
Varianty lidských chromozomů a jejich význam z pohledu klinické genetiky
Variants of human chromosomes and their significance from the point of view of clinical genetics
The first association of a chromosomal anomaly (trisomy of chromosome 21) with a pathological phenotype (Down syndrome) was discovered more than 50 years ago. Since that time clinical genetics itself has changed a great deal. Today we know of many numerical and structural abnormalities of the human karyotype, which are causal for different clinical syndromes. However – not each of the chromosomal abnormalities is believed to be pathological. Today we know a number of so called heteromorphisms – variants of the human karyotype that do not affect the phenotype of their carriers. Nearly all of these variants are inherited from one of the parents. The chromosomes with the areas of constitutive heterochromatin (1, 9, 16 and Y) and acrocentric chromosomes (13, 14, 15, 21 and 22) are commonly involved. Although these findings are neither uncommon nor clinically significant, the interpretation of the findings by cytogeneticists and clinical geneticists can be sometimes difficult. Also – whenever there is a complete cytogenetic formula – including the variant – in the clinical report provided to the patient, it is necessary to inform the patient very clearly about the benign character of this finding in order to prevent unnecessary anxiousness.
Keywords:
karyotype, chromosomes, heteromorphism, heterochromatine.
Authors:
A. Šípek jr. 1; R. Mihalová 1; A. Panczak 1; L. Celbová 1; A. Šípek 2,3,4; I. Hazdrová 2; V. Gregor 2,3,5
Authors‘ workplace:
Ústav biologie a lékařské genetiky 1. LF UK a VFN, Praha, Přednostka: doc. MUDr. Milada Kohoutová, CSc.
1; Oddělení lékařské genetiky, Thomayerova nemocnice, Praha, Primář: MUDr. Vladimír Gregor
2; Screeningové centrum Praha – Sanatorium PRONATAL, Odborný vedoucí: doc. MUDr. Tonko Mardešič, CSc.
3; Ústav obecné biologie a genetiky 3. LF UK, Praha, Přednostka: doc. MUDr. Marie Černá, CSc.
4; Katedra lékařské genetiky, Institut postgraduálního vzdělávání ve zdravotnictví, Praha, Vedoucí: prof. MUDr. Petr Goetz, CSc.
5
Published in:
Prakt. Lék. 2012; 92(4): 205-209
Category:
Of different specialties
Overview
První souvislost chromozomové anomálie (trizomie chromozomu 21) a patologického fenotypu (Downova syndromu) byla objevena již před více než 50 lety. Od té doby prodělala klinická genetika významný rozvoj. Do dnešního dne byla popsána celá řada numerických i strukturních abnormalit lidského karyotypu, které jsou přímo odpovědné za různé klinické syndromy. Ne každá odchylka ve stavbě lidských chromozomů se však ukázala být patologickou. Dnes již známe celou řadu tzv. heteromorfismů – variant lidského karyotypu, které nikterak neovlivňují fenotyp svého nositele. Prakticky vždy jsou tyto varianty zděděné od jednoho z rodičů. Nejčastěji různé varianty zasahují chromozomy s oblastmi konstitutivního heterochromatinu (chromozomy 1, 9, 16 a Y) a akrocentrické chromozomy (13, 14, 15, 21 a 22). Přestože nejde o klinicky významné ani vzácné nálezy, může být vyhodnocení cytogenetikem i interpretace nálezu klinikem někdy zatíženo určitou nejistotou. Pokud je ve zprávě určené pro pacienta uveden kompletní cytogenetický nález i s variantou, je důležité pečlivé vysvětlení benigní povahy tohoto nálezu, aby se předešlo zbytečnému stresování pacienta.
Klíčová slova:
karyotyp, chromozomy, heteromorfismus, heterochromatin.
Úvod
V roce 1956 uveřejnili Tjio a Levan svou přelomovou práci, ve které jako první správně určili počet chromozomů v lidském karyotypu – tedy 23 párů neboli 46 chromozomů celkem (14). Další významné objevy na poli lidské cytogenetiky učinil francouzský genetik a pediatr Jerome Lejeune, který objasnil chromozomovou etiologii Downova syndromu (trizomie chromozomu 21) a syndromu Cri-du-chat (delece krátkého ramene chromozomu 5) (9, 10). S rozvojem metod klinické cytogenetiky, zejména různých barvících a pruhovacích metod, byly postupně objevovány další abnormality chromozomů, které byli cytogenetici schopni odhalit, čímž výrazným způsobem zlepšili diagnostické možnosti klinické genetiky jako takové. Ne každá odchylka od normální stavby chromozomů však byla spojena s patologickým fenotypem. Postupem času byla identifikována celá řada chromozomových odchylek, které se vyskytují i u zcela zdravých jedinců, aniž by je nosičství této varianty nějakým způsobem limitovalo. Takovéto odchylky se již od sedmdesátých let 20. století označují jako chromozomové varianty či také heteromorfismy (15). I dnes se s těmito odchylkami setkáváme v klinické cytogenetické praxi zcela běžně (4). V poslední době je navíc problematika chromozomových variant rozšířena o fenotypově nevýznamné varianty, které byly odhaleny díky častějšímu využití molekulárně-cytogenetických metod s vysokým rozlišením (např. array-CGH) (8).
Varianty heterochromatinových oblastí
Heterochromatin představuje transkripčně neaktivní oblast chromatinu. Klasická cytogenetika rozlišuje fakultativní heterochromatin, což jsou oblasti specificky inaktivované v konkrétním typu buněk (s ohledem na rozdílnou genovou expresi) a heterochromatin konstitutivní, který zahrnuje oblasti konstantě inaktivované ve všech buňkách organismu. V lidském karyotypu existují chromozomy, pro které je rozsáhlejší oblast konstitutivního heterochromatinu typická – jedná se o dlouhá ramena chromozomů 1, 9, 16 a Y (6).
Tyto oblasti jsou tvořeny především různými typy opakujících se – repetitivních DNA sekvencí. Typické jsou sekvence tzv. satelitní DNA – například β-satelitní DNA, α-satelitní DNA či III-satelitní DNA, které se v těchto oblastech s určitou variabilitou vyskytují (3, 16). Absence kódující DNA v těchto oblastech vedla k dodnes zastávanému názoru, že různé varianty uspořádání těchto oblastí nemají žádné klinické dopady (4, 6) (obr. 1). Repetitivní sekvence v těchto oblastech jsou navíc i příčinou vysoké variability těchto oblastí (2, 12).
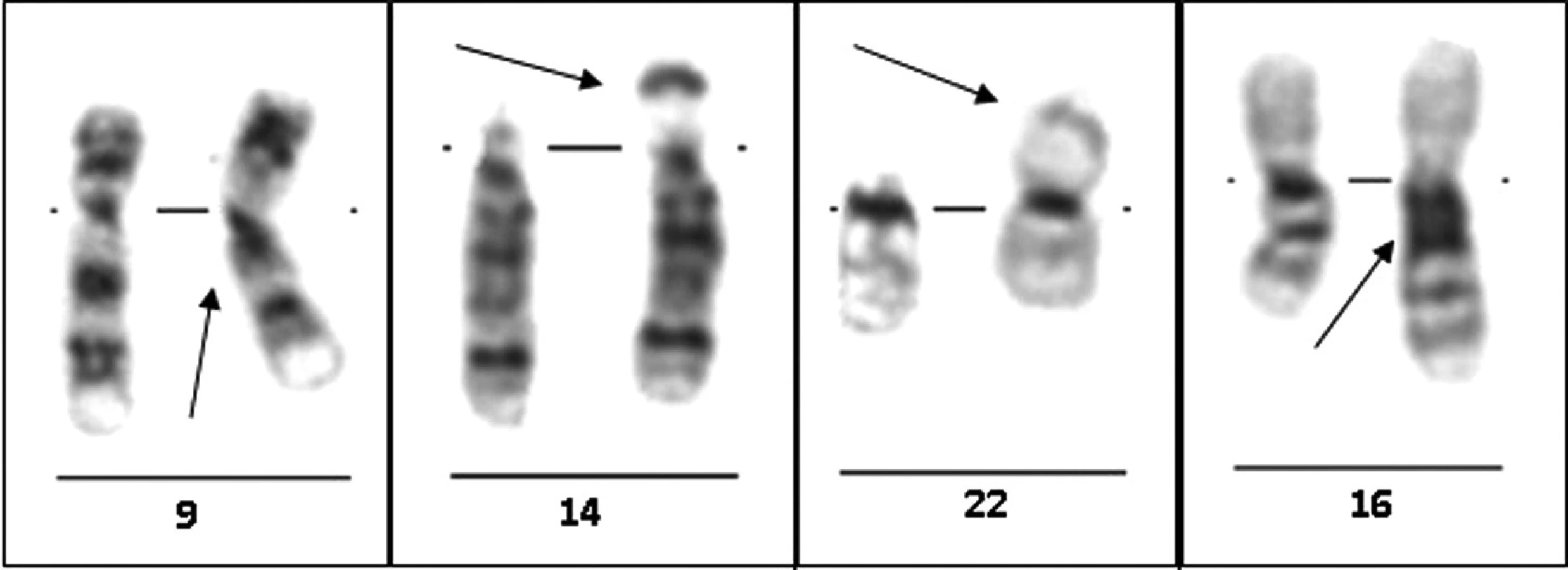
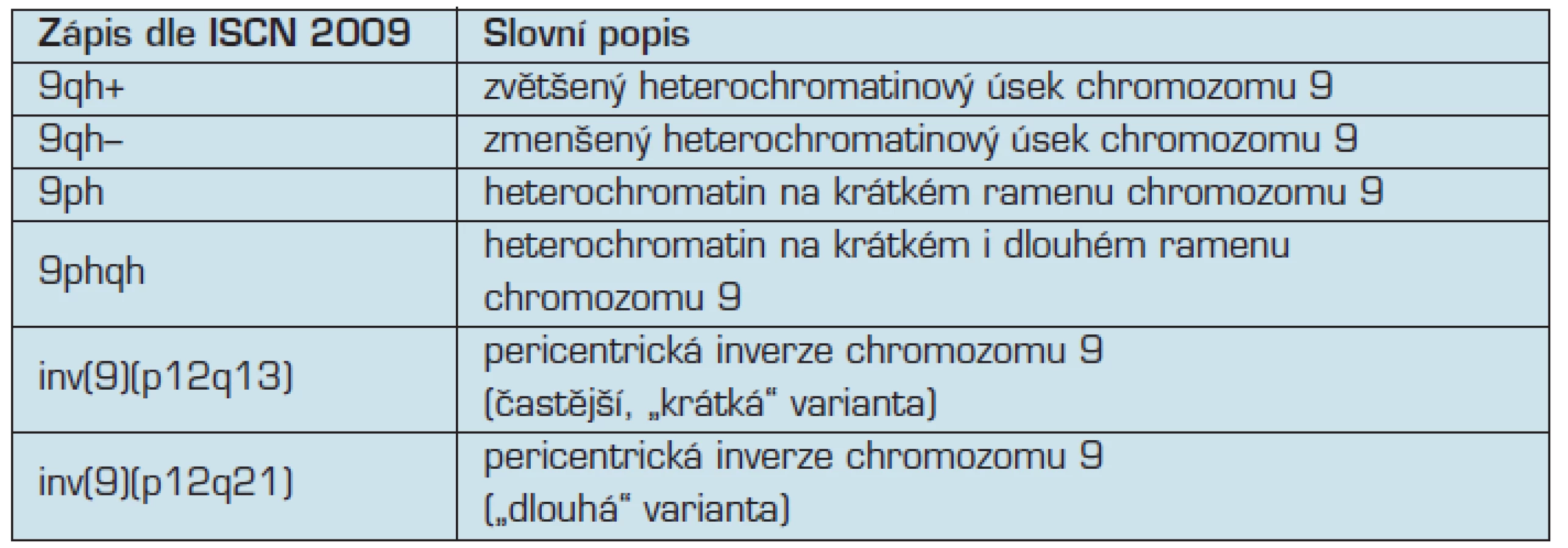
Variabilní uspořádání těchto oblastí se nejčastěji projevuje jako tzv. délkové varianty. Mezinárodní cytogenetická nomenklatura pro tyto varianty používá označení qh+ (zvětšený heterochromatinový úsek) či qh– (zmenšený heterochromatinový úsek) (11). Nejvíce variabilní je heterochromatinová oblast na chromozomu 9 (12), kde se vyskytují i další typy variant, například 9ph varianta (přesun heterochromatinu na krátké rameno chromozomu 9), 9phqh varianta (heterochromatin zasahující na krátké i dlouhé rameno) a nakonec pericentrické inverze heterochromatinu chromozomu 9 (tab. 1) (6, 11).
Varianty krátkých ramen akrocentrických chromozomů
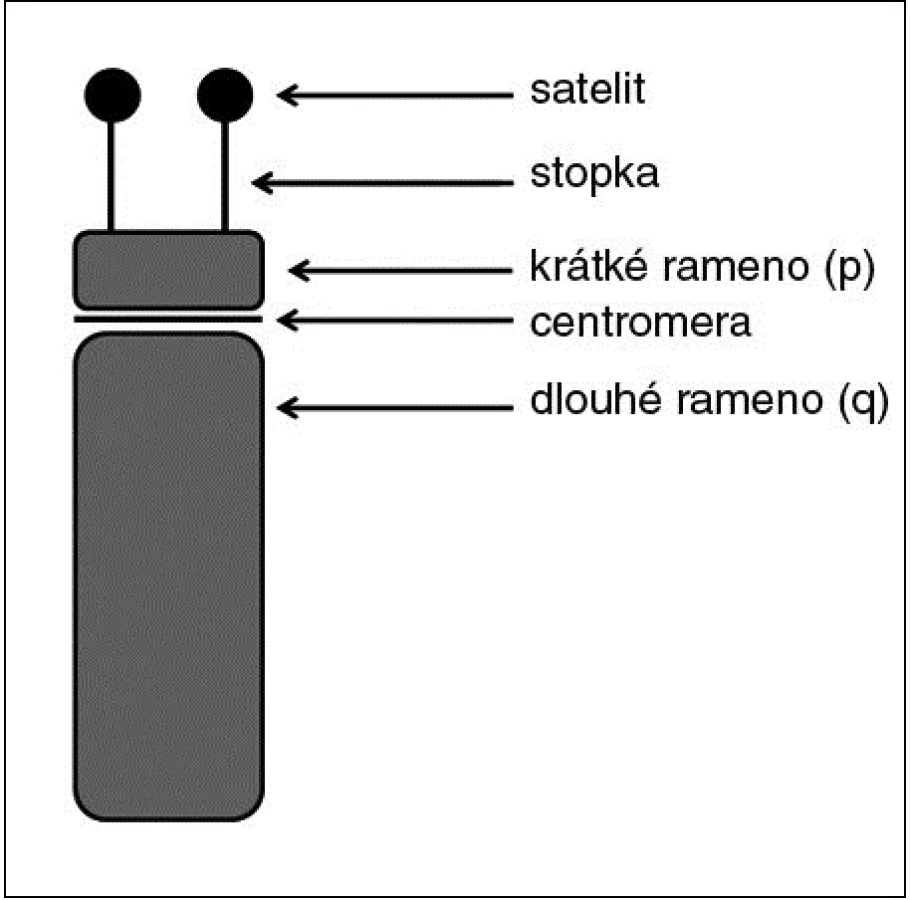
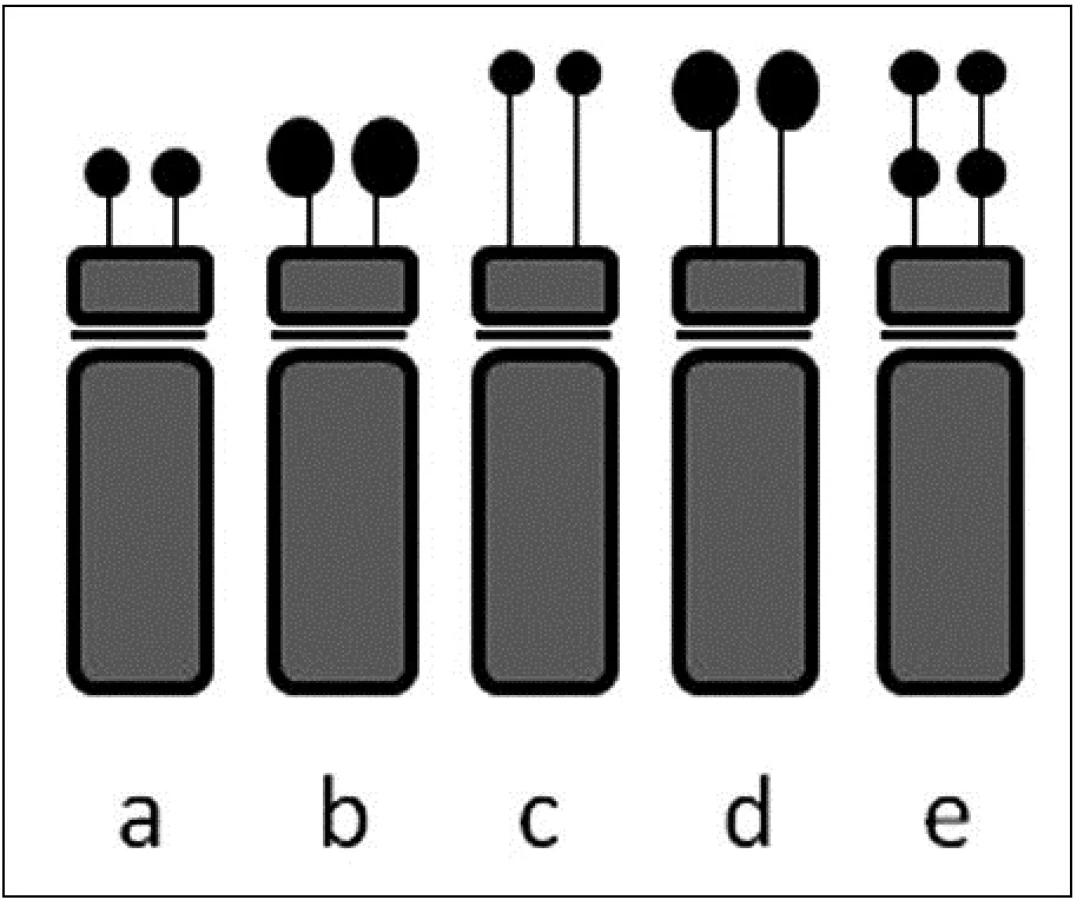
Akrocentrické chromozomy jsou chromozomy s velmi krátkými krátkými rameny zakončenými satelity (obr. 2). V lidském karyotypu patří mezi akrocentry chromozomy skupiny D (13, 14 a 15) a chromozomy skupiny G (21 a 22) (6). Typicky se na krátkém ramenu akrocentrického chromozomu vyskytují tyto struktury: a) samotné krátké rameno, tvořené převážně repetitivními sekvencemi (satelitní DNA); b) stopka (stalk), obsahující geny pro 18S a 28S ribozomální RNA; c) vlastní satelit tvořený opět repetitivními sekvencemi (3, 16). Repetitivní sekvence opět zajišťují výraznou variabilitu těchto oblastí, přičemž ani tyto varianty nejsou spojeny s žádnými klinickými projevy (4). Variabilita se týká jak délkových či velikostních variant jednotlivých částí krátkého ramena (například zvětšené satelity = ps+), tak i variant numerických (například vícenásobné satelity = pss) (3, 11). Příklady těchto variant shrnuje obrázek 3.
Inverze chromozomů jako varianty lidského karyotypu
Inverze je strukturní chromozomová aberace, která vzniká jako následek dvou zlomů na jednom chromozomu otočením uvolněného fragmentu o 180° a jeho opětovným připojením v invertované pozici (6). Podle vztahu invertovaného úseku k centromeře chromozomu navíc rozlišujeme inverze pericentrické (invertovaný úsek obsahuje centromeru) a inverze paracentrické (invertovaný úsek centromeru neobsahuje) (6). Inverze nejsou zcela benigními odchylkami, v některých případech jde dokonce o nebalancované aberace, kdy samotný mechanismus vzniku inverze naruší kontinuitu genetické informace, a způsobí tak rozvoj patologického fenotypu – zmiňován je například kauzální vztah inv(16)(p13.3q13) a Rubinsteinova-Taybiho syndromu (6). Pericentrické i paracentrické inverze (i když jsou balancované) navíc skýtají ještě jedno skryté riziko. Přítomnost inverze na jednom chromozomu z páru může vést k obtížnému párování chromozomů v průběhu meiotického dělení (v průběhu párování homologních chromozomů vznikají smyčky) a v extrémním případě i k narušení tohoto procesu (dochází ke vzniku rekombinovaných chromozomů, a tím pádem i ke vzniku gamet s nebalancovanou genetickou výbavou) (1, 2). Přítomnost inverze u jednoho z rodičů tak může vést k zásadním komplikacím v průběhu reprodukce, neboť vývoj zygoty s nebalancovanou genetickou výbavou končí často spontánním potratem, v extrémním případě i narozením potomka s chromozomovou aberací a fenotypovým postižením (1, 2).
Existují ovšem i takové inverze, které se vyskytují v populaci relativně často a jejich přítomnost není spojena ani s narušením genů u jejich nositele ani s rizikem vzniku rekombinovaných chromozomů pro jeho potomky. Tyto inverze jsou v drtivé většině případů zděděné od jednoho z rodičů a považovány za varianty lidského karyotypu (4, 6). Modelovým případem je inverze heterochromatinu na chromozomu 9 (různého rozsahu), která se vyskytuje u 1 až 3 % populace (16). Za „bezpečné“ inverze jsou považovány i některé inverze na dalších chromozomech, které se ovšem vyskytují vzácněji než inv(9), například inv(2)(p11.2q13), inv(10)(p11.2q21.2) a některé další (tab. 2) (4, 6).
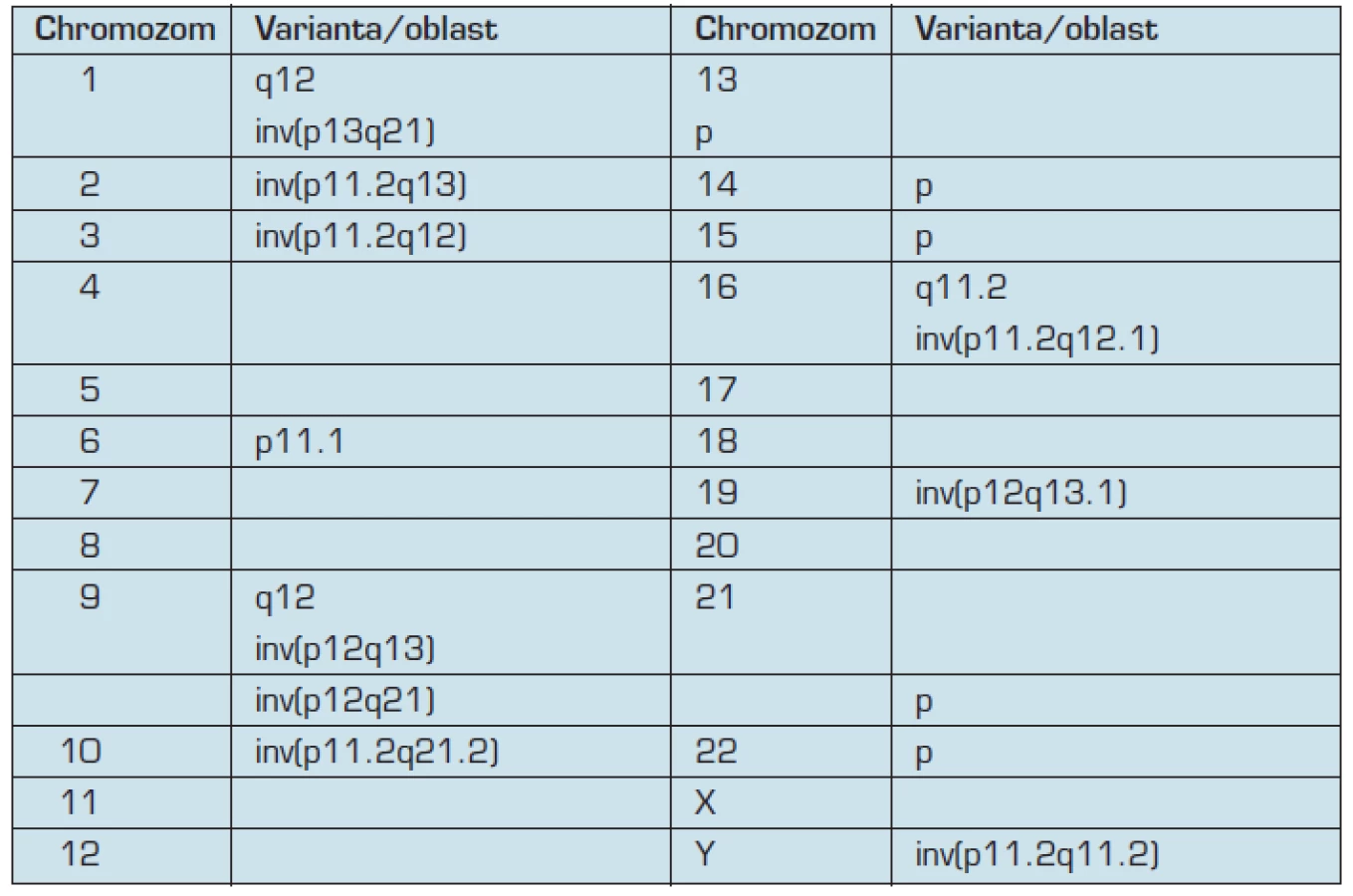
Další typy variant
Uvedený výčet tří nejčastějších skupin variant zdaleka není konečný. Zahrnuje však nejčastější odchylky, se kterými se může ve zprávách každý lékař setkat. Existuje celá řada dalších odchylek, které byly pozorovány u fenotypově normálních jedinců – řadí se sem například fragilita určitých oblastí, ale vzácně i některé delece, duplikace či inzerce, byť jsou takovéto případy zmiňovány velmi vzácně (6, 8). Dále jsou to také nové molekulárně-cytogenetické metody s vysokým rozlišením (příkladem je například metoda array-CGH), díky kterým byla odhalena celá řada nových (v běžném cytogenetickém vyšetření neviditelných) patologických změn, ale také variant, které u svých nositelů žádné fenotypové projevy nezpůsobují (8). Typickým příkladem fenotypově nevýznamné strukturní odchylky může být duplikace na chromozomu 15 v oblasti 15q11.2-q13 neobsahující kritickou oblast pro syndrom Prader-Willi (6). Detailní přehled všech těchto variant bohužel zcela přesahuje rozsah tohoto sdělení.
Interpretace nálezu cytogenetikem
Zhodnocení karyotypu je úkolem klinického cytogenetika (tab. 3). Z jeho pohledu mohou uvedené varianty někdy znamenat pro zhodnocení karyotypu určité dilema. Důvodů je zde hned několik:
- Posouzení délkové varianty (délkové varianty heterochromatinových oblastí či délkové varianty krátkých ramen akrocentrických chromozomů) je do velké míry závislé na subjektivním názoru hodnotícího cytogenetika. Extrémní délkové varianty (ať již ve smyslu výrazného prodloužení či naopak zkrácení) budou pravděpodobně správně vyhodnoceny ve většině případů, ovšem u hraničních rozsahů je finální rozhodnutí až příliš závislé na zvyklostech a zkušenostech konkrétního hodnotitele (respektive na zvyklostech konkrétní laboratoře). Byly sice popsány určité postupy, pomocí kterých je toto vyhodnocování možné provádět exaktněji (16), nicméně jejich aplikace do klinické praxe je otázkou jednotlivých laboratoří. Z tohoto důvodu mohou být četnosti délkových variant v různých laboratořích jen obtížně porovnavatelné (4). Na druhou stranu, varianty ze skupiny bezpečných inverzí, jakou je například inv(9)(p12q13), jsou jednoznačně identifikovatelné a četnost jejich nálezů je proto snáze interpretovatelná.
- Určité extrémní formy některých variant mohou být snadno zaměnitelné s chromozomovými patologiemi, které již fenotypové projevy mít mohou. Například nález připomínající extrémně zvětšené satelity některého z akrocentrických chromozomů může ve skutečnosti odpovídat translokaci zcela jiného genetického materiálu na tento chromozom (12). Vyhodnocení konkrétního nálezu jako neškodné varianty tak může být leckdy velmi obtížné a vyžádat si provedení dodatečných vyšetření (4, 12). Zvláště v rámci prenatální diagnostiky může mít nesprávné vyhodnocení „varianty“ fatální následky.
- S řadou variant se klinický cytogenetik setkává relativně běžně. Existují ovšem varianty, které se vyskytují relativně vzácně – například některé typy klinicky nevýznamných inverzí. Přestože je literatura uvádí jako fenotypově nevýznamné odchylky, cytogenetik se s nimi setkává relativně vzácně a jeho rozhodování tím může být ovlivněno. Některé odchylky tak nebudou popsány jako varianty, nýbrž jako balancované přestavby (tudíž přestavby bez očekávaného fenotypového projevu pro svého nositele). Tento jemný rozdíl hraje určitou úlohu při dalším přístupu k nositeli této odchylky (například přítomnost balancované přestavby u probanda typicky ověřujeme vyšetřením karyotypu rodičů, zatímco v případě běžných variant se takovéto ověření neprovádí).
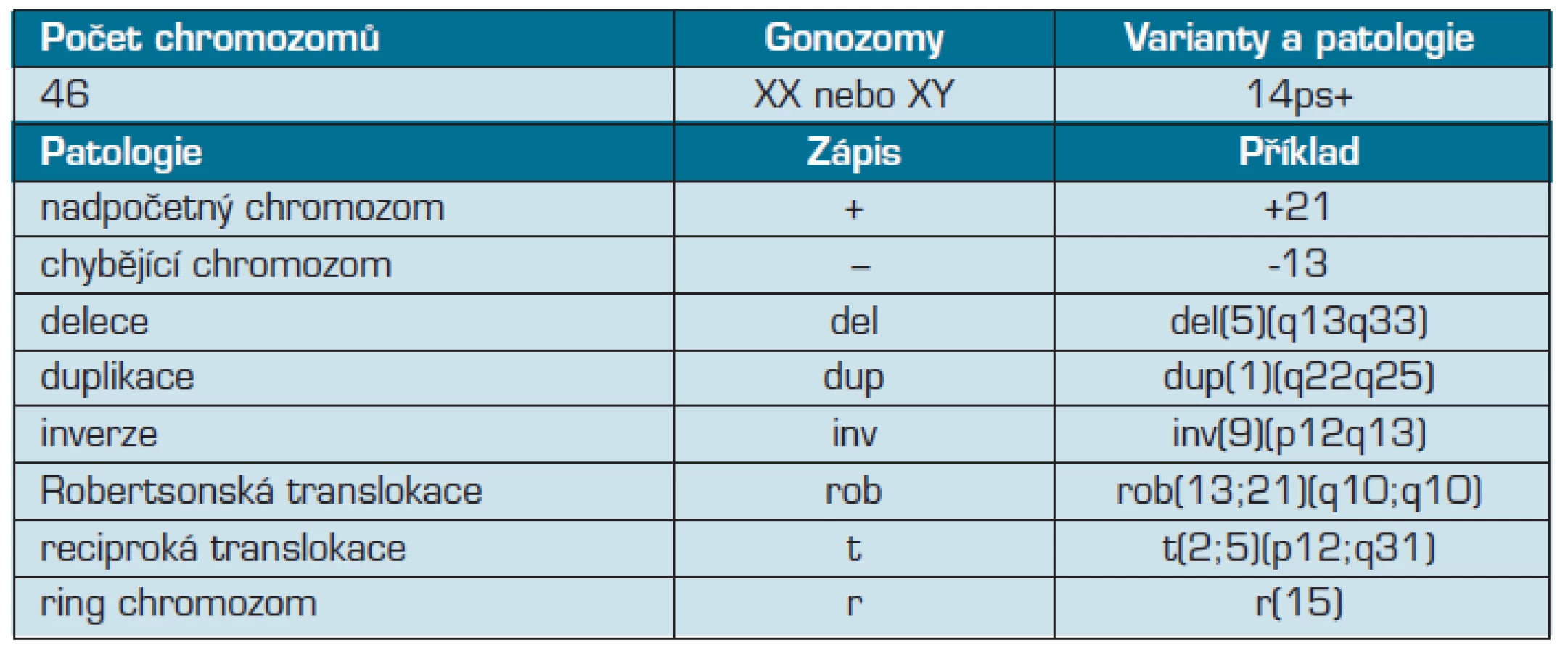
V rozhodování o zhodnocení a vydání výsledku u konkrétní identifikované varianty pomáhá cytogenetikům mezinárodní cytogenetická nomenklatura – ISCN (11) a doporučení různých odborných společností (European Cytogeneticists Association, ECA) (5).
Interpretace nálezu klinickým genetikem
Jak již bylo v textu mnohokrát zmíněno, veškeré uvedené varianty jsou považovány za odchylky ve stavbě karyotypu bez fenotypových projevů. Z pohledu klinika-negenetika (např. pediatra, gynekologa či neurologa) je tak takováto informace leckdy až nadbytečná, případně i matoucí. V souvislosti s tím řada odborných společností doporučuje nálezy fenotypově nevýznamných variant do zprávy pro kliniky neuvádět (5). Pokud je nález určité varianty ve zprávě o cytogenetickém vyšetření přesto uveden, musí být zcela benigní povaha této varianty pacientovi pečlivě vysvětlena, a to zejména v případě, kdy bylo cytogenetické vyšetření prováděno za účelem objasnění příčiny patologického stavu. Neprovedené či nedostatečné vysvětlení „abnormálního“ nálezu může u pacienta vyvolat zbytečné obavy.
Z pohledu klinického genetika tedy tyto varianty nejsou významným nálezem, přesto jsou situace, kdy se nad takovýmto nálezem musí hlouběji zamyslet. V případě vyšetřování dospělých osob není interpretace chromozomových variant většinou problematická. Určité riziko je spojené například s nosičstvím chromozomových inverzí, které nejsou na seznamu tzv. „bezpečných“ inverzí, neboť tyto abnormality již mohou být spojeny se vznikem nebalancované přestavby u potomka (1, 2). Obtížná může být interpretace některých chromozomálních variant odhalených o u plodu v rámci prenatální diagnostiky. V případě pochybností o benigní povaze nálezu je vhodné ozřejmit původ varianty vyšetřením karyotypu rodičů, případně doplňujícím molekulárně-cytogenetickým vyšetřením (6, 12). Dnes aktuální otázkou je rovněž (ne)doporučení osob s variantami chromozomů k darování gamet. Přestože existuje několik zmínek o možné spojitosti chromozomových heteromorfismů (zejména heterochromatinových variant chromozomu 9) a poruch reprodukce (13), studie sledující úspěšnost metod asistované reprodukce u osob s chromozomovými variantami neprokázala žádný rozdíl v úspěšnosti mezi skupinou osob s těmito variantami a bez nich (7).
Závěr
V lidském karyotypu existuje celá řada variabilních oblastí. Řadu klinicky nevýznamných variant je možné pozorovat i v průběhu standardního cytogenetického vyšetření. Vzhledem k klinickému významu těchto variant, nejsou tyto odchylky ve zprávě o cytogenetickém vyšetření často vůbec zaznamenávány. Pokud jsou někdy uvedeny, potom je především důležité poučit pacienta o jejich povaze, aby se tato „odchylka“ od normálního výsledku nestala důvodem zbytečné nejistoty či dokonce anxiety pacienta.
MUDr. Antonín Šípek jr.
Ústav biologie a lékařské genetiky 1. LF UK a VFN
Albertov 4
128 01 Praha 2
E-mail: antonin.sipek@lf1.cuni.cz
Sources
1. Balíček, P. Paracentrické inverze lidských chromozómů a jejich rizika. Čas. lék. čes., 2004, 143(1), s. 35–38.
2. Balíček, P. Pericentrické inverze lidských chromozomů a jejich rizika. Čas. lék čes., 2001, 140(2), s. 38–42.
3. Bhasin, M.K. Human population cytogenetics: A review. Int J Hum Genet, 2005, 5(2), p. 83–152.
4. Brothman, A.R., Schnedier, N.R., Saikevych, I., et al. Cytogenetic heteromorphisms: survey results and reporting practices of giemsa-band regions that we have pondered for years. Arch Pathol Lab Med, 2006, 130(7), p. 947–949.
5. E.C.A. Permanent Working Group for Cytogenetics and Society, Cytogenetic Guidelines and Quality Assurance: A common European framework for quality assessment for constitutional and acquired cytogenetic investigations, 2007, 33 p. Dostupné z: http://www.biologia.uniba.it/eca/ NEWSLETTER/NS-17/Guidelines.pdf.
6. Gardner, R.J. McK., Sutherland, G.R. Chromosome abnormalities and genetic counseling. 3rd ed. Oxford: Oxford University Press, 2004, 577 p.
7. Hong, Y., Zhou, Y.W., Tao, J., et al. Do polymorphic variants of chromosomes affect the outcome of in vitro fertilization and embryo transfer treatment? Hum Reprod, 2011, 26(4), p. 933–940.
8. Kowalczyk, M., Srebniak, M., Tomaszewska, A. Chromosome abnormalities without phenotypic consequences. J Appl Genet, 2007, 48(2), p. 157–166.
9. Lejeune, J., Gautier, M., Turpin, R. Etude des chromosomes somatiques de neuf enfants mongoliens. C R Hebd Seances Acad Sci, 1959, 248(11), p. 1721–1722.
10. Lejeune, J., Lafourcade, J., Berger, R., et al. Trois cas de délétion partielle du bras court d’un chromosome 5. C R Hebd Seances Acad Sci, 1963, 257(18), p. 3098–3102.
11. Shaffer, L.G., Slovak, M.L., Campbell, L.J. ISCN 2009: An international system for human cytogenetic nomenclature (2009). Basel: Karger, 2009, 138 p.
12. Starke, H., Mrasek, K., Liehr, T. Three cases with enlarged acrocentric p-arms and two cases with cryptic partial trisomies. J Histochem Cytochem, 2005, 53(3), p. 359–360.
13. Starke, H., Seidel, J., Henn, W., et al. Homologous sequences at human chromosome 9 bands p12 and q13-21.1 are involved in different patterns of pericentric rearrangements. Eur J Hum Genet, 2002, 10(12), p. 790–800.
14. Tjio, J.H., Levan, A. The chromosome number of man. Hereditas, 1956, 42, p. 1–6.
15. Verma, R.S., Dosik, H. Human chromosomal heteromorphisms: nature and clinical significance. Int Rev Cytol, 1980, 62, p. 361–333.
16. Wyandt, H.E., Tonk, V.S., et al. Atlas of human chromosome heteromorphisms. Dordrecht: Kluwer Academic Press, 2004, 3000 p.
Labels
General practitioner for children and adolescents General practitioner for adultsArticle was published in
General Practitioner

2012 Issue 4
- Memantine in Dementia Therapy – Current Findings and Possible Future Applications
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Memantine Eases Daily Life for Patients and Caregivers
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
Most read in this issue
- Astma a výživa: stravovací doporučení pro prevenci a léčbu astmatu
- Varianty lidských chromozomů a jejich význam z pohledu klinické genetiky
- Jaké je v České republice riziko onemocnění legionelózou?
- Význam histologické verifikace metastáz tumorů