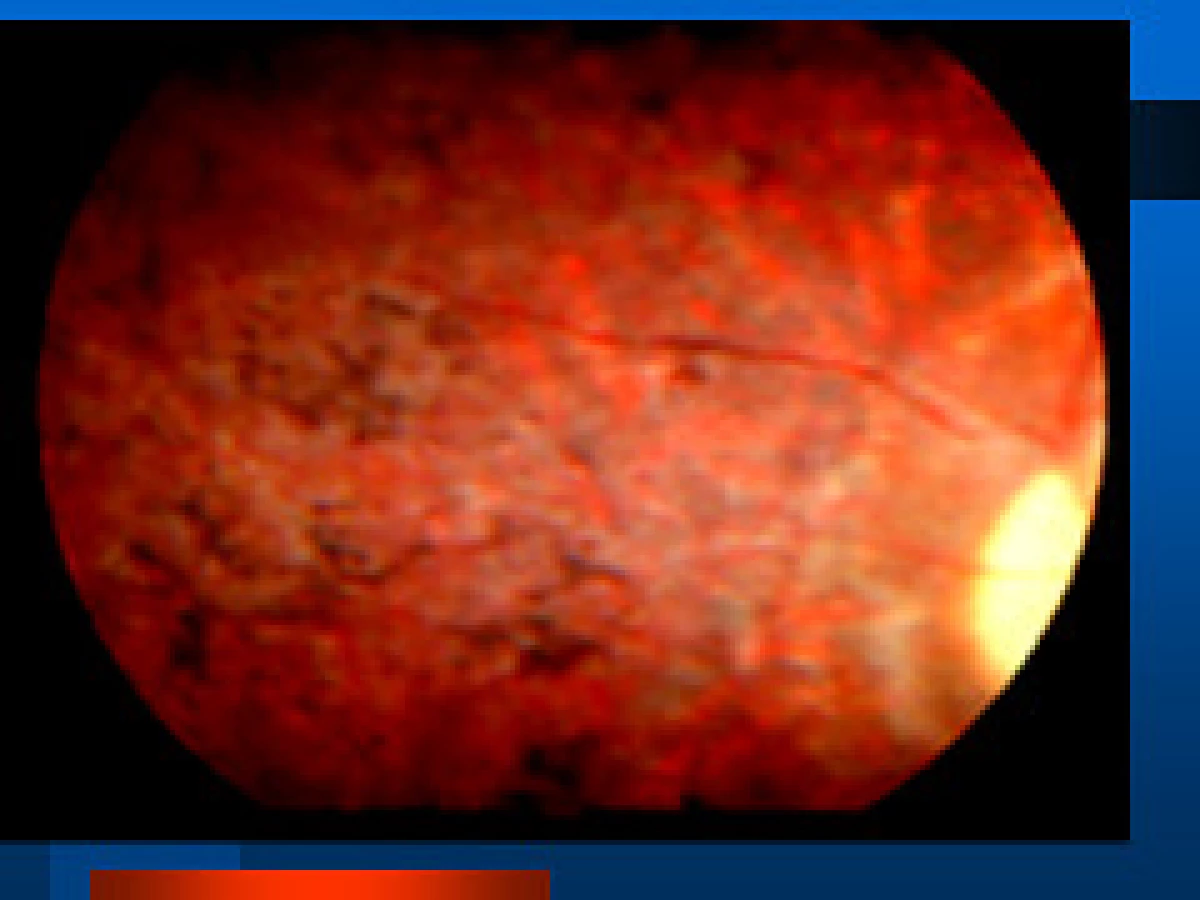
Prenatálna diagnostika je možná iba u infantilnej formy zistením absencie peroxisómov v choriových klkoch v prvom až druhom trimestri. V liečbe je diéta bez chlorofylu a fytolu v snahe redukovať hladinu kyseliny fytánovej plazmaferézou.
Diskusia
Metabolické choroby sú dôsledkom insuficiencie enzýmov nutných na katalýzu nespočetného množstva biochemických reakcií v tele. Tieto procesy majú vždy kongenitálny a najčastejšie autosómovo recesívne dedičný prenos v poslednej dobe s odhalovanou génovou podstatou [1–7]. Vyznačujú sa alternatívne v rôznych kombináciách:
- tvorbou anomálneho metabolického produktu
- nadprodukciou normálneho metabolického produktu
- insuficienciou normálneho metabolického produktu
- kombináciou predošlých faktorov
Diagnostika týchto porúch si vyžaduje bezpodmienečne spoluprácu mnohých medicínskych odborov a snahu o rýchlu diagnostiku, ktorá u mnohých foriem metabolických chorôb dokáže stabilizovať celkový stav pacienta a taktiež poskytnúť genetickú konzultáciu [8].
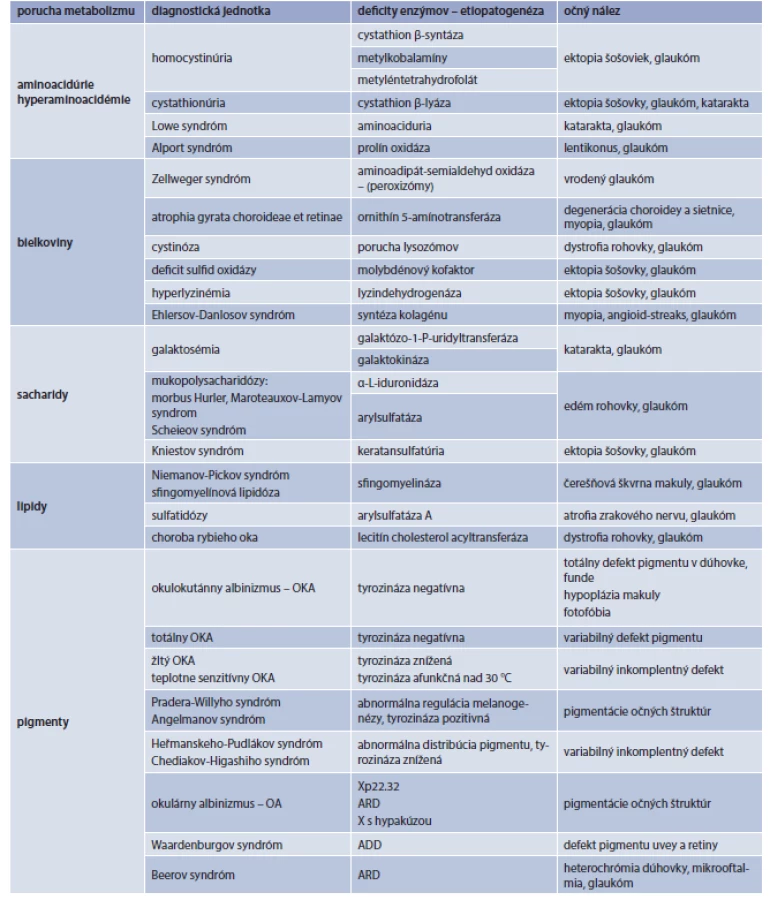
Dedičné poruchy metabolizmu s možnou manifestáciou niektorých závažných očných ochorení ako glaukóm, katarakta, myopia, atrofia zrakového nervu a dystrofia sietnice ilustruje tab.
Záver
Metabolické choroby majú pomerne často i očnú symptomatológiu, a to nezriedka primárnu, ktorá vedie k správnej diagnóze systémovej poruchy. Na ilustrácii viacerých pacientov Kliniky detskej oftalmológie je zdôraznená potreba rýchlej diagnostiky a interdisciplinárnej spolupráce.
Doručené do redakcie 1. 1. 2017
Prijaté po recenzii 15. 2. 2017
prof. MUDr. Anton Gerinec, CSc.
gerinec@dfnsp.sk
www.dfnsp.sk
Sources
1. Aragão RE, Ramos RM, Pereira FB et al. ‘Cherry red spot’ in a patient with Tay-Sachs disease: case report. Arq Bras Oftalmol 2009; 72(4): 537–539.
2. Bökenkamp A, Ludwig M. The oculocerebrorenal syndrome of Lowe: an update. Pediatr Nephrol 2016; 31(12): 2201–2212.
3. Gerinec A. Detská oftalmológia. Osveta: Martin 2005. ISBN 80–8063–181–6.
4. Janzen N, Illsinger S, Meyer U et al. Early cataract formation due to galactokinase deficiency: impact of newborn screening. Arch Med Res 2011; 42(7): 608–612. Dostupné z DOI: <http://dx.doi.org/10.1016/j.arcmed.2011.11.004>.
5. Martínez-Gutiérrez JD, Mencía-Gutiérrez E, Gracia-García-Miguel Tet al. Classical familial homocystinuria in an adult presenting as an isolated lens subluxation. Int Ophthalmol 2011; 31(3): 227–232. Dostupné z DOI: <http://dx.doi.org/10.1007/s10792–011–9444-x>.
6. Sanabria D, Groot H, Guzmán J et al. An overview of oculocutaneous albinism: TYR gene mutations in five Colombian individuals. Biomedica 2012; 32(2): 269–276. Dostupné z DOI: <http://dx.doi.org/10.1590/S0120–41572012000300015>.
7. van der Tol L, Sminia ML, Hollak CE et al. Cornea verticillata supports a diagnosis of Fabry disease in non-classical phenotypes: results from the Dutch cohort and a systematic review. Br J Ophthalmol 2016; 100(1): 3–8. Dostupné z DOI: <http://dx.doi.org/10.1136/bjophthalmol-2014–306433>.
8. Tylki-Szymańska A. Mucopolysaccharidosis type II, Hunter’s syndrome. Pediatr Endocrinol Rev 2014; 12(Suppl 1): 107–113.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Forum Diabetologicum

2017 Issue 1
Most read in this issue
- Angio OCT – nová neinvazívna zobrazovacia vyšetrovacia metóda diagnostiky a monitoringu diabetickej retinopatie
- Autoimunita a diabetes mellitus
- Metabolické choroby a oko v detskom veku
- Diabetes mellitus a dermatologické ochorenia