
Infekce jaterní cysty při polycystóze jater jako zdroj sepse
Hepatic cyst infection as a source of sepsis in liver polycystosis
Liver cysts are commonly identified by imaging methods. Polycystic liver disease is diagnosed when there are ten or more cysts and often develops as part of polycystic kidney disease or an isolated polycystic liver disorder. This disease is usually asymptomatic and does not require treatment. Impairment of renal function is one of the main symptoms in patients with cystic kidney disease. Complications of polycystosis, such as cyst bleeding, portal hypertension, icterus, and hepatic failure, are rare. A hepatic cyst infection that requires both antibiotic therapy and radiological or surgical intervention is a serious complication due to the high failure rate of separate antibiotic therapy. Quinolones are the recommended antibiotic therapy. Medications, such as somatostatin analogues, mTOR inhibitors, and ursodeoxycholic acid, have only been used in clinical trials. Arterial embolization of the hepatic artery or percutaneous aspiration and subsequent sclerotherapy of the cyst can aid interventional radiology. Several clinical classifications listed in this article can help clinicians decide whether to perform surgical therapy or liver transplantation. We present the case of a 60-year-old man with hereditary polycystosis of the kidneys and liver who was treated for sepsis due to hepatic cyst infection. Although parenteral antibiotic therapy reduced his fever and improved his laboratory findings, improvement of subjective problems occurred after the radiological intervention. Due to disease progression and the risk of the cyst infection recurring, the patient attended a consultation at a transplant center and liver transplantation was indicated.
Key words:
polycystic liver disease – sepsis – antibiotic therapy
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE „uniform requirements“ for biomedical papers.
Submitted:
5. 1. 2018
Accepted:
8. 2. 2018
Authors:
G. Růžičková; L. Gabalec
Authors‘ workplace:
Interní oddělení – Endoskopie, Orlickoústecká nemocnice, Nemocnice Pardubického kraje, a. s., Ústí nad Orlicí
Published in:
Gastroent Hepatol 2018; 72(2): 122-128
Category:
Hepatology: Case Report
doi:
https://doi.org/10.14735/amgh2018122
Overview
Cysty jater jsou častým nálezem na zobrazovacích metodách. Polycystické postižení jater popisujeme při počtu minimálně 10 cyst. Vyskytuje se nejčastěji jako součást polycystického onemocnění ledvin nebo jako izolované polycystické postižení jater. Onemocnění je obvykle asymptomatické a nevyžaduje léčbu. U pacientů s postižením ledvin bývá v popředí klinického obrazu renální selhání. Komplikace polycystózy jsou vzácné, jedná se o krvácení do cysty, vznik portální hypertenze, ikterus a jaterní selhání. Závažnou komplikací je infekce jaterní cysty, která vyžaduje jak antibiotickou terapii, tak radiologickou nebo chirurgickou intervenci z důvodu vysokého procenta selhání samotné antibiotické léčby. Z antibiotik je doporučeno podávání chinolonů. Medikamentózní léčba ať analogy somatostatinu, m-TOR inhibitory či ursodeoxycholovou kyselinou je zatím zkoušena pouze v klinických studiích. Intervenční radiologie může pomoci arteriální embolizací hepatické arterie nebo perkutánní aspirací a následnou skleroterapií cysty. Existuje několik klinických klasifikací uvedených v článku, které lze použít k rozhodnutí o nutnosti chirurgického řešení a případně transplantace jater. Prezentujeme případ 60letého muže s dědičnou polycystózou ledvin a jater, léčeného pro septický stav při infekci jaterní cysty. Parenterální antibiotická terapie sice vedla k ústupu febrilií a úpravě laboratorních hodnot, k ústupu subjektivních obtíží však došlo až po odsátí obsahu infikované cysty. Pacient byl vzhledem k pokročilosti onemocnění a riziku recidivy infekce cyst indikován k transplantaci jater.
Klíčová slova:
polycystóza jater – sepse – antibiotická léčba
Úvod
Polycystóza jater (PLD – polycystic liver disease) je autozomálně dominantně (AD) dědičná porucha, která je obvykle součástí dominantního polycystického postižení ledvin (ADPKD – autosomal dominant polycystic kidney disease) s incidencí 1/400–1/1 000 nebo méně často AD dědičného izolovaného polycystického postižení jater (PCLD – polycystic liver disease) s incidencí 1/100 000–1/1 000 000 nebo se může jednat o Von Meyenburgovy komplexy (VMC) – s odlišnou incidencí v různých studiích 1/18–1/145 nebo 7–60/1 000 [1].
Jde o vývojovou anomálii, při které jsou játra a ledviny prostoupeny četnými cystami. Cysty se vyskytují i v pankreatu, slezině, plicích nebo ovariích. Cysty jater sestávají ze stěny s biliárním epitelem, který reaguje na sekretin, obsah cyst je podobný plazmě a je bohatý na γ-glutamyltransferázu (GGT) a imunoglobulin IgA, jen raritně obsahuje žluč. Cysty mohou být obkrouženy vazivem a jejich zvětšování je způsobeno sekrecí zadrženého biliárního epitelu.
Asymptomatičtí pacienti mívají zcela normální hodnoty jaterních testů, u symptomatických nacházíme mírnou elevaci alkalické fosfatázy (ALP) a celkového bilirubinu, asi u 60–70 % symptomatických pacientů je zvýšena i hladina GGT [2]. Léčba ve většině případů není nutná, onemocnění je obvykle symptomatické, jen v případě velkých cyst nebo bolestivého tlaku či dyspepsie lze provést odsátí tekutiny pod sonografickou kontrolou, vzácně je indikována chirurgická intervence. Komplikace jsou vzácné. Může dojít ke kompresi biliárního stromu s cholestázou nebo ke krvácení do cysty. Raritní jsou spontánní infekce cysty. Mechanizmus jejich vzniku není znám [3].
Prognóza onemocnění je příznivá, pokud je současně přítomna polycystóza ledvin, pak tato obvykle rozhoduje o osudu nemocných.
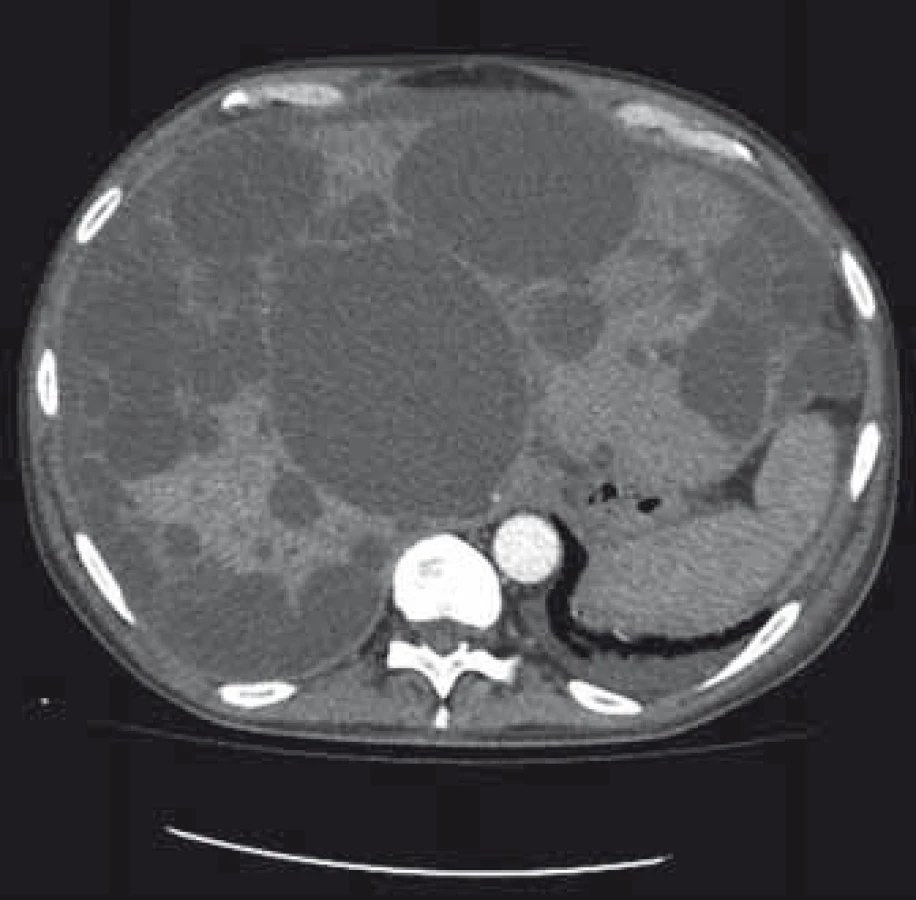
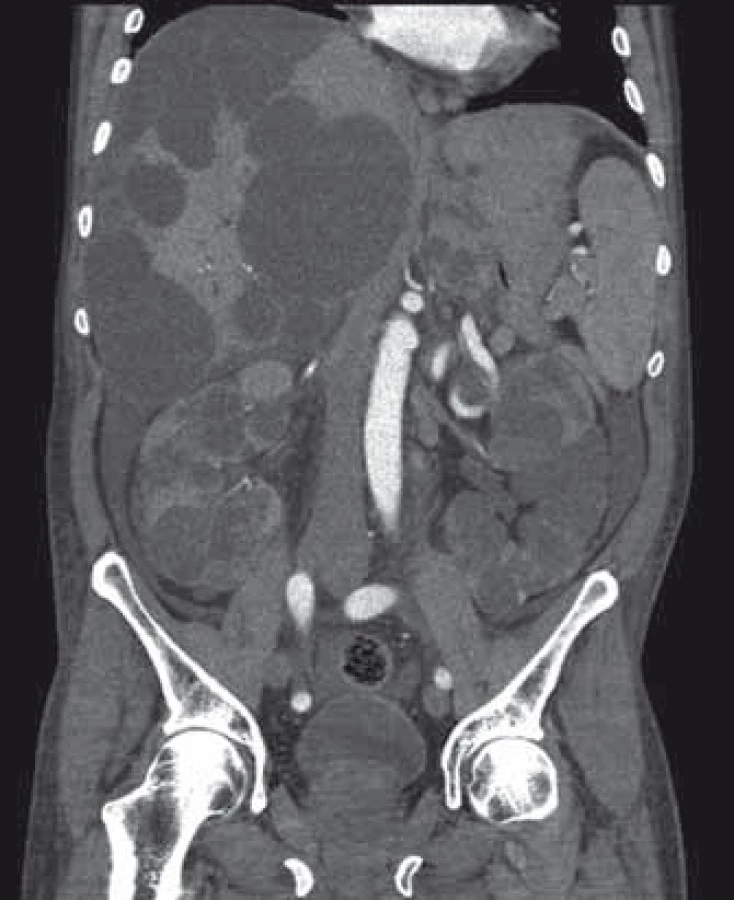
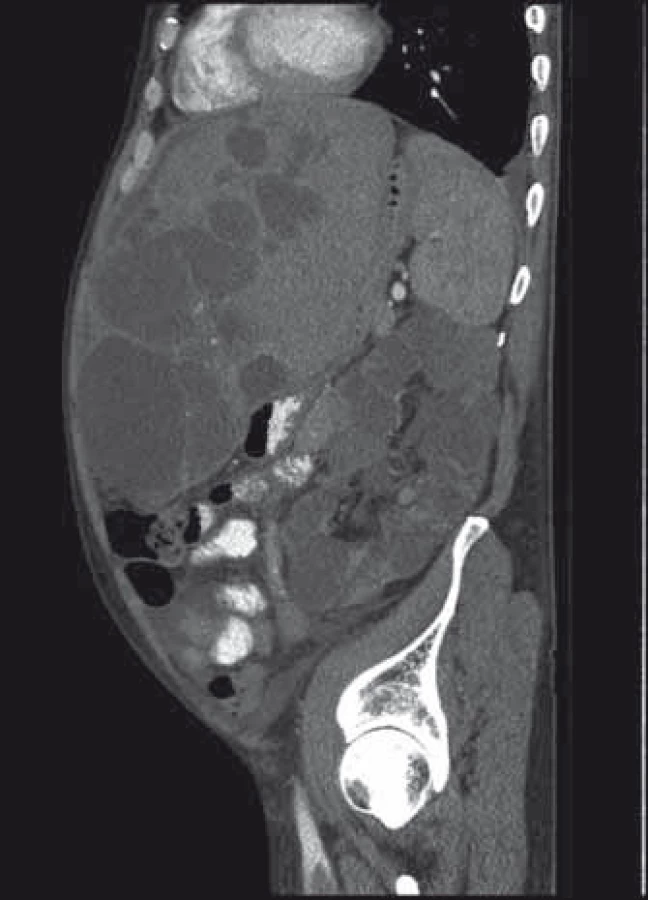
Popis případu
Jedná se o 60letého muže, který je pro polycystózu ledvin sledovaný nefrologem, hodnoty renálních funkcí jsou na úrovni CKD (chronic kidney disease) III. stadia s hodnotami kreatininu v séru kolem 150 µmol/l – hodnoty jsou stabilní poslední 4 roky. Pacient má prokázaný vrozený trombofilní stav – heterozygotní Leidenskou mutaci faktoru V se snížením rezistence k aktivovanému proteinu C (APC). Před 4 lety prodělal masivní plicní embolii léčenou standardně antikoagulační terapií, bez trombolýzy, následně byl s odstupem 1 měsíce hospitalizován pro ataku postantibiotické klostridiové kolitidy. Pacient je dále léčen pro hypertenzi a benigní hypertrofii prostaty.
Pacient byl přijat na interní oddělení pro 6 dní trvající febrilie, zimnice, třesavky, celkovou slabost. Ambulantně byl léčen od 2. dne obtíží aminopenicilinem (Amoksiklav 625 mg po 8 hod) bez efektu. Klinicky byl subfebrilní, dehydratovaný, s tachykardií 110/min a krevním tlakem 92/60 mm Hg. Zdroj infektu z klinického vyšetření nebyl zřejmý. Patologickým nálezem byla objemná tuhá rezistence v celé horní 1/3 břicha. Laboratorně byla patrna elevace C reaktivního proteinu – CRP 285 mg/l (norma do 5 mg/l), vysoký prokalcitonin 3,19 µg/l (norma do 0,10 ug/l), ale leukocyty byly v normě – 7,8 × 109/l, v diferenciálním rozpočtu neutrofily 79 %. Došlo k zvýšení hodnot kreatitininu na 205 µmol/l a urey na 14,5 mmol/l. Nález v moči a sedimentu byl nezánětlivý. Hodnoty bilirubinu byly hraniční 18,7 µmol/l, aminotransferázy (ALT, AST) v normě, GGT 6,67 µkat/l, ALP 2,80 µkat/l. Na rentgenu (RTG) plic byla zobrazena vysoko uložená bránice vpravo a zmnožené pruhy parahilózně až vzhledu počínajícího infiltrátu. Ve srovnání se starší dokumentací se jedná o chronické změny. Na sonografickém vyšetření břicha objemná játra prostoupená mnohočetnými anechogenitami až 8 cm, ledviny zvětšené – 140 mm vpravo a 180 mm vlevo prostoupené mnohočetnými anechogenitami velikosti až 50 mm, všechny čiré, neprokázáno městnání v uropoetickém systému, malý ascites perihepaticky. Pacient byl léčen parenterálně cefotaximem (1 g à 6 hod), parenterálně rehydratován, pro anamnézu trombofilního stavu byl zajištěn preventivní dávkou nízkomolekulárního heparinu. Byla podávána probiotika, nutriční podpora – sipping. Kultivačně byl opakovaně negativní nález v moči, krku i v hemokulturách. Při antibiotické (ATB) terapii došlo k ústupu teplot, laboratorně pozvolna klesala zánětlivá aktivita. Pro trvající bolesti břicha v oblasti epigastria bylo provedeno kontrolní sonografické vyšetření břicha, kde bylo identifikováno několik cyst v levém laloku jater se zahuštěným obsahem, největší o velikosti 85 mm. K ozřejmění nálezu na játrech provedeno vyšetření výpočetní tomografií (CT) břicha, kde byl popsán bizarní obraz PLD a polycystózy ledvin, s téměř kompletním útlakem v. cava inferior cystami, s podezřením na infekci cyst v levém laloku jater. Po domluvě s radiologem jsme indikovali probatorní punkci cysty pod CT kontrolou. Tato byla provedena 8. den hospitalizace s odsátím 140 ml zkaleného obsahu. Při punkci vedlejší cysty byl její obsah čirý. Materiál byl odeslán na kultivaci v běžných zkumavkách a kultivačních lahvičkách na hemokultury, patogenní agens však ani tak nebylo prokázáno, cytologicky byl v punktátu popsán obraz hnisavého zánětu. Stav jsme uzavřeli jako sepsi při infekci jaterní cysty vzhledem ke klinickému a cytologickému nálezu a zobrazovacím metodám. Po punkci cysty došlo k výrazné úlevě od obtíží, bolesti břicha ustoupily, došlo k rychlému zlepšování celkového stavu pacienta, který byl před punkcí značně limitován bolestí i v základních činnostech. Laboratorně se hodnoty N katabolitů stabilizovaly na pacientových obvyklých hodnotách, CRP pozvolna klesalo, v týdnu před propuštěním však pokles CRP zpomaluje, proto ještě před propuštěním byla provedena změna ATB terapie z cefalosporinu, který byl podáván celkem 14 dní v původní dávce, na chinolon (ciprofloxacin 400 mg à 12 hod), který byl 3 dny podáván parenterálně a dále ponechán v perorální formě v postupně se snižující dávce (500 mg à 12 hod na 2 týdny, pak à 24 hod na 2 týdny) k dlouhodobé udržovací ATB léčbě. CRP bylo při propuštění 50,9 mg/l. Při ambulantní kontrole 3 týdny po propuštění byl pacient v dobrém stavu, afebrilní, bez obtíží krom mírných tlaků v břiše po jídle, CRP 24,5 mg/l, nadále byl ponechán na ATB terapii chinolonem – ciprofloxacinem v dávce 250 mg/24 hod.
Pacient byl referován do IKEM jako kandidát transplantace jater a ledvin.
Diskuze
Hlavním úmyslem autorů tohoto článku bylo vytvoření přehledu o problematice polycystických onemocnění, který sami v době léčby pacienta s infekcí jaterní cysty postrádali. Algoritmus diagnostiky a léčby infikované cysty je uveden na schématu 1 a 2.


Jaterní cysty jsou běžným nálezem při sonografickém vyšetření břicha [4]. O PLD hovoříme, pokud má pacient v játrech více než 20 cyst [1]. Nověji stačí k diagnóze více než 10 cyst [5]. PLD byla poprvé popsána roku 1856 Bristowem u pacientů se současnou ADPKD [3]. Až v roce 2003 byly popsány další typy polycystózy, z nichž některé se vyskytují samostatně bez postižení ledvin. Polycystická onemocnění jater jsou vzácné poruchy vzniklé na podkladě strukturálních změn během vývoje žlučového stromu, kdy dochází k oddělení duktálních struktur od biliárního stromu, které vyústí ve vznik cyst. Tyto struktury jsou patrné již velmi časně, ale jsou asymptomatické až do dospělosti, kdy dochází k jejich růstu.
V současnosti rozeznáváme tři typy polycystického onemocnění jater: 1. VMC (biliární hamartomy, jaterní cystické hamartomy) charakterizované přítomností malých nodulárních cystických lézí, onemocnění není dědičné; 2. izolované polycystické onemocnění jater – AD dědičné s přítomností mnohočetných jaterních cyst (PLD) a 3. AD dědičné polycystické onemocnění ledvin postihující výskytem mnohočetných cyst ledviny i játra (ADPKD) [6]. U všech tří typů onemocnění jsou cysty jater odděleny od žlučového stromu, jehož architektura není postižena. VMC, také nazývané mikrohamartomy, jsou benigní cystické uzly rozptýlené v celých játrech, obvykle jsou lokalizovány interlobulárně v oblasti periferních žlučovodů pod Glissonovým pouzdem. VMC se objevuje samostatně nebo současně s PCLD a ADPKD. Cystické struktury zpočátku komunikují s periferním biliárním stromem, ale v průběhu vývoje se oddělují. VMC jsou většinou zjištěny náhodně zobrazovacími metodami, obvykle zůstávají během života němé a nevyžadují další dovyšetřování. V laboratorním nálezu se mohou vyskytovat mírné odchylky od normy, signifikantní hepatomegalie je při VMC vzácná. Výjimečně může vzniknout ikterus a cholangitida [1].
Polycystické postižení jater a autozomálně dominantní polycystické postižení ledvin
Hlavním klinickým projevem při PCLD a nejčastější extrarenální manifestací při ADPKD jsou jaterní cysty. Cysty vznikají ze žlučovodů středního kalibru. Histologicky jsou ohraničeny kuboidními oploštělými epitelovými buňkami obklopenými fibrózním stromatem. Mohou být omezeny pouze na jeden nebo více segmentů jater nebo mohou postihovat játra v celém rozsahu.
Fenotyp PCLD je omezen pouze na játra. U pacientů s ADPKD můžeme radiologicky identifikovat přítomnost cyst v dalších orgánech – pankreatu (9 %), semenných váčcích (43 %), tyto projevy bývají asymptomatické. Arachnoidální cysty jsou pozorovány u 8 % pacientů s ADPKD, výjimečně mohou vést ke vzniku subdurálního hematomu.
Přítomnost velkých a početných cyst vede často k hepatomegalii. Prevalence jaterních cyst u pacientů s ADPKD ve věku 15–45 let je 83 %, u 35–46letých pacientů až 94 %. Ženy s ADPKD nebo PCLD obvykle mívají závažnější fenotyp, obzvláště ty s anamnézou vícečetné gravidity a dlouhodobé expozici estrogenům, expozice estrogenu v průběhu těhotenství, užívání orální antikoncepce nebo estrogenové substituční terapie může urychlit progresi nemoci. Ženy mívají průměrně větší objem jater a bývají mladší v době prezentace onemocnění. Všeobecně pacienti s PCLD mívají více příznaků a jaterních (liver-related) komplikací než pacienti s ADPKD. Cysty jater se zřídka projevují v raném dětství, výjimečně byly zaznamenány symptomatické případy u mladých dospělých s ADPKD.
Odhaduje se, že asi 1/2 pacientů s pokročilým onemocněním prodělala některou z komplikací – krvácení do cysty, rupturu nebo infekci cysty, častější jsou u pacientů s ADPKD než u PCLD [1,3].
Krvácení do jaterní cysty se typicky projevuje jako akutní bolest v pravém horním kvadrantu břicha. Příznaky vznikají akutně, progredují v prvním dnu (dnech), ale spontánně ustupují. Charakter bolesti může být i kolikovitý a doprovázený zvracením. Diagnózu určí ultrasonografie nebo magnetická rezonance (MR). Ruptura cysty je vzácnou komplikací, která se projevuje akutním záchvatem bolesti. Hemodynamicky významné krvácení je raritní. Pokud pokračuje intraperitoneální leak, je nutná chirurgická intervence ke kontrole hemostázy [1,7].
Prevalence infekce jaterních cyst u ADPKD je 3 % u pacientů s end-stage renálním selháním, ale méně než 1 % u pacientů v nižších stadiích renálního selhání. Příčinou infekce jaterní cysty může být předchozí abdominální operace, chronická hemodialýza, diabetes mellitus nebo imunosupresivní léčba, často je však příčina vzniku infekce nejasná [8,9].
Infekce jaterních cyst je závažnou komplikací s náročnou léčbou a vysokým rizikem recidivy. Aktuální diagnostická kritéria zahrnují klinické obtíže, laboratorní a radiologické parametry vč. zvýšené citlivosti břicha, horečku nad 38 °C trvající déle než 3 dny, zvýšený CRP a prokázanou nepřítomnost krvácení do cysty na CT. Zesílení stěny jaterní cysty a heterogenní obsah (debris – sediment) jsou vždy podezřelé z infekce. Infekce jaterních cyst může být příčinou život ohrožující sepse, častěji ale způsobuje opakované epizody horečnatých stavů bez jiné zjištěné příčiny. Přínos MR zobrazení je neznámý, CT má nízkou senzitivitu i specificitu pro průkaz infekce cysty. K potvrzení infekce jaterní cysty je možné provést pozitronovou emisní tomografii (PET) se značenými leukocyty [8,10]. Pacienti s PCLD mohou mít v období infekce jaterní cysty výrazně zvýšený marker CA 19-9, s poklesem po léčbě [11]. Detekce neutrofilů a infekčního agens v aspirátu cystické tekutiny potvrzuje infekci cysty a umožňuje cílenou ATB léčbu.
Pro ATB léčbu infekce jaterní cysty při polycystóze neexistují evidence-based doporučení. Skupina nizozemských autorů analyzovala 41 publikovaných článků zahrnující 54 případů infekce jaterní cysty. Nejčastěji užívanými skupinami ATB byly chinolony (34 %) a cefalosporiny (34 %), peniciliny (31 %) a/nebo aminoglykosidy (29 %) v nejrůznějších kombinacích [8]. Samotná ATB terapie však ve vysokém procentu selhává, zřejmě z důvodu horšího průniku ATB do cysty a intervenční výkon (drenáž nebo fenestrace) je nutnou součástí léčby [12].
V pokročilém stadiu polycystického onemocnění může docházet ke vzniku portální hypertenze při snížení průtoku jaterními žilami (hepatic vein outflow) nebo kompresí portální žíly objemnými cystami. Příznaky omezení průtoku jaterními žilami (HVOO – hepatic vein outflow obstruction) jsou abdominální bolesti, hepatomegalie a ascites. U HVOO často nacházíme trombózu jaterních žil. Útlakový syndrom při hepatomegalii může postihnout i v. cava inferior, kdy dochází ke zvýšení tlaku v renálních žilách a vzniku nebo progresi ascitu a otoků dolních končetin. Sekundární komplikací portální hypertenze je vznik jaterní fibrózy, jícnových varixů, splenomegalie nebo ascitu, tyto komplikace jsou popsány u starších pacientů.
Další možnou komplikací je ikterus jako projev jaterního selhání, pozorovaného v pokročilých stadiích onemocnění [13].
V kardiovaskulární oblasti můžeme u pacientů s ADPKD nalézt hypertenzi, intrakraniální aneuryzmata, arteriální aneuryzmata, dilataci kořene aorty, aneuryzma abdominální aorty nebo těžké chlopenní vady, nejvyšší prevalenci má prolaps mitrální chlopně – u pacientů s ADPKD je to až 25–41,2 %, při PCLD 0–10,5 % [14]. Proto je doporučeno časné hodnocení kardiovaskulárních rizikových faktorů u pacientů s ADPKD, v literatuře je doporučován screening intrakraniálních aneuryzmat u rodinných příslušníků MR angiografií [15].
K odhadu závažnosti PLD můžeme použít několik klinických klasifikací:
- Gigotova klasifikace – členění vytvořené k identifikaci nejlepších kandidátů pro chirurgickou fenestraci cyst (tab. 1) [16].
- Quianova klasifikace – hodnotí závažnost onemocnění podle počtu cyst a přítomnosti symptomatické hepatomegalie, je používána v souvislosti s rodinným screeningem (tab. 2) [17].
- Schnelldorferova klasifikace – pomáhá rozlišit pacienty, kteří profitují z resekční nebo transplantační léčby (tab. 3) [18].

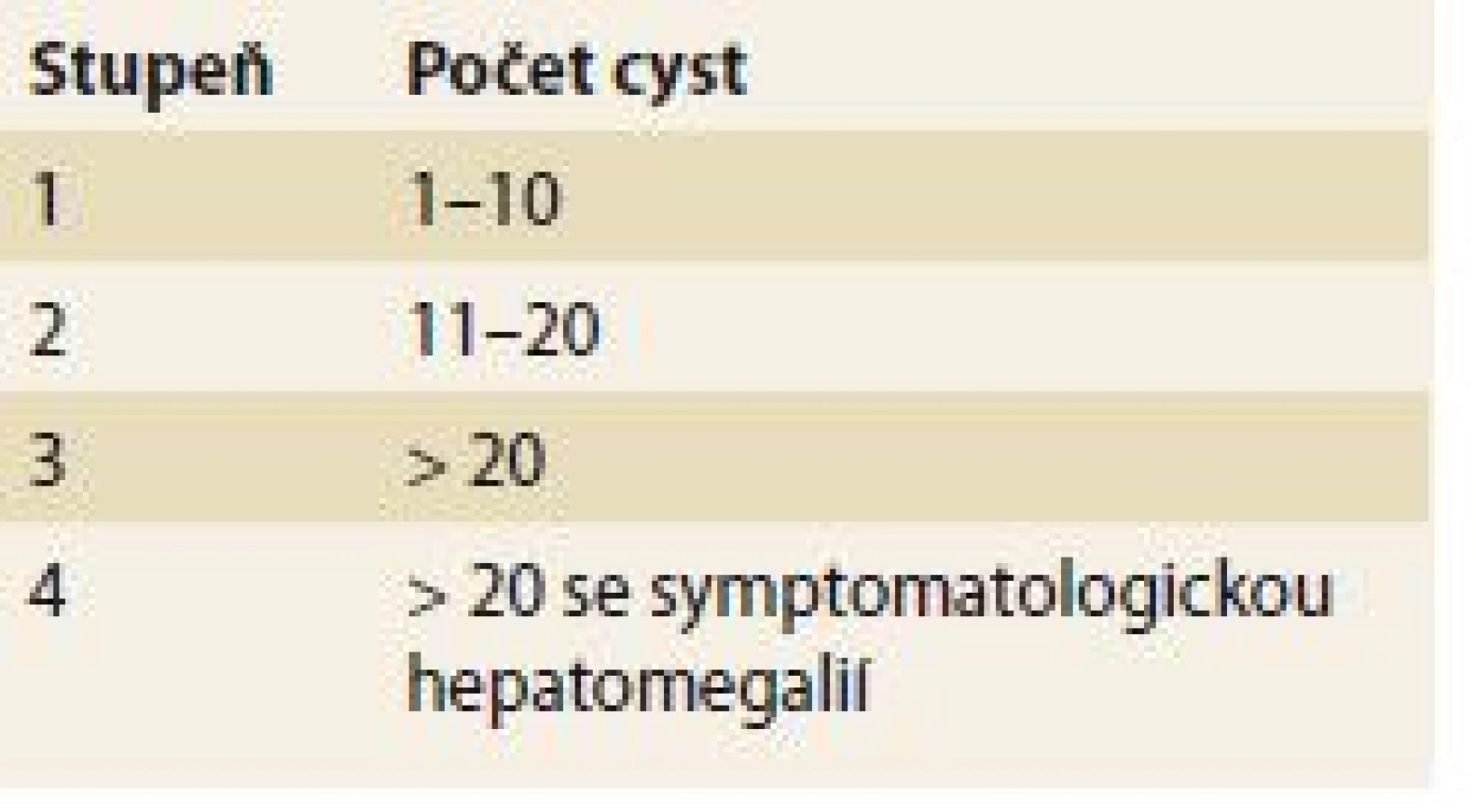
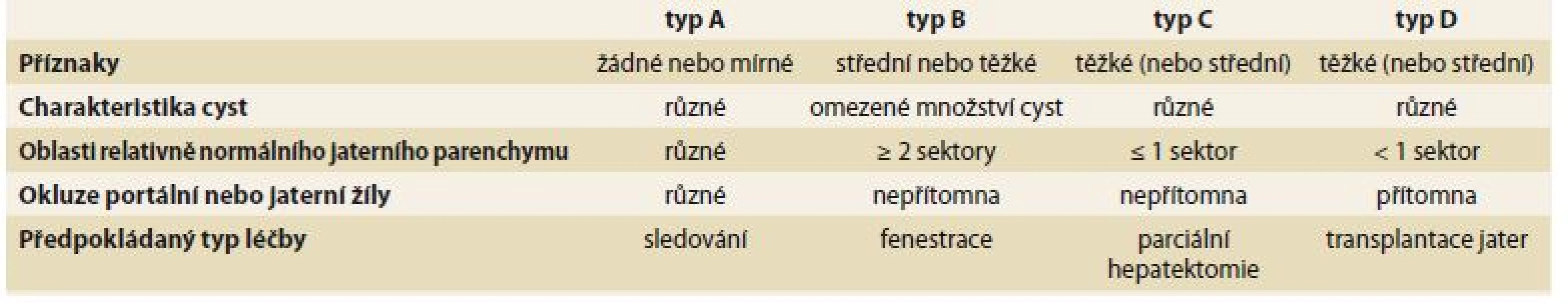
Nechirugická léčba může být použita u symptomatických pacientů s postižením II. nebo III. typu dle Gigota.
Analoga somatostatinu redukují sekreci tekutin v trávicím ústroji a proliferaci mnoha typů buněk vč. cholangiocytů. V klinických studiích byla prokázána signifikantní redukce objemu jater po 6–12měsíční terapii lanreotidem v porovnání s placebem. Redukce objemu jater však byla pouze 3–5 % a závažnost abdominálních obtíží nebyla významně ovlivněna. Terapie analogy somatostatinu je určena pouze po vybranou skupinu pacientů se symptomatickou PLD s vysokým rizikem chirurgické intervence. U pacientů s ADPKD byl při léčbě analogy somatostatinu pozorován vyšší výskyt infekcí jaterních cyst. Proto byla předchozí infekce jaterních cyst považována za vylučovací kritérium pro studii [19].
V klinických studiích u pacientů s ADPKD byly testovány imunosupresivní a antiproliferativní účinky inhibitorů m-TOR (mammalian target of rapamycin) – everolimu a sirolimu, terapeutický efekt nebyl prokázán a použití mimo klinické studie není doporučeno [20,21]. Užití kyseliny ursodeoxycholové v randomizované klinické studii prokázalo, že není schopna redukovat objem jater u PCLD, ale u podskupiny pacientů ADPKD redukuje růst cyst [22].
Intervenční radiologie
Arteriální embolizace by mohla být přínosná pro pacienty s pokročilým onemocněním, kdy větve hepatické arterie zásobující jaterní segmenty nahrazené cystami jsou ošetřeny transkatetrovou embolizací s použitím microcoils nebo částic polyvinyl alkoholu o průměru 150–250 µm. Největší soubor zahrnoval 30 pacientů, kdy byla prokázána redukce objemu cyst (cca o 2 cm3), po několika měsících došlo ke zmírnění abdominálních symptomů bez závažných komplikací [23].
Perkutánní skleroterapie se provádí pod radiologickou kontrolou s aspirací obsahu cysty a následnou injekcí sklerotizačního agens (používá se etanol, etanolamin oleát, minocyklin nebo tetracyklin), které inhibuje reakumulaci tekutiny zničením vnitřního epitelu cysty. Je obvykle vhodná u symptomatických pacientů klasifikovaných do typu I dle Gigota, obvykle pokud je průměr cysty větší než 5 cm. Ačkoli i jednotlivé ošetření bývá úspěšné, někteří pacienti vyžadují opakování postupu. Většina pacientů podstupujících perkutánní aspiraci a skleroterapii udává zlepšení obtíží v následujícím období po výkonu, ale jen u 20 % dochází k parciální nebo plné regresi onemocnění [24].
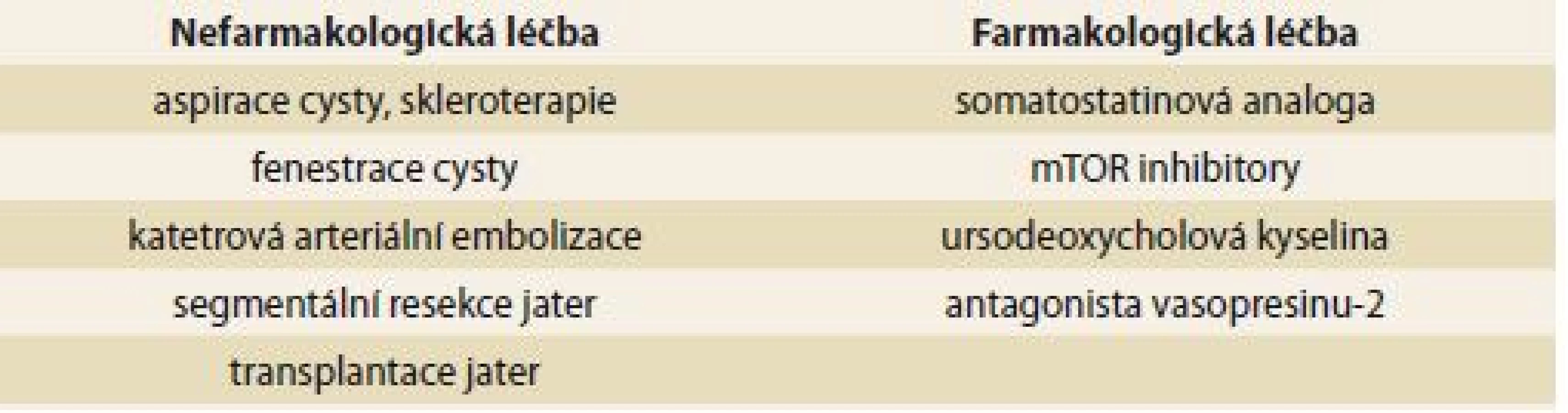
Rozhodnutí o typu chirurgické léčby u pacientů s polycystózou je vždy pro klinika výzvou. Neexistuje konsenzus pro výběrová kritéria a optimální načasování a způsob chirurgické léčby. Současné možnosti zahrnují fenestraci, parciální resekci jater a ortotopickou transplantaci jater (OLT – orthotopic liver transplantation). Fenestrace a parciální resekce jater je možno zvažovat u pacientů typu I a II dle Gigota, u typu III jsou tyto techniky neefektivní a jedinou léčebnou možností je OLT. OLT je indikována u pacientů s obtěžující symptomatologií snižující jejich performance status a kvalitu života [7]. Možnosti neintervenční a intervenční léčby jsou uvedeny v tab. 4 [21].
Závěr
Polycystické onemocnění jater a ledvin je vrozené s postupnou progresí velikostí cyst. Obvykle probíhá nenápadně a bez závažných komplikací. Při velkém objemu cyst, bolestivém tlaku či dyspepsii lze provést punkci s odsátím cysty pod ultrasonografickou či CT kontrolou. Infekční komplikace jaterních cyst jsou velmi raritní. Pro ATB léčbu pacientů s infekcí cysty jater neexistují evidence-based doporučení. Nizozemští autoři doporučují chinolony jako terapii první volby a při selhání ATB léčby perkutánní drenáž [8].
Pacienti s dědičnou PLD a ledvin jsou limitováni stavem renálních funkcí a jsou indikováni k transplantaci obvykle z důvodu renálního selhání, nikoli pro selhání jaterních funkcí. K transplantaci z hepatologické indikace může dojít při útlakovém syndromu nebo prokázané infekci cysty. Chirurgické řešení – laparoskopické fenestrace, kombinované neanatomické jaterní resekce doplněné fenestrací, které se provádí v hepatobiliárních chirurgických centrech, je možnou alternativou u symptomatických nemocných. Nechirurgickou možností by mohla být intervenční radiologie nebo subkutánní aspirace a sklerotizace, medikamentózní ovlivnění progrese cyst probíhá zatím jen na úrovni klinických studií.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Doručeno: 5. 1. 2018
Přijato: 8. 2. 2018
MUDr. Gabriela Růžičková
Interní oddělení – Endoskopie
Orlickoústecká nemocnice
Nemocnice Pardubického kraje, a. s.
Čs. armády 1076, 562 18 Ústí nad Orlicí
Sources
1. Cnossen WR, Drenth JP. Polycystic liver disease: an overview of pathogenesis, clinical manifestations and management. Orphanet J Rare Dis 2014; 9 : 69. doi: 10.1186/1750-1172-9-69.
2. Lin HP, Ho WC, Lee CC et al. Infected simple hepatic cysts – 3 cases report. J Intern Med Taiwan 2009; 20 (4): 373–377.
3. Abu-Wasel B, Walsh C, Keough V et al. Pathophysiology, epidemiology, classification and treatment options for polycystic liver diseases. World J Gastroenterol 2013; 19 (35): 5775–5786. doi: 10.3748/wjg.v19.i35.5775.
4. Krechler T, Kaláb M, Brodanová M et al. Diagnosis and treatment of hepatic cysts. Vnitr Lek 2000; 46 (7): 395–397.
5. D‘Agnolo HM, Kievit W, van Munster KN et al. Center is an important indicator for choice of invasive therapy in polycystic liver disease. Transpl Int 2017; 30 (1): 76–82. doi: 10.1111/tri. 12875.
6. Redston MS, Wanless IR. The hepatic von Meyenburg complex: prevalence and association with hepatic and renal cysts among 2843 autopsies [corrected]. Mod Pathol 1996; 9 (3): 233–237.
7. Russell RT, Pinson CW. Surgical management of polycystic liver disease. World J Gastroenterol 2007; 13 (38): 5052–5059.
8. Lantinga MA, Geudens A, Gevers TJ et al. Systematic review: the management of hepatic cyst infection. Aliment Pharmacol Ther 2015; 41 (3): 253–261. doi: 10.1111/apt.13047.
9. Husa P, Freibergerová M, Svačinka R et al. Liver abscesses – one of possible causes of fever of unknown origin. Klin Mikrobiol Infekc Lek 2009; 15 (2): 58–64.
10. Lantinga MA, Drenth JP, Gevers TJ. Diagnostic criteria in renal and hepatic cyst infection. Nephrol Dial Transplant 2015; 30 (5): 744–751. doi: 10.1093/ndt/gfu227.
11. Kanaan N, Goffin E, Pirson Y et al. Carbohydrate antigen 19-9 as a diagnostic marker for hepatic cyst infection in autosomal dominant polycystic kidney disease. Am J Kidney Dis 2010; 55 (5): 916–922. doi: 10.1053/j.ajkd.2009.12. 023.
12. Holánek M, Pokrivčák T, Ševela K et al. Infekce jaterní cysty u pacienta s autozomálně dominantní polycystickou chorobou ledvin (ADPKD). Interní Med 2014; 16 (3): 116–118.
13. Grams J, Teh SH, Torres VE et al. Inferior vena cava stenting: a safe and effective treatment for intractable ascites in patients with polycystic liver disease. J Gastrointest Surg 2007; 11 (8): 985–990. doi: 10.1007/s11605-007-0182-3.
14. Gevers TJ, de Koning DB, van Dijk AP et al. Low prevalence of cardiac valve abnormalities in patients with autosomal dominant polycystic liver disease. Liver Int 2012; 32 (4): 690–692. doi: 10.1111/j.1478-3231.2011.02683.x.
15. Schievink WI, Spetzler RF. Screening for intracranial aneurysms in patients with isolated polycystic liver disease. J Neurosurg 1998; 89 (5): 719–721. doi: 10.3171/jns.1998.89.5. 0719.
16. Gigot JF, Jadoul P, Que F et al. Adult polycystic liver disease: is fenestration the most adequate operation for long-term management? Ann Surg 1997; 225 (3): 286–294.
17. Qian Q, Li A, King BF et al. Clinical profile of autosomal dominant polycystic liver disease. Hepatology 2003; 37 (1): 164–171. doi: 10.1053/jhep.2003.50006.
18. Schnelldorfer T, Torres VE, Zakaria S et al. Polycystic liver disease: a critical appraisal of hepatic resection, cyst fenestration, and liver transplantation. Ann Surg 2009; 250 (1): 112–118. doi: 10.1097/SLA.0b013e3181ad 83dc.
19. Lantinga MA, D‘Agnolo HM, Casteleijn NF et al. Hepatic cyst infection during use of the somatostatin analog lanreotide in autosomal dominant polycystic kidney disease: an interim analysis of the randomized open-label multicenter DIPAK-1 study. Drug Saf 2017; 40 (2): 153–167. doi: 10.1007/s40264-016-0486-x.
20. Walz G, Budde K, Mannaa M et al. Everolimus in patients with autosomal dominant polycystic kidney disease. N Engl J Med 2010; 363 (9): 830–840. doi: 10.1056/NEJMoa1003 491.
21. Wong MY, McCaughan GW, Strasser SI. An update on the pathophysiology and management of polycystic liver disease. Expert Rev Gastroenterol Hepatol 2017; 11 (6): 569–581. doi: 10.1080/17474124.2017.1309280.
22. D‘Agnolo HM, Kievit W, Takkenberg RB et al. Ursodeoxycholic acid in advanced polycystic liver disease: a phase 2 multicenter randomized controlled trial. J Hepatol 2016; 65 (3): 601–607. doi: 10.1016/j.jhep.2016.05.009.
23. Takei R, Ubara Y, Hoshino J et al. Percutaneous transcatheter hepatic artery embolization for liver cysts in autosomal dominant polycystic kidney disease. Am J Kidney Dis 2007; 49 (6): 744–752. doi: 10.1053/j.ajkd.2007.03. 018.
24. Drenth JP, Chrispijn M, Nagorney DM et al. Medical and surgical treatment options for polycystic liver disease. Hepatology 2010; 52 (6): 2223–2230. doi: 10.1002/hep.24036.
Labels
Paediatric gastroenterology Gastroenterology and hepatology SurgeryArticle was published in
Gastroenterology and Hepatology

2018 Issue 2
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Spasmolytic Effect of Metamizole
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- The Importance of Limosilactobacillus reuteri in Administration to Diabetics with Gingivitis
Most read in this issue
- Terlipresin – stále nepostradatelný ve dvou indikacích
- Infekce jaterní cysty při polycystóze jater jako zdroj sepse
- Neobvyklá příčina zvětšování břicha
- Akutní poškození ledvin u pacientů s akutní pankreatitidou