
Trombotická mikroangiopatie z pohledu nefrologa
Trombotic microangiopathy – a nephrologists‘ view
Thrombotic microangiopathy (TMA) is a syndrome characterised by haemolytic anaemia with fragmented red blood cells (schistocytes) in peripheral blood, thrombocytopenia caused by increased platelet aggregation and consumption, resulting in a disturbance of microcirculation caused by a formation of microthrombi. Vascular occlusion causes tissue and organ ischaemia that results in functional disorders of various organs, mainly the central nervous system, kidneys, heart, lungs and gut. Idiopathic TMAs include two etiologically distinct categories known as thrombotic thrombocytopenic purpura and haemolytic uremic syndrome. Secondary TMAs may be caused by certain drugs, infections, malignancies, systemic diseases, or associated with pregnancy. TMA is a rare disease; the reported incidence rates of idiopathic TMA range from one to four patients per million persons year. However, due to the heterogeneity of clinical symptoms, any physician can come across patients suffering from TMA. Timely diagnosis and appropriate treatment are crucial for the outcome of the patients, otherwise the affected patients are threatened with rapid progressive decline of vital functions. This article aims to provide a brief overview of thrombotic microangiopathies and highlights certain novel diagnostic and therapeutic approaches. In addition, the article helps increase general awareness of these diseases, specifically in view of possible difficulties in diagnosis and classification of clinical categories of TMAs.
Key words:
haemolytic uremic syndrome – thrombotic thrombocytopenic purpura – thrombocytopenia – haemolytic anaemia – plasma exchange – eculizumab
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE „uniform requirements“ for biomedical papers.
Submitted:
10. 6. 2015
Accepted:
17. 6. 2015
:
J. Vachek 1; O. Zakiyanov 1; V. Motáň 2; V. Tesař 1
:
Klinika nefrologie 1. LF UK a VFN v Praze
1; Transfuzní oddělení, Oddělení klinické hematologie, Nemocnice České Budějovice a. s.
2
:
Gastroent Hepatol 2015; 69(3): 262-266
:
Chapters from Internal Medicine: Case report
prolekare.web.journal.doi_sk:
https://doi.org/10.14735/amgh2015262
Trombotická mikroangiopatie (TMA) je syndrom charakterizovaný hemolytickou anémií s fragmentací erytrocytů v periferní krvi, trombocytopenií způsobenou zvýšenou agregací trombocytů a jejich konzumpcí, poruchami mikrocirkulace vyvolanými přítomností mikrotrombů. Okluze cév vede k ischemii tkání a orgánů, což vede k difuznímu funkčnímu postižení různých orgánů, nejčastěji pak centrální nervové soustavy, ledvin, srdce, plic a střeva. Mezi klasické trombotické mikroangiopatie řadíme dvě etiologicky odlišné klinické jednotky – trombotickou trombocytopenickou purpuru a hemolyticko-uremický syndrom. Sekundární TMA mohou být způsobeny některými léky, infekcemi, maligními onemocněními, systémovými chorobami nebo mohou souviset s těhotenstvím. Jedná se o vzácná onemocnění – v případě klasických TMA s udávanou incidencí 1 – 4 případy/ milion obyvatel ročně. Vzhledem k různorodosti klinických projevů se však může s trombotickou mikroangiopatií setkat kterýkoli lékař. Pro osud pacienta je rozhodující včasná diagnóza a adekvátní terapie, v opačném případě je nemocný ohrožen rychlým selháním životních funkcí. Cílem sdělení je podat stručný přehled o trombotických mikroangiopatiích, upozornit na některé novinky v diagnostice a terapii a přispět ke zvýšení povědomí o těchto onemocněních, u nichž lze předpokládat, že nejsou vždy správně diagnosticky zařazena.
Klíčová slova:
hemolyticko-uremický syndrom – trombotická trombocytopenická purpura – trombocytopenie – hemolytická anémie – výměnná plazmaferéza – eculizumab
Mezi klasické trombotické mikroangiopatie (TMA) se řadí dvě málo častá, ale závažná onemocnění – trombotická-trombocytopenická purpura (Moschcowitzův syndrom – TTP) a hemolyticko-uremický syndrom (Gasserův syndrom – HUS) s dalšími klinickými podjednotkami (tab. 1) [1,2]. V posledních letech byl učiněn značný pokrok v poznání patogeneze těchto etiologicky různorodých onemocnění chápaných dříve jako jedna klinická jednotka (tzv. TTP/ HUS). To umožnilo kromě odhadnutí prognózy nemoci i zavedení diferencované terapie, jež se dosud řídila spíše empirií a v řadě případů byla spojena s neuspokojivými výsledky, vysokou mortalitou a závažnými komplikacemi [1,3].
![Zjednodušené rozdělení trombotických mikroangiopatií [1,3–5].
Tab. 1. Simplified distribution of thrombotic microangiopathies [1,3–5].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/10ae186f72daf555def46e31989d397b.png)
Nejvýznamnějším pokrokem je zavedení inhibitorů aktivace komplementu (eculizumab), jež dramaticky zlepšily prognózu některých nemocných [4].
Vedle těchto primárních forem TMA existují i sekundární formy, které mohou být vyvolány léky, infekcemi, maligními onemocněními nebo mohou souviset s těhotenstvím.
Vyšetřovací metody
V anamnéze se zaměřujeme především na následující údaje: všechna prodělaná onemocnění, možné vyvolávající příčiny (nádory, infekce, systémová onemocnění, transplantace, těhotenství, podrobná léková anamnéza). Nezapomínáme na rodinnou anamnézu pro možnost hereditární formy onemocnění [1,6 – 8].
Laboratorně vyšetřujeme krevní obraz s diferenciálním rozpočtem včetně retikulocytů, fragmentocytů (schistocytů), haptoglobinu, volného hemoglobinu, bilirubinu a Coombsova testu. První známkou hemolýzy může být vzestup sérové laktátdehydrogenázy (LDH). K potvrzení nebo vyloučení orgánového postižení indikujeme podle klinického obrazu zpravidla vyšetření uvedená v tab. 2.
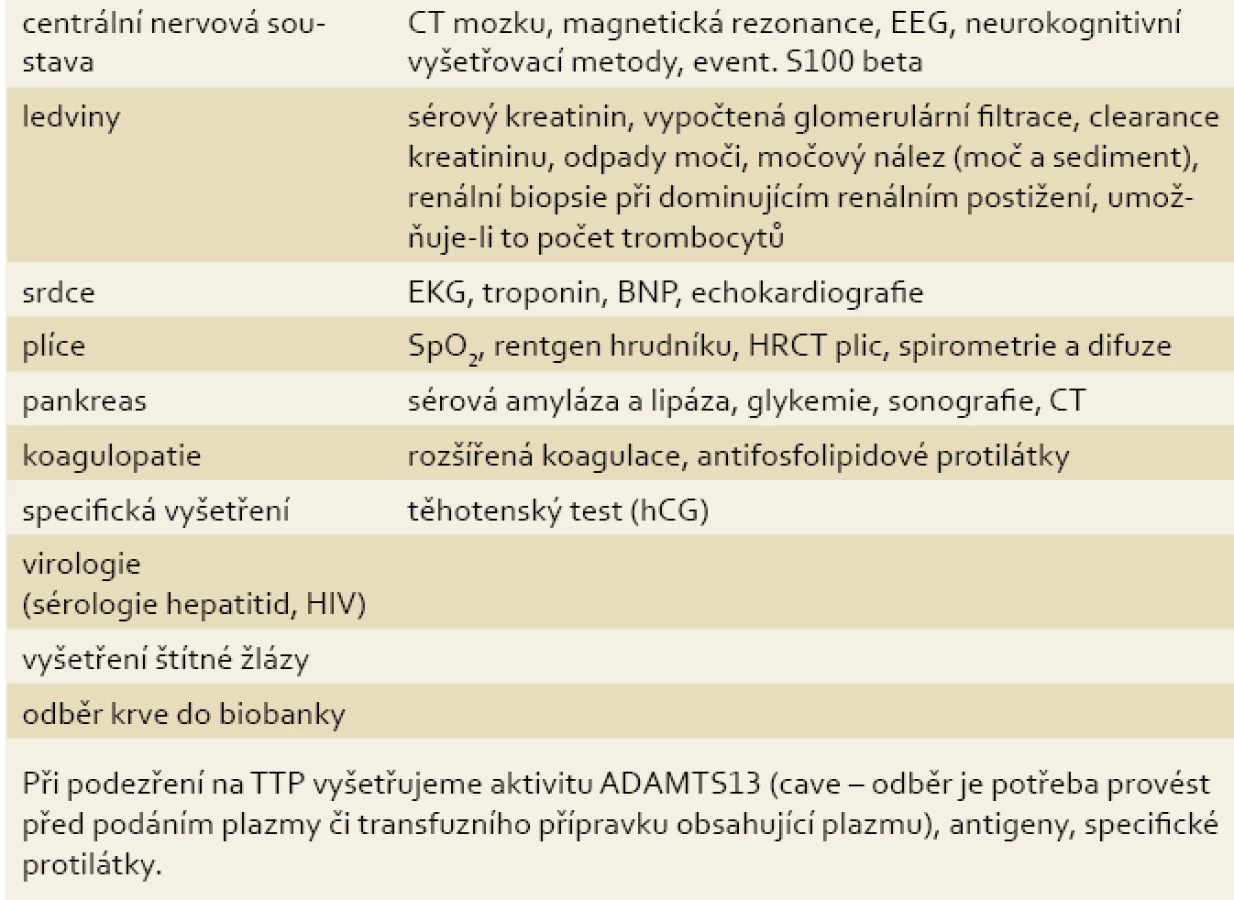
Při podezření na TTP vyšetřujeme aktivitu ADAMTS13 (cave – odběr je potřeba provést před podáním plazmy či transfuzního přípravku obsahujícího plazmu), antigeny, specifické protilátky a inhibitory, dále aktivitu von Willebrandova faktoru; při suspekci na HUS provádíme bakteriologická vyšetření, průkaz toxinů (E. coli, Shigella) a dále vyšetření komplementu (C3, C4, CH50 a další složky). Rozsah vyšetření konzultujeme se specializovaným hematologem [1,3,8].
Principy terapie TMA
Z etablovaných terapeutických možností máme k dispozici výměnnou plazmaferézu. Efekt spočívá v eliminaci protilátek a imunokomplexů, doplňujeme ADAMTS13 a normální von Willebrandův faktor, jde o iniciální terapii všech forem TMA s výjimkou typického HUS [9]. Zároveň metoda umožňuje podání velkých objemů plazmy. Plazmaferézy se provádějí denně do normalizace počtu trombocytů a LDH. Alternativou je podávání čerstvě mražené plazmy, kdy takto doplňujeme ADAMTS13 a komplement, této možnosti se využívá zvláště při kongenitálním deficitu. Podávání plazmy je spojeno mj. s riziky alergických a infekčních komplikací, preventivně před terapií podáváme antihistaminika a glukokortikoidy [1 – 3,8].
Mechanizmem účinku glukokortikoidů a rituximabu je imunosuprese, ta je účinná při přítomnosti autoprotilátek proti ADAMTS13 a komplementu. Rituximab je u TTP velmi efektivní, remise je dosaženo u většiny pacientů během tří týdnů [8,9]. Pro úplnost je třeba zmínit možnost splenektomie, jež má za následek eliminaci paměťových a T buněk, provádí se jen v případě refrakterní TTP. Podpůrná terapie zahrnuje intenzívní péči, náhradu funkce ledvin zpravidla hemodialýzou a transfuze erytrocytů [8]. Individuálně lze zvážit podávání protidestičkových léků (kyselina acetylsalicylová, clopidogrel, prasugrel) k potlačení trombogeneze. Naopak podávání trombokoncentrátů lze přirovnat k přilévání oleje do ohně a mělo by být omezeno jen na zvláště těžké případy trombocytopenie.
Pro léčbu atypického hemolyticko-uremického syndromu je k dispozici nové léčivo eculizumab [4,10,11] – humanizovaná monoklonální protilátka proti komplementárnímu faktoru C5, která rychle normalizuje laboratorní parametry a zlepšuje renální funkci. Problémem je extrémně vysoká cena, nutnost dlouhodobého podávání a zvýšené riziko infekčních komplikací (především meningokokové meningitidy) – jde o důsledek blokády terminální cesty aktivace komplementu [11].
Kazuistika
Pětačtyřicetiletá nemocná bez závažnější osobní anamnézy byla přijata během vánočních svátků na krajské gastroenterologické pracoviště pro abdominalgie se zvracením a mírnou hepatopatií charakteru parenchymatózní hepatální léze. Během několika hodin po přijetí došlo k rozvoji oligurie, po vyloučení subrenální obstrukce byla indikována akutní dialýza. Pro podezření na rychle progredující glomerulonefritidu byla pacientka přeložena na naši kliniku k určení diagnózy a k terapii. Laboratorně byla zjištěna progredující anémie (Hb 80 g/ l) a trombocytopenie (vstupně 67 × 109/ l), současně byly zachyceny zvýšené schistocyty (fragmentocyty) a rovněž známky hemolýzy (elevace LDH, vyšší bilirubin, spotřebovaný haptoglobin, zvýšené retikulocyty, negativní Coombsův test, INR a aPTT v normě), neurologická symptomatika přítomna nebyla. Na základě klinického obrazu a výsledků laboratorních vyšetření byla vyslovena suspekce na TMA, byly zahájeny plazmaferézy proti plazmě a podány kortikoidy (metylprednizolon iniciálně 250 mg/ den s postupným převodem na p.o. formu). Pro trvající renální selhávání a hyperhydrataci bylo pokračováno v hemodialyzační léčbě.
Před zahájením plazmaferéz byl proveden odběr plazmy do citrátu za účelem dalšího vyšetření (ADAMTS13, membránový kofaktorový protein). Protože však pacientka byla přijata ve sváteční den, byl vzorek poslán do laboratoře se žádostí o zamrazení, aby bylo možné jej vyšetřit nejbližší pracovní den – bohužel však došlo k jeho znehodnocení. Vyšetření dalšího vzorku, odebraného po zahájení plazmaferéz, již nebylo validní.
Již po šesti plazmaferézách došlo k normalizaci trombocytů, jaterních enzymů, obnovení diurézy a celkové stabilizaci stavu, pacientka byla bez jakýchkoli krvácivých projevů. Částečné zlepšení renálních funkcí umožnilo snížení frekvence hemodialýz na dvakrát týdně s vizí postupného prodlužování mezidialyzačních intervalů, pacientka byla v celkově dobrém stavu propuštěna do péče hemodialyzačního střediska krajské nemocnice a pravidelně kontrolována na našem pracovišti. Případ pacientky byl uzavřen jako atypický HUS s dobrou odpovědí na terapii výměnnými plazmaferézami.
Po třech měsících od přijetí do nemocnice se hladina sérového kreatininu stabilizovala na 148 umol/ l, clearance kreatininu byla 48 ml/ min – ustálila se tedy ve stadiu CKD 3A, bylo tedy možné hemodialyzační léčbu ukončit. K další kontrole na naše pracoviště pacientka již nepřišla a nepodařilo se nám ji kontaktovat ani přes spádové hemodialyzační středisko, její další osud nám není znám.
Závěr
TMA je charakterizována hemolytickou anémií s fragmentací erytrocytů v periferní krvi, trombocytopenií způsobenou zvýšenou agregací trombocytů a jejich konzumpcí a poruchami mikrocirkulace trombotické etiologie s následným difuzním funkčním postižením CNS, ledvin, srdce, plic a střeva. TMA patří ke vzácným onemocněním, lze se však oprávněně domnívat, že v některých případech ujdou pozornosti. Z pohledu nefrologa je nutné zdůraznit, že k rutinnímu laboratornímu vyšetření pacienta se zvýšenou hladinou sérového kreatininu patří i stanovení LDH (laktátdehydrogenázy), protože může jít o první projev hemolýzy. Pak je nutné v rámci diferenciální diagnostiky pomýšlet i na onemocnění z okruhu TMA, např. HUS. K dalším varovným (nicméně nespecifickým) klinickým příznakům, jejichž přítomnost by měla rovněž vést k podezření na TMA, patří bolesti hlavy, kvantitativní nebo kvalitativní poruchy vědomí, případně jiné neurologické symptomy; dále jakékoli dysfunkce srdce, plic nebo střeva a krvácivé projevy (sufuze, petechie). Samozřejmostí by měl být pečlivý odběr vegetativní anamnézy a dotaz na prodělané průjmy, dále je nutné při odběru anamnézy a vyšetřování pacienta zvažovat i další možné příčiny TMA (např. léky, nádorové onemocnění, systémové choroby, infekci HIV). Pacienta s TMA je třeba co nejdříve odeslat na specializované pracoviště, které zahájí co nejdříve léčbu a doplní další potřebná vyšetření.
Výměnná plazmaferéza a substituce čerstvě mraženou plazmou může vést k restauraci renálních funkcí, naopak při neadekvátní léčbě je vysoké riziko fatálního vyústění. Přestože plazmaferéza a substituce plazmou vede u velké části pacientů k výraznému zlepšení stavu, jde často jen o symptomatický postup, navíc spojený s některými riziky. Pro léčbu některých nemocných s atypickým HUS je k dispozici nový lék eculizumab, další terapeutické modality jsou zatím ve fázi klinických studií.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Doručeno: 10. 6. 2015
Přijato: 17. 6. 2015
MU Dr. Jan Vachek
Klinika nefrologie
1. LF UK a VFN v Praze
U nemocnice 2 128 08 Praha 2
jan.vachek@gmail.cz

MUDr. Jan Vachek
MU Dr. Jan Vachek se narodil v roce 1981 v Praze, studium na 3. lékařské fakultě UK ukončil v roce 2007. Část studia absolvoval v rámci programu Erasmus/ Sokrates na berlínské lékařské fakultě Charité, kromě toho se zúčastnil několika dalších pre - a postgraduálních studijních pobytů a stáží. Po studiu pracoval na interním oddělení pražské nemocnice ve Vysočanech – Clinicum a.s., po dvou letech přešel na Kliniku nefrologie 1. LF UK a Všeobecné fakultní nemocnice, kde pracuje dosud. Kromě toho externě spolupracuje s hemodialyzačním střediskem a interním oddělením Klatovské nemocnice, a.s., hemodialyzačním střediskem B. Braun Avitum a obecně prospěšnou společností Alzheimercentrum, vykonává pohotovostní služby na Společném interním příjmu Všeobecné fakultní nemocnice. Pod vedením prof. Tesaře a MU Dr. Ciferské se věnuje postgraduálnímu studiu v oboru farmakologie a toxikologie. Atestoval s pochvalou z vnitřního lékařství v roce 2013. Ve stejném roce vydal monografii Farmakoterapie v těhotenství a při kojení (Vachek, Tesař, Zakiyanov, Maxová), oceněnou Českou internistickou společností, je autorem publikace o historii inzulinu (Příběh inzulínu, 2009). Publikuje v domácím i zahraničním odborném tisku.
Sources
1. Nangaku M, Nishi H, Fujita T. Pathogenesis and prognosis of thrombotic microangiopathy. Clin Exp Nephrol 2007; 11(2): 107 – 114.
2. Tsai HM. Current concepts in thrombotic thrombocytopenic purpura. Ann Rev Med 2006; 57 : 419 – 436.
3. Adler M, Kremer Hovinga JA, Lämmle B. Thrombotic thrombocytopenic purpuran – an often missed diagnosis. Rev Med Suisse 2014; 10(452): 2280 – 2284.
4. Legendre CM, Licht C, Muus P et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med 2013; 368(23): 2169 – 2181. doi: 10.1056/ NEJMoa1208981.
5. Bennett CL, Weinberg PD, Rozenberg-Ben-Dror K et al. Thrombotic thrombocytopenic purpura associated with ticlopidine. A review of 60 cases. Ann Intern Med 1998; 128(7): 541 – 544.
6. Knoebl P. Blood coagulation disorders in septic patients. Wien Med Wochenschr 2010; 160(5 – 6): 129 – 138. doi: 10.1007/ s10354-009-0738-9.
7. Merayo-Chalico J, Demichelis-Gómez R, Rajme-López S et al. Risk factors and clinical profile of thrombotic thrombo-cytopenic purpura in systemic lupus erythematosus patients. Is this a distinctive clinical entity in the thrombotic microangiopathy spectrum?: a case control study. Thromb Res 2014;134(5): 1020 – 1027. doi: 10.1016/ j.thromres.2014.09.005.
8. Scheiring J, Rosales A, Zimmerhackl LB. Clinical practice. Today’s understanding of the haemolytic uraemic syndrome. Eur J Pediatr 2010; 169(1): 7–13. doi: 10.1007/ s00431-009-1039-4.
9. Khandelwal P, Gupta A, Sinha A et al. Effect of plasma exchange and immunosuppressive medications on antibody titers and outcome in anti-complement factor Hantibody-associated hemolytic uremic syndrome. Pediatr Nephrol 2015; 30(3): 451 – 457. doi: 10.1007/ s00467-014-2948-7.
10. Delmas Y, Vendrely B, Clouzeau B et al. Outbreak of Escherichia coli O104:H4 haemolytic uraemic syndrome in France: outcome with eculizumab. Nephrol Dial Transplant 2014; 29(3): 565 – 572. doi: 10.1093/ ndt/ gft470.
11. Cullinan N, Gorman KM, Riordan M et al. Case report: Benefits and challenges of long-term eculizumab in atypical hemolytic uremic syndrome. Pediatrics 2015; 135(6): e1506 – e1509. doi: 10.1542/ peds.2014-3503.
Labels
Paediatric gastroenterology Gastroenterology and hepatology SurgeryArticle was published in
Gastroenterology and Hepatology

2015 Issue 3
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
- The Importance of Limosilactobacillus reuteri in Administration to Diabetics with Gingivitis
Most read in this issue
- Saccharomyces boulardii – probiotic yeast from Indochina
- Groove (paraduodenal) pancreatitis – a case series of six patients
- Recommendation of surgical treatment in patients with inflammatory bowel diseases – part 2: Crohn´s disease
- Trombotic microangiopathy – a nephrologists‘ view