
Hepatopatie jako první projev systémové AL amyloidózy
Hepatopathy as the first manifestation of systemic AL amyloidosis
Amyloidosis is considered a rare disease characterized by the deposition of insoluble protein – amyloid, into the extracellular space of tissues, which damages their functions. There are about 30 types of amyloidosis, of which the most common is systemic AL amyloidosis, which we include among monoclonal gammopathies. At first, symptoms may be nonspecific, such as fatigue, loss of appetite, and weight loss, or there may be symptoms of damage to specific organs, such as the heart and kidneys and nervous system, but also liver, soft tissue, or GIT. In our case, we document a patient who first presented at our gastroenterology clinic. It is important to consider the possibility of amyloidosis after all other common hepatopathies have been excluded. The diagnosis of this patient was prolonged for several months mainly because the patient hesitated to undergo a liver biopsy, which afterwards clearly confirmed the diagnosis. The patient’s status temporarily improved when symptomatic therapy was implemented. After a targeted examination, damage to other organs was also detected, but the treatment no longer had any influence on the infaust prognosis of the patient. Early diagnosis and treatment is important for amyloidosis, because it can have a significant impact on patient outcome. The problem is that, even with clear indications, this disease is often neglected in diagnostic reports.
Key words:
amyloidosis – hepatomegaly – biopsy
Submitted: 2. 5. 2018
Accepted: 17. 9. 2018
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE „uniform requirements“ for biomedical papers.
Authors:
Fusková J.; Miča T.
Authors‘ workplace:
Gastroenterologická ambulance, Nemocnice ATLAS, a. s., Zlín
Published in:
Gastroent Hepatol 2018; 72(6): 508-511
Category:
Hepatology: Case Report
doi:
https://doi.org/10.14735/amgh2018508
Overview
Amyloidóza je považována za vzácné onemocnění, které je charakterizováno ukládáním nerozpustného proteinu – amyloidu – do extracelulárního prostoru tkání a poškozuje jejich funkci. Existuje cca 30 typů amyloidóz, z nichž nejčastější je systémová AL amyloidóza, kterou řadíme mezi monoklonální gamapatie. Projevy mohou být nejdříve nespecifické, jako je únava, nechutenství či váhový úbytek, nebo plynoucí z postižení jednotlivých orgánů – srdce nebo ledvin, nervového systému, ale i jater, měkkých tkání či gastrointestinálního traktu. Na našem případu dokumentujeme, že pacient s touto diagnózou se může primárně objevit i v ambulanci gastroenterologa, a proto je důležité po vyloučení všech dalších běžných hepatopatií vzít do úvahy i možnost amyloidózy. Naše diagnóza byla o několik měsíců protrahována zejména z toho důvodu, že pacient váhal s provedením jaterní biopsie, která diagnózu jednoznačně potvrdila, jelikož při zavedení symptomatické terapie došlo přechodně ke zlepšení stavu pacienta. Po cíleném vyšetření bylo zjištěno postižení i dalších orgánů, ale zahájená léčba již neměla na infaustní prognózu pacienta žádný vliv. U amyloidózy je důležitá včasná diagnostika a zahájená léčba, která významně ovlivňuje další stav pacienta. Problémem je, že i při jasných příznacích bývá v diagnostické rozvaze toto onemocnění často opomíjeno.
Klíčová slova:
amyloidóza – hepatomegalie – biopsie
Úvod
Amyloidóza je vzácné onemocnění charakterizované ukládáním nerozpustného fibrilárního proteinu (amyloidu) do extracelulární matrix různých tkání a orgánů. Termín amyloid poprvé použil Virchow v roce 1854 pro patologický extracelulární materiál v játrech [1]. Současný klasifikační systém zahrnuje více než 30 typů amyloidóz.
Amyloidóza se vyskytuje i ve formě lokalizované, pouze s ložiskovou depozicí amyloidu na jeden orgán, ale mnohem častější je forma systémová. Nejčastější je systémová AL amyloidóza (ALA) (70 %), méně častá je její ložisková forma (19 %). Dále se vyskytují typy např. senilní (4 %) a hereditární (4 %) a AA amyloidóza (3 %), dříve označovaná jako sekundární nebo reaktivní, vznikající na podkladě jiných chronických zánětlivých onemocnění, v gastroenterologii např. u Crohnovy choroby [2,3]. Při systémové ALA dochází k produkci volných lehkých řetězců imunoglobulinů (FLC – free light chains) abnormálními klony plazmatických buněk v kostní dřeni. Tyto rozpustné řetězce kolují krevním oběhem, následně vycestovávají do orgánů a tkání, ve kterých tvoří amyloidová depozita odolná vůči proteolýze díky své terciární struktuře ve formě beta skládaného listu. Častěji se vyskytuje produkce lehkých řetězců typu lambda než kappa, a to v poměru 3 : 1. V řadě případů je systémová ALA asociována s hematologickými nádorovými onemocněními, nejčastěji mnohočetným myelomem (10– 20 %).
Příznaky onemocnění mohou být jednak nespecifické – únava, slabost, malátnost, nechutenství, váhový úbytek, a jednak vyplývající z postižení jednotlivých orgánů. Nejčastěji postiženým orgánem bývá srdce (60 %) a ledviny (70 %) [2,4]. Naše kazuistika popisuje případ muže, u něhož klinicky dominovalo postižení jaterní, jehož obvyklými symptomy jsou zvětšení a bolesti břicha, hepatomegalie a portální hypertenze spojená s krvácením do horní části gastrointestinálního traktu (GIT). K diagnostice ALA je nezbytné provedení tkáňové biopsie postiženého orgánu s průkazem amyloidových depozit, v tomto případě jater [2,5,6].
Kazuistika
Muž, 66 let, odeslán praktickým lékařem v červenci 2016 do naší ambulance na gastroskopii pro nauzeu, váhový úbytek a nechutenství. V anamnéze dále udávána alkoholická nemoc jater, cíleným dotazem pacient přiznává konzumaci asi dvou piv denně a blíže nespecifikované množství destilátů. Gastroskopicky zjištěna refluxní ezofagitida I. stupně dle Savary-Miller, dále aftózní antrumgastropatie a na přechodu antra a těla žaludku patrna na velké kurvatuře jizva cca 40 mm. Z té byl odebrán vzorek na histologii, kde byla popsána chronická inaktivní gastritida středního stupně, barvení na Helicobacter pylori negativní. Vzhledem k hepatopatii udávané v přiložené dokumentaci doplněna sérologie na našem pracovišti, kde jsme zjistili elevace: alaninaminotransferáza (ALT) 1,43 µkat/ l, aspartátaminotransferáza (AST) 1,92 µkat/ l, gama-glutamyltransferáza (GGT) 19,75 µkat/ l, alkalická fosfatáza (ALP – alkaline phosphatase) 8,17 µkat/ l; jinak ostatní laboratoř bez pozoruhodností (urea, kreatinin, bilirubin, sérová amyláza, albumin, INR, krevní obraz s diferenciálem). Do léčby jsme zavedli pantoprazol v dávce 2 × 40 mg/den, dále byl pacient poučen o nevhodnosti pití alkoholických nápojů. Pacienta jsme pozvali ke kontrole v září 2016, stěžuje si na progredující únavu, nechutenství a tlaky v pravém podžebří, pocity plnosti, jakýkoli abúzus od předchozí návštěvy popírá. Pohmatově játra přesahují jaterní oblouk o celou dlaň v medioklavikulární čáře, tuhé až kamenné konzistence. V laboratoři další elevace GGT (28,14 µkat/ l) a ALP (15,22 µkat/ l), tumor markery AFP, CEA v normě, CA 19-9 64,2 U/ ml. Doplněna sérologie infekčních a autoimunitních hepatitid, která byla negativní, ceruloplazmin, hladina sérového železa, feritinu a transferinu v normě. Ve stejné době provedena kontrolní gastroskopie, kde je již viditelná jen neostrá z-linie, jinak makroskopicky normální nález až do D2. Na sono břicha popsána hepatomegalie a cirhotická přestavba jater, bez známek obstrukce žlučových cest, jinak normální nález. CT břicha s nálezem hepatomegalie, nehomogenní mapovité sycení jaterního parenchymu, bez jednoznačných ložiskových změn, stav po uzávěru v. lienalis, nahrazena kolaterálním oběhem, sycení sleziny snížené s vícečetnými drobnými hyperdenzními okrsky, známky portální hypertenze, zmnožené lymfatické uzliny v ligamentum hepatoduodenale, menší ascites perihepaticky. Vzhledem k výše uvedeným okolnostem se již nezdá pravděpodobná pouze alkoholická etiologie zvýšení aktivity jaterních enzymů, která byla udávána v dosavadní dokumentaci. S pacientem byla probrána nutnost provedení necílené jaterní biopsie, kterou ovšem i přes intervenci rodiny odkládá. Do léčby je empiricky nasazena kyselina ursodeoxycholová v dávce 1 000 mg/ den, spironolakton 50 mg/ den a ponechána terapie inhibitory protonové pumpy. V listopadu 2016, tj. po 2 měsících, je pacient subjektivně bez větších obtíží, přetrvává jen nechutenství, objektivně ovšem dochází k dalšímu zvětšení jater, která vyplňují prakticky celé epigastrium a v pravé přední axilární čáře dosahují až k lopatě kosti kyčelní. Laboratorně nově hypoalbuminemie 27,2 g/ l, hyponatremie 134 mmol/ l, naopak pokles aktivity jaterních enzymů (ALT 0,81 µkat/ l, AST 1,82 µkat/ l, GGT 6,09 µkat/ l, ALP 7,73 µkat/ l), v krevním obraze posun v červené řadě doprava (ery 5,97 tera/ l, hematokrit 0,54, hemoglobin 179 g/ l). Pacient další došetření v této fázi opět odmítá. V lednu 2017 byl přivezen rodinou pro zhoršení stavu, progresi únavy, další váhový úbytek, nechutenství, nárůst objemu břicha, dušnost a brnění dolní poloviny těla. V laboratoři již přítomna azotemie (urea 9,1 mmol/ l, kreatinin 140 umol/ l), albumin 20,7 g/ l. Konzultován hematolog, byla jednoznačně doporučena biopsie jater k objasnění etiologie hepatopatie s hepatomegalií a indikována pozitronová emisní tomografie PET/ CT, kde byl výsledek bez průkazu patologické ložiskové depozice fluordeoxyglukózy v rozsahu celotělového vyšetření. Pacient je nakonec k biopsii jater v únoru 2017 přesvědčen. Histopatolog popisuje obraz amyloidózy s významnou redukcí jaterního parenchymu (obr. 1), v rámci diferenciální diagnózy se jedná o primární systémovou ALA, imunohistochemicky pozitivní prealbumin, AA-amyloid negativní, AL-amyloid lambda řetězce pozitivní, kappa negativní. Tento výsledek byl potvrzen i druhým čtením. Pacient byl odeslán k zahájení léčby na hematoonkologii FN Brno, doplněna další vyšetření. Sternální punkce potvrzuje jako primární diagnózu mnohočetný myelom. Echokardiografie s nedilatovanou levou komorou, těžkou koncentrickou hypertrofií stěn při postižení amyloidem, bez lokální poruchy kinetiky s ejekční frakcí 55 %, porucha relaxace, postižena i stěna pravé komory, která je hypertrofická se zachovalou systolickou funkcí. Zahájena cílená terapie cyklofosfamidem (aplikovány dvě dávky), opakovaně provedena hemodialýza. Přes zavedenou léčbu ale dochází k rychlé progresi stavu a pacient umírá v březnu 2017 v důsledku srdečního selhání.
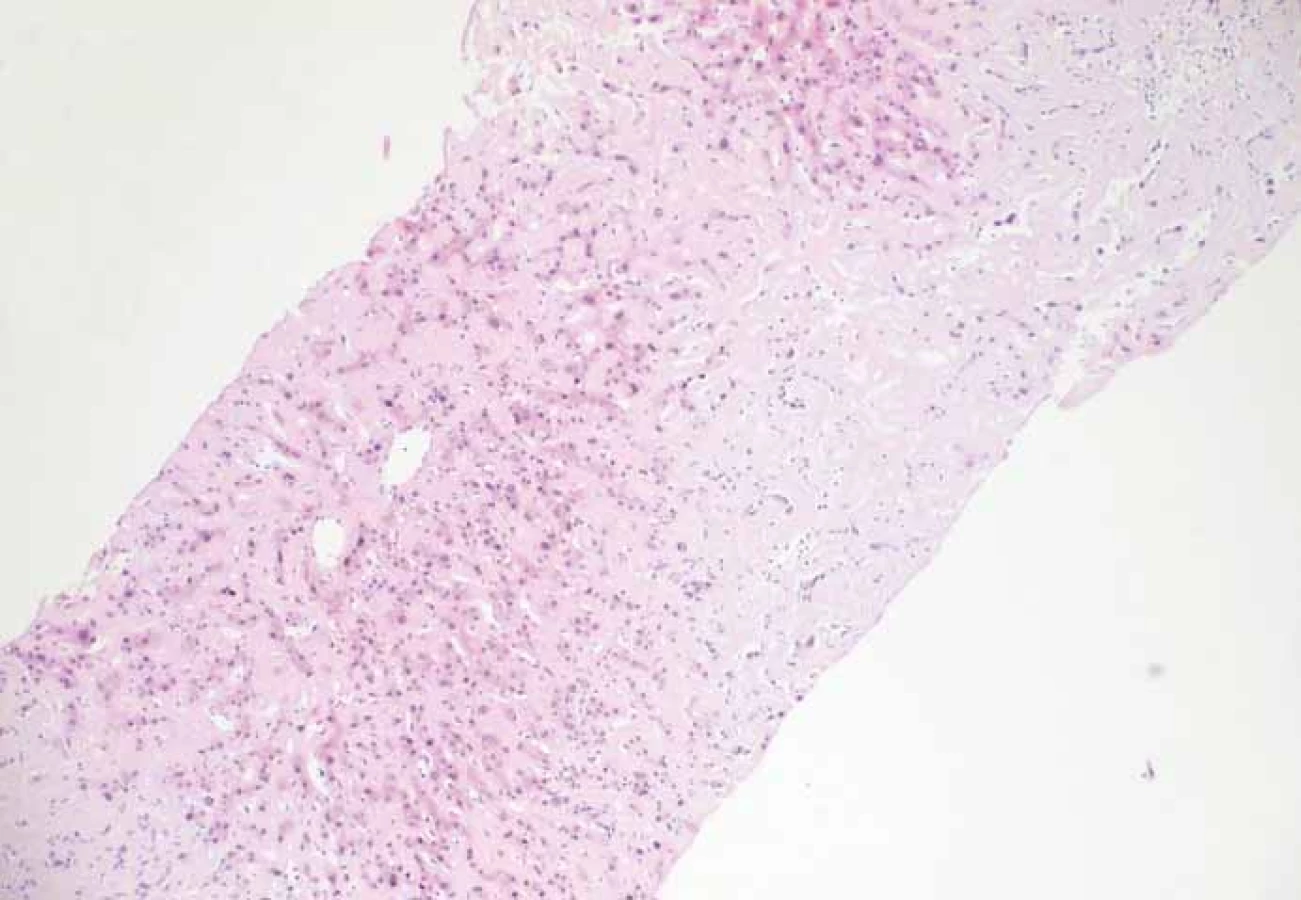
Diskuze
Amyloidóza je relativně vzácné onemocnění, v Evropě s incidencí 8– 10 případů/ 1 milion obyvatel/ rok, v USA 5– 12 případů/ 1 milion obyvatel/ rok [7,8]. Postihuje především jedince vyššího věku, obvykle mezi 50– 70 lety, častěji muže [2].
ALA může postihnout kterýkoli orgán, klinická symptomatologie bývá proto velmi různorodá. Nejčastěji postiženými orgány jsou ledviny, srdce, nervový systém, játra, GIT ale postihuje i měkké tkáně. V době stanovení diagnózy je u 70 % osob prokazatelné postižení nejméně dvou orgánů [1].
Postižení jater se vyskytuje cca u 20 % nemocných s přítomností únavy, váhového úbytku, anorexie, zvětšování břicha, hepatomegalie, splenomegalie, portální hypertenze s ascitem. Výjimečným a nepříznivým projevem je cholestatická žloutenka. V laboratoři se běžně vyskytuje elevace GGT a ALP, hypoalbuminemie, popisována je i hyperlipidemie. Velikost jater ani jejich funkce ovšem nemusí korelovat se závažností zasažení orgánu amyloidem. Mikroskopicky se nalézají depozita amyloidu periportálně, perivaskulárně i peribiliárně a v intrahepatálních žlučovodech s konečnou významnou redukcí funkčních hepatocytů [2,5,6]. Postižení GIT je provázeno nespecifickými příznaky jako nauzea, zvracení, refluxní obtíže, bolesti břicha, postprandiální průjem až malabsorpční syndrom, zácpa, makroglosie, ale infiltrace GIT amyloidem může být i zdrojem život ohrožujícího krvácení, jehož příčinou jsou jícnové varixy, amyloidové eroze či vředy nebo střevní obstrukce [2,5,9].
Pro amyloidózu je typické postižení ledvin projevující se zpravidla výraznou neselektivní proteinurií charakteru nefrotického syndromu (NS). V důsledku těžké hypoalbuminemie se objevují otoky až anasarka, pleurální, perikardiální výpotky a ascites. Příčinou NS jsou depozita amyloidu uložená subendoteliálně v cévách a mezangiu glomerulů [10]. Procentuálně druhým nejčastějším projevem je postižení srdce s nálezem restriktivní hypertrofické kardiomyopatie při echokardiografii, časté jsou i arytmie. Pro odlišení od hypertrofické kardiomyopatie hypertenzní etiologie je možné doplnit magnetickou rezonanci. Nejprve se projevují poruchy diastolické funkce a později i systolické vedoucí k městnavému srdečnímu selhání. Srdeční amyloidóza má nejhorší prognózu s mediánem přežití u neléčeného pacienta okolo 6 měsíců od počátku příznaků srdečního selhání [11,12]. Postižení nervového systému se projevuje periferní a autonomní neuropatií, které mohou diagnóze amyloidózy předcházet i řadu let. Při periferní neuropatii dominuje postižení dolních končetin charakteru axonální, distální, smíšené a symetrické polyneuropatie s dysesteziemi, parestezií až syndromem neklidných nohou. Senzitivní neuropatie bývá symetrická a někdy značně bolestivá, motorická neuropatie je vzácná. Autonomní neuropatie způsobuje poruchy motility GIT, dysgeuzii, podílí se na rozvoji posturální hypotenze, erektilní dysfunkce a anhidrózy [2]. Z ostatních příznaků jsou to např. kožní purpura, kašel, dušnost, hemoptýza při postižení plic nebo chrapot [2]. U našeho pacienta dominovalo klinicky postižení jaterní, až mnohem později, s odstupem několika měsíců, se manifestovalo postižení ledvin a neuropatie.
Nezbytné je tedy provedení tkáňové biopsie s průkazem amyloidových depozit a jejich typizace. Léčba se u různých typů amyloidů totiž od sebe zásadně liší. Depozita amyloidu lze rozpoznat již v základním barvení hematoxylin eozinem, ke specifickému znázornění amyloidu se používá barvení konžskou červení (obr. 2). V polarizačním mikroskopu pak takto nabarvené vzorky vykazují tzv. dvojlom a dichroizmus [2]. Odběr vzorků může být proveden z necílené biopsie trávicího traktu nejčastěji z oblasti rekta, ale i jazyka a malých slinných žláz, alternativou je odběr podkožního tuku v okolí pupku. Pokud je výsledek negativní, pak se provádí cílená biopsie z postiženého orgánu (ledviny, srdce, nerv, játra). V kostní dřeni se prokazuje přítomnost monoklonální plazmocelulární populace [13]. Stanovení definitivní diagnózy bývá často obtížné a může trvat i několik let. V našem případě trvala diagnóza několik měsíců, jelikož pacient s provedením jaterní biopsie dlouhodobě váhal. V odebrané histologii byl průkaz ALA jednoznačný, vč. stanovení subtypizace, nicméně při odběru vzorku sliznice žaludku cca 6 měsíců před jaterní biopsií depozita amyloidu zachycena nebyla.
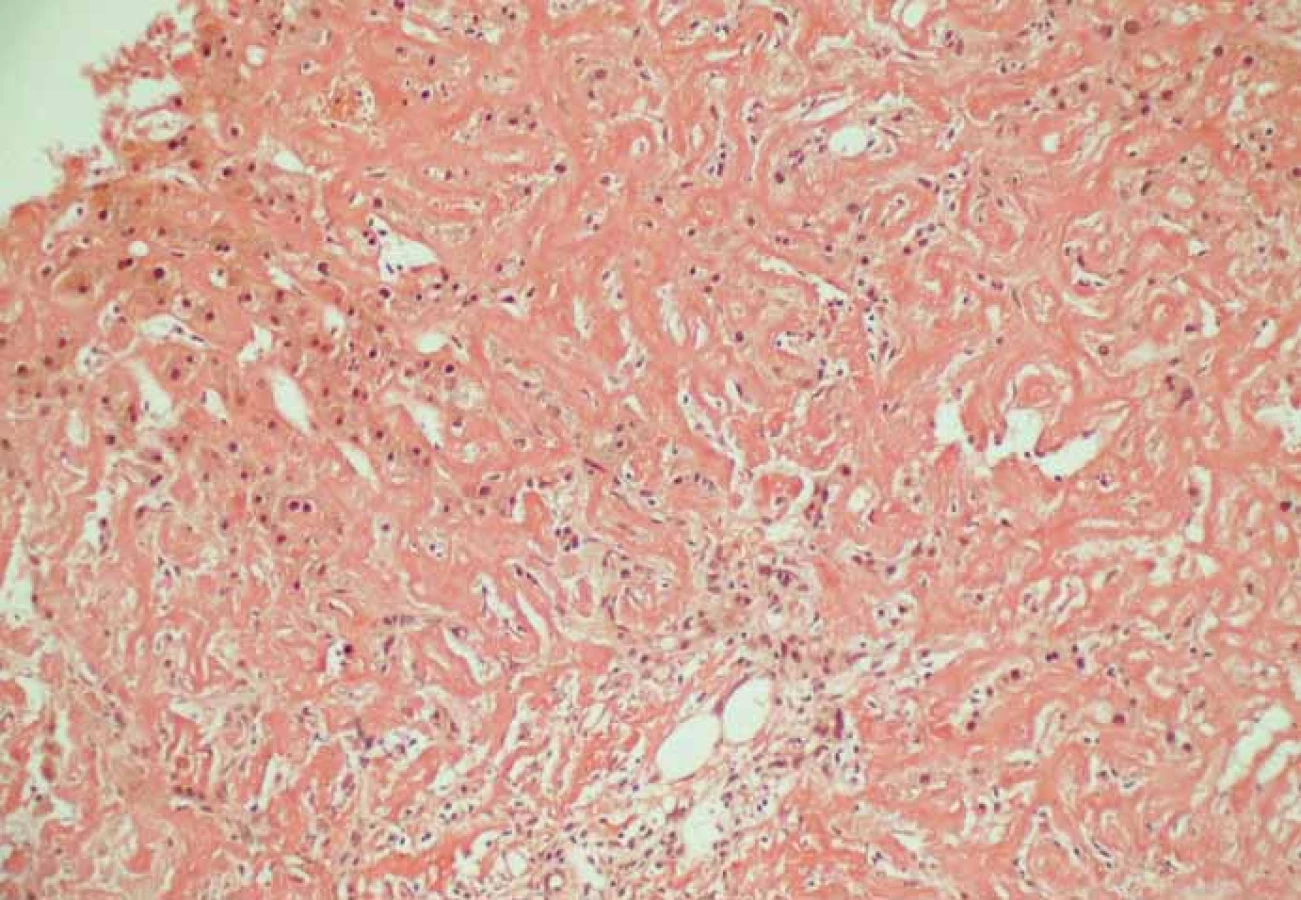
Specifická léčba ALA je obdobná jako u mnohočetného myelomu, zahrnuje dvě varianty, a to kombinaci alkylačních cytostatik s kortikoidy (nejrozšířenější režim melfalan + dexamethazon) nebo vysokodávkovou chemoterapii s následnou autologní transplantací periferních kmenových buněk. Cílem je suprese klonu plazmatických buněk v kostní dřeni a tím snížení produkce FLC. Kromě výše uvedené terapie se relativně nově uplatňují i léky označované jako imunomodulátory, které cíleně ovlivňují funkce plazmatických buněk jako např. thalidomid, lenalidomid, bertezomib. Symptomatická léčba se odvíjí od postižení jednotlivých orgánů. Za hematologickou remisi onemocnění je považována normalizace koncentrace FLC v séru. Jedním z nejdůležitějších faktorů účinnosti léčby je včasné stanovení diagnózy a rozsah orgánového postižení [10,13]. Průměrný medián přežití u pacientů s ALA je 36 měsíců. Významným prognostickým faktorem je stanovení sérové hladiny srdečních biomarkerů (Tnt, TnI, NT-pro BNP), neboť závažnost postižení srdce i u asymptomatických jedinců je významným limitujícím faktorem [2].
Závěr
V kazuistice je popsán případ systémové ALA s postižením mnohočetných orgánů. Sdělením chceme upozornit na toto vzácné onemocnění, jehož diagnostika spadá dominantně do rukou nefrologa nebo kardiologa, a vzácně se může primárně objevit i v ambulanci gastroenterologa [4,9,14]. Proto je vhodné u pacienta s hepatomegalií a elevací ALP po vyloučení všech běžných jaterních chorob, jako jsou alkoholická, virová, autoimunitní onemocnění jater, Wilsonova choroba, lékové postižení a hepatocelulární karcinom, do diagnostické rozvahy vzít i postižení amyloidem. Včasné stanovení správné diagnózy, rozsahu orgánového postižení a strategie léčby může pacientovi zásadně prodloužit život.
Doručeno: 2. 5. 2018
Přijato: 17. 9. 2018
MU Dr. Jana Fusková
Gastroenterologická ambulance, Nemocnice ATLAS, a.s.
třída Tomáše Bati 5135
760 01 Zlín
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Sources
1. Adam Z, Krejčí M, Vorlíček J et al. Hematologie, přehled maligních hematologický nemocí. 2. vyd. Praha: Grada Publishing 2008 : 250– 255.
2. Ščudla V, Pika T, Látalová P et al. Diagnostika a stratifikace systémové AL amyloidózy ve světle „Doporučení České myelomové skupiny 2013“. Klin Biochem Metab 2014; 22(43): No. 2, 49– 60.
3. Kroupa R, Dastych M, Šenkyřík M et al. Systémová AL-amyloidóza s dominující klinickou manifestací v trávicím traktu. Vnitř Lék 2005; 51(5): 588– 592.
4. Zahradová L. Systémová amyloidóza, Onkológia (Bratislava) 2016; 11(6): 361– 364.
5. Petre S, Shah IA, Gilani N. Review article: gastrointestinal amyloidosis – clinical features, diagnosis and therapy. Aliment Pharmacol Ther 2008; 27(11): 1006– 1016. doi: 10.1111/ j.1365-2036.2008.03682.x.
6. Wang YD, Zhao CY, Yin HZ. Primary hepatic amyloidosis: a mini literature review and five cases report. Ann Hepatol 2012; 11(5): 721– 727.
7. Kyle RA, Linos A, Beard CM et al. Incidence and natural history of primary systemic amyloidosis in Olmsted county, Minnesota, 1950 through 1989. Blood 1992; 79(7): 1817– 1822.
8. Comenzo RL, Reece D, Palladini G et al. Consensus guidelines for the conduct and reporting of clinical trials in systemic light-chain amyloidosis. Leukemia 2012; 26(11): 2317– 2325. doi: 10.1038/ leu.2012.100.
9. Husová L, Hušek K, Čáslava T et al. Primární amyloidoza jako neobvyklá příčina krvácení z trávicího ústrojí. Čes a Slov gastroent 2000; 54(3): 112– 116.
10. Ryšavá R. Systémové amyloidózy a jejich léčba. Praha: Maxdorf Jessenius 2013 : 48– 95.
11. Kováčik F, Táborský M, Hutyra M. Srdeční amyloidóza – kazuistika. Interv Akut Kardiol 2016; 15(2): 145– 147.
12. Hassan W, Al-Segrani H, Mourad W et al. Amyloid heart disease. New frontiers and insights in pathophysiology, diagnosis, and management. Tex Heart Inst J 2005; 32(2): 178– 184.
13. Merlini G, Seldin DC, Gertz MA. Amyloidosis: pathogenetis and new therapeutic options. J Clin Oncol 2011; 29(14): 1924– 1933. doi: 10.1200/ JCO.2010.32.2271.
14. Kohoutová D, Tichý M, Maisnar V et al. Systémová AL-amyloidóza – kazuistika. Klin Biochem Metab 2007; 15(36): No. 2, 91– 94.
Labels
Paediatric gastroenterology Gastroenterology and hepatology SurgeryArticle was published in
Gastroenterology and Hepatology

2018 Issue 6
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
- The Importance of Limosilactobacillus reuteri in Administration to Diabetics with Gingivitis
Most read in this issue
- Meckelův divertikl jako příčina náhlé příhody břišní
- Hepatopatie jako první projev systémové AL amyloidózy
- Wilsonova choroba v dětském věku – dvě kazuistiky
- Endoskopická léčba rektálního syndromu při lipomu rekta technikou „loop and let go“