
Korelace fenotypu a genotypu u českých pacientů s komplexem tuberózní sklerózy
Genotype/phenotype correlation in Czech tuberous sclerosis patients
Tuberous sclerosis complex is a genetic disorder that is manifested by benign tumours in many tissues and organs (particularly of the skin, brain, kidney, heart). Pathogenic mutations are in TSC1 and TSC2 gene. This study is focused on phenotype/genotype correlation. In the group of 99 probands were found 75 mutations in TSC2 gene and 24 mutations in TSC1 gene. Phenotype/genotype analysis of a group of 118 patients revealed that TSC2 mutations are associated with more severe phenotypes than TSC1 mutations. Furthermore, there were found significant correlations between findings of hypomelanotic spots and the incidence of cortical tubers, the facial skin involvement (angiofibromas) and evidence of renal angiomyolipomas, between hamartoma of the retina and heart rhabdomyoma, or between epilepsy and mental retardation in the group of patients with TSC2 mutations. Conversely, in individuals with TSC1 mutations have been rarely observed angiomylipomas and multiple renal cysts, fibrous plaques forehead and hamartoma of the retina. No statistically significant correlation was observed between different types of mutations and specific phenotypes.
Keywords:
complex of tuberous sclerosis – TSC1 gene – TSC2 gene – sequencing, phenotype/ genotype correlation
:
H. Filipová 1; R. Vrtěl 1; R. Vodička 1; B. Petrák 2; V. Curtisová 1; M. Procházka 1
:
Ústav lékařské genetiky FN a LF UP, Olomouc
Přednosta: prof. MUDr. Martin Procházka, Ph. D.
1; Klinika dětské neurologie 2. LF a FN Motol, Praha
Přednosta: prof. MUDr. Vladimír Komárek, CSc.
2
:
Prakt. Lék. 2016; 96(4): 185-189
:
Of different specialties
Komplex tuberózní sklerózy je genetické onemocnění, které se projevuje benigními tumory v mnoha tkáních a orgánech (především na kůži, mozku, ledvinách a srdci). Příčinou tuberózní sklerózy jsou mutace v TSC1 a TSC2 genu. Studie je zaměřena na fenotyp/genotypovou korelaci. Ve skupině 99 probandů bylo nalezeno 75 mutací v TSC2 genu a 24 mutací v TSC1 genu. Fenotyp/genotypová analýza souboru 118 pacientů odhalila, že TSC2 mutace jsou spjaty se závažnějším fenotypovým projevem než mutace TSC1. Dále byly zjištěny významné korelace mezi nálezem hypomelanotických skvrn a výskytem kortikálních tuberů, mezi postižením kůže faciálními angiofibromy a nálezem renálních angiomyolipomů, mezi hamartomy sítnice a srdečním rhabdomyomem či mezi epilepsií a mentální retardací ve skupině pacientů s TSC2 mutacemi. Naopak u jedinců s TSC1 mutacemi byly vzácně pozorovány angiomyolipomy a vícečetné renální cysty, fibrózní plaky čela a hamartomy sítnice. Žádná statisticky významná korelace nebyla zaznamenána mezi jednotlivými typy mutací a konkrétním fenotypovým projevem.
Klíčová slova:
komplex tuberózní sklerózy – TSC1 gen – TSC2 gen, sekvenování – fenotyp/genotypová korelace
ÚVOD
Komplex tuberózní sklerózy (TSC) se vyznačuje autozomálně dominantním typem dědičnosti, lokusovou heterogenitou, extrémní variabilitou klinických projevů a prevalencí odhadovanou na 1/6000 až 1/10000 (9). Charakteristickým rysem onemocnění je tvorba benigních tumorů (hamartomů) v mnoha tkáních. Nejčastěji bývá zasažena kůže, mozek, srdce a ledviny (5). Příčinou TSC jsou mutace v TSC tumor-supresorovým genech. TSC1 gen (9q34) kóduje protein hamartin (8), gen TSC2 (16p13.3) tuberin (3). Produkty obou genů společně vytvářejí komplex, který v rámci mTORC1 (mammalian target of rapamycin complex 1) signalizace funguje jako inhibitor buněčného růstu a proliferace. Jestliže, nastane mutace v jednom z TSC genů, je tvorba či funkce tohoto komplexu narušena, čímž je mTOR signalizace aberantně konstitutivně aktivována a dochází k vývoji různých hamartomů (4).
V roce 2012 byla diagnostická kritéria pro TSC revidována a jejich součástí jsou nyní jak klinická, tak genetická kritéria. Klinická diagnostická kritéria zahrnují níže uvedené minoritní a majoritní znaky. Při nálezu dvou majoritních znaků či jednoho majoritního a zároveň dvou a více znaků minoritních je splněna podmínka pro stanovení definitivní diagnózy TSC. Možná diagnóza TSC je dána přítomností pouze jednoho majoritního znaku či výskytem dvou a více minoritních znaků. Výjimku, kdy nelze stanovit definitivní diagnózu, představuje situace současného výskytu lymfangioleiomyomatózy a angiomyolipomů bez přítomnosti dalších znaků TSC (6).
Majoritní znaky TSC
- hypomelanotické skvrny (tři a více, alespoň 5 mm v průměru)
- angiofibromy (tři a více) či fibrózní plaky na kůži hlavy
- unguální fibromy (dvě a více)
- šagrénová skvrna
- vícečetné sítnicové hamartomy
- kortikální dysplazie (kortikální tubery, cerebrální radiální migrační dráhy bílé hmoty)
- subependymální noduly (SEN)
- subependymální obrovskobuněčný astrocytom (SEGA)
- srdeční rhabdomyom
- lymfangioleiomyomatóza (LAM)
- angiomyolipomy (dva a více) (AML)
Minoritní znaky TSC
- kožní léze typu „confetti“
- jamky zubní skloviny (tři a více)
- intraorální fibromy (dva a více)
- bezbarvá skvrna na sítnici
- vícečetné renální cysty
- nerenální hamartomy
Kauzální mutace bývá odhalena až u 85 % TSC pacientů, přičemž přibližně dvě třetiny případů jsou způsobeny de novo mutacemi v TSC genech (7). Výsledky z doposud provedených fenotyp/genotypových korelací v souborech pacientů z různých zemí dokládají, že TSC2 mutace jsou obvykle asociovány s těžším fenotypem onemocnění než TSC1 mutace (1). U pacientů s de novo mutacemi v genu TSC1 byl, oproti přibližně stejně starým sporadickým TSC2 případům, prokázán např. menší počet subependymálních nodulů a kortikálních tuberů, nižší frekvence záchvatů, menší výskyt středně těžké či těžké mentální retardace rovněž méně závažněji byly postiženy ledviny (2).
SOUBOR PACIENTŮ
Analyzovaný soubor tvořilo 118 pacientů, přičemž probandů bylo 99 a 19 případů reprezentovalo tuberózní sklerózou postižené rodinné příslušníky některých probandů. DNA vzorky či periferní krev pacientů pocházely z genetických (popřípadě neurologických) pracovišť z různých míst České republiky. Na základě dostupných informací o klinickém obrazu a dle diagnostických kritérií všichni pacienti (kromě jednoho) splňovali podmínky pro stanovení definitivní (95 osob) nebo možné (22 osob) diagnózy TSC. Do studie byli zahrnuti i jedinci, u nichž byl znám pouze jeden klinický údaj. Věkový rozptyl v souboru: 2 dny – 65 let.
METODIKA
Izolace DNA ze vzorků periferní krve byla provedena Millerovou vysolovací metodou dle běžného postupu.
Rozsáhlé delece byly zachyceny metodou MLPA (multiplex ligation-dependent probe amplification) pomocí TSC2 P046-C1 sond a při dodržení doporučeného postupu firmy MRC-Holland. K separaci byl využit genetický sekvenátor 3130 (Applied Biosystems) a k vyhodnocení dat software GeneMapper a Coffalyser.
K detekci mutací malého rozsahu bylo použito několik přístupů. U části pacientů byla analýza TSC genů provedena kombinací skenovací techniky denaturační gradientové gelové elektroforézy (DGGE) a následného sekvenování vytipovaného aberantního fragmentu (exonu) Sangerovou metodou. Při DGGE byly využívány čtyři (20–65 %, 30–70 %, 40–80 % a 45–75 %) denaturační gradienty polyakrylamidového gelu. Separace amplikonů jednotlivých exonů TSC genů včetně pozitivních a negativních kontrolních vzorků probíhala při podmínkách 60 °C/88 V/1600 Vh v systému DCodeTM, BIO-RAD.
U většiny pacientů byla pro odhalení mutace zvolena strategie přímého Sangerova sekvenování obou genů. Standardním postupem byly osekvenovány všechny kódující exony TSC genů a rovněž úseky sekvence zahrnující intron/exonové rozhraní. K separaci byl využit genetický analyzátor 3130 (Applied Biosystems) a k vyhodnocení dat software Gen Scan Analysis a SeqScanner.
U další části vyšetřovaných TSC osob byla mutace zjištěna technikou masivního paralelního sekvenování (MPS) prostřednictvím platformy Ion Torrent na přístroji Ion PGM. Knihovna byla navržena tak, aby bylo zajištěno pokrytí všech 23 exonů TSC1 a 42 exonů TSC2 genu a rovněž oblastí intron/exonového rozhraní. Postup byl proveden zcela v souladu s následujícími protokoly: Ion AmpliSeqTM Library Preparation (Pub. no. MAN0006735 a zkrácená verze postupu MAN0007524, Rev. 3); Ion PGMTM Hi-QTM OT2 Kit (Pub. no. MAN0010902) a jeho zkrácená verze (Cat. no. A27739, Pub. no. MAN0010903, Rev. A.0); Ion PGMTM Hi-QTM Sequencing Kit (Cat. no. A25592, Pub. no. MAN0009816, Rev. C.0) Software k vyhodnocení získaných dat byl následující: Ion ReporterTM, Illumina Genome Viewer, Illumina Variant Studio a NextGENe. Nález mutace metodou MPS byl vždy konfirmován Sangerovým sekvenováním. Při sekvenační analýze byly využívány tyto NCBI referenční sekvence: genomická sekvence TSC1 genu NG_012386.1 a sekvence pro jeho transkript NM_000368.4, pro gen TSC2 genomická sekvence NG_005895.1 a pro jeho transkript NM_000548.3. Potvrzení identifikovaných mutací bylo rovněž provedeno Sangerovým sekvenováním i na DNA vzorcích z nezávislého odběru.
Při statistickém zpracování byla využita neparametrická Spearmanova korelace. Korelace je považována za statisticky významnou pro hodnoty p < 0,05.
VÝSLEDKY
Analýza DNA probandů
Ve skupině 99 probandů bylo identifikováno 3krát více mutací v genu TSC2 (75 mutací) než v genu TSC1 (24 mutací). Sporadický výskyt zastupovalo 40 osob, 17 jedinců představovalo familiární formu výskytu a u 42 postižených nebylo možné tento údaj stanovit. U čtyř pacientů byla odhalena mutace ve formě mozaiky. Téměř každý proband byl nositelem jiné mutace, pouze šest různých mutací se vyskytovalo u více probandů. Syndrom přilehlých genů (TSC2/PKD1) byl zjištěn u čtyř probandů, přičemž u tří jedinců byl deletován celý gen TSC2. Spektrum mutací nalezených v TSC1 a v TSC2 genech ve skupině probandů je zachyceno v grafech 1 a 2.
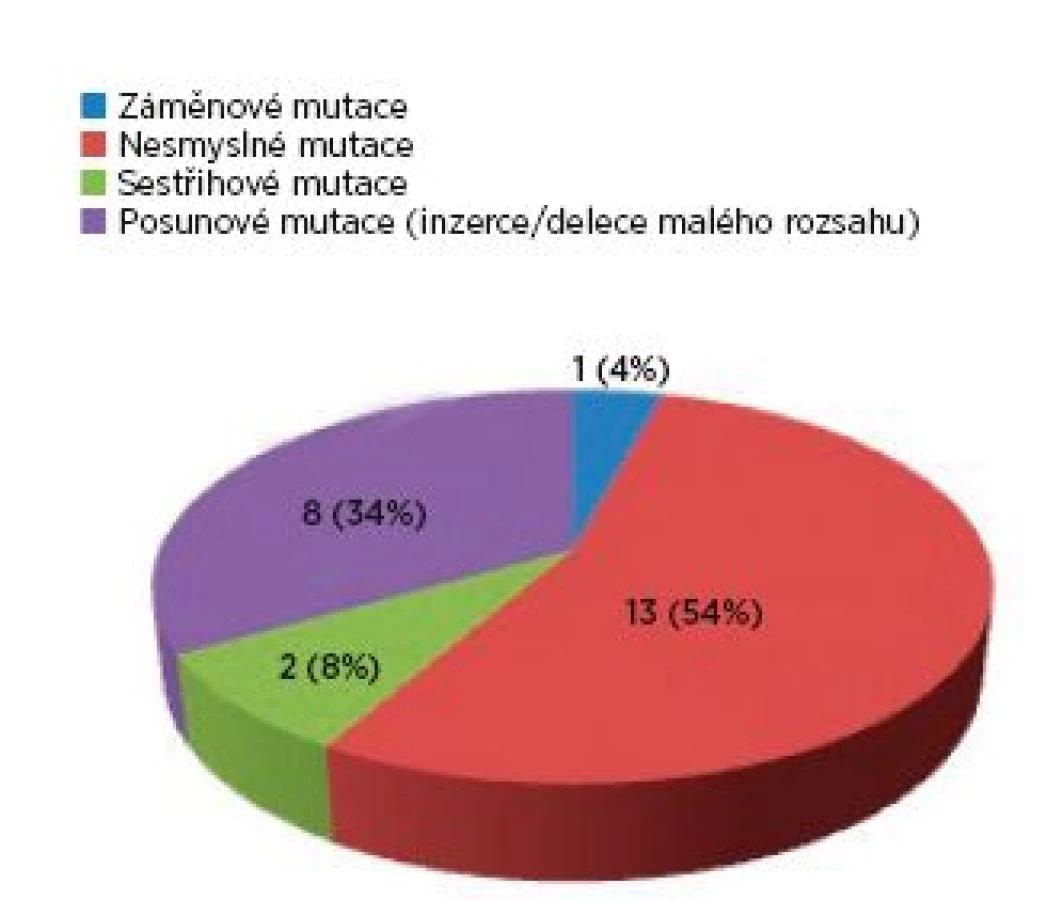
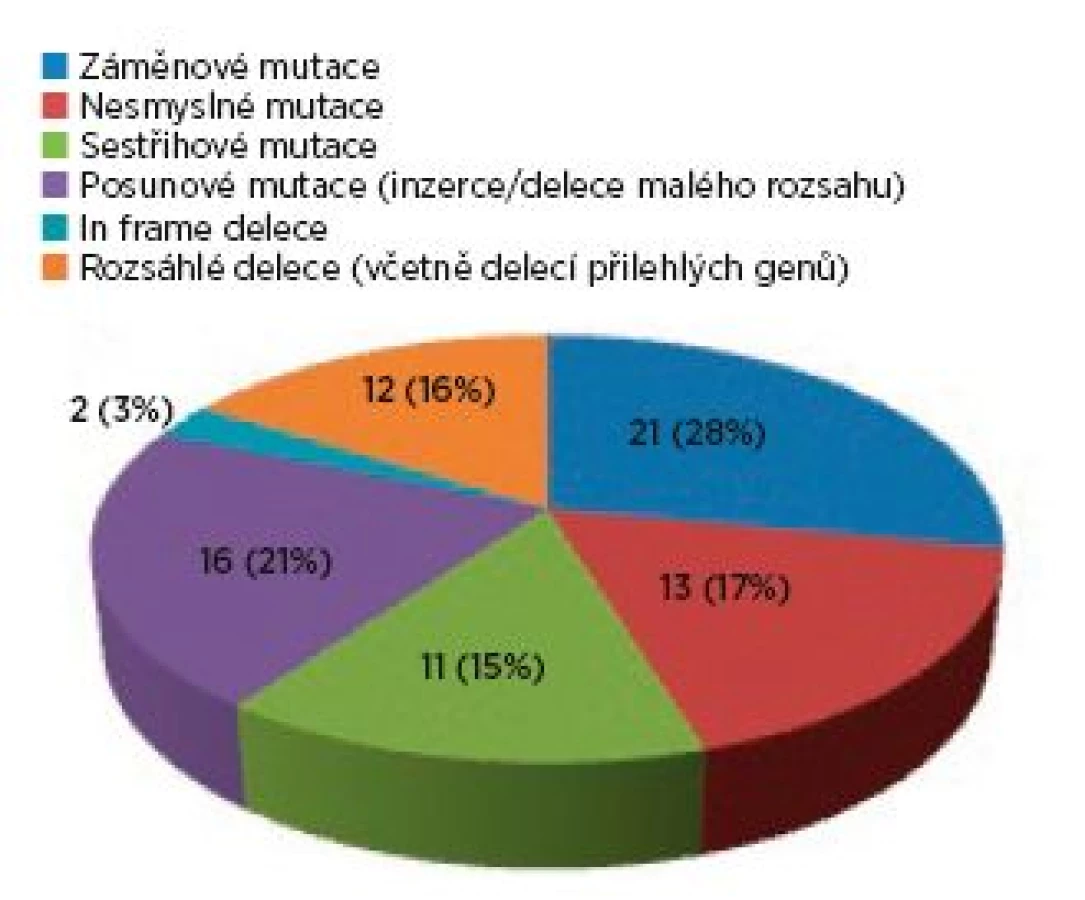
Fenotyp/genotypová korelace
Pro zjištění korelace fenotypu a genotypu byla využita klinická data od 118 TSC pacientů (výše zmíněných 99 probandů + 19 příbuzných postižených tuberózní sklerózou). Hlavní limitací analýzy byly nedostatečné informace o klinickém obrazu některých pacientů, a proto se počty vyšetřených osob pro jednotlivé sledované TSC znaky lišily.
V tabulce 1 je shrnut celkový záchyt TSC znaků a přidružených projevů onemocnění a rovněž jejich výskyt jak ve skupině pacientů s mutacemi v genu TSC2, tak s mutacemi genu TSC1. Celkově TSC2 mutace byly spjaty s vyšší četností nálezů TSC znaků u nemocných než TSC1 mutace.
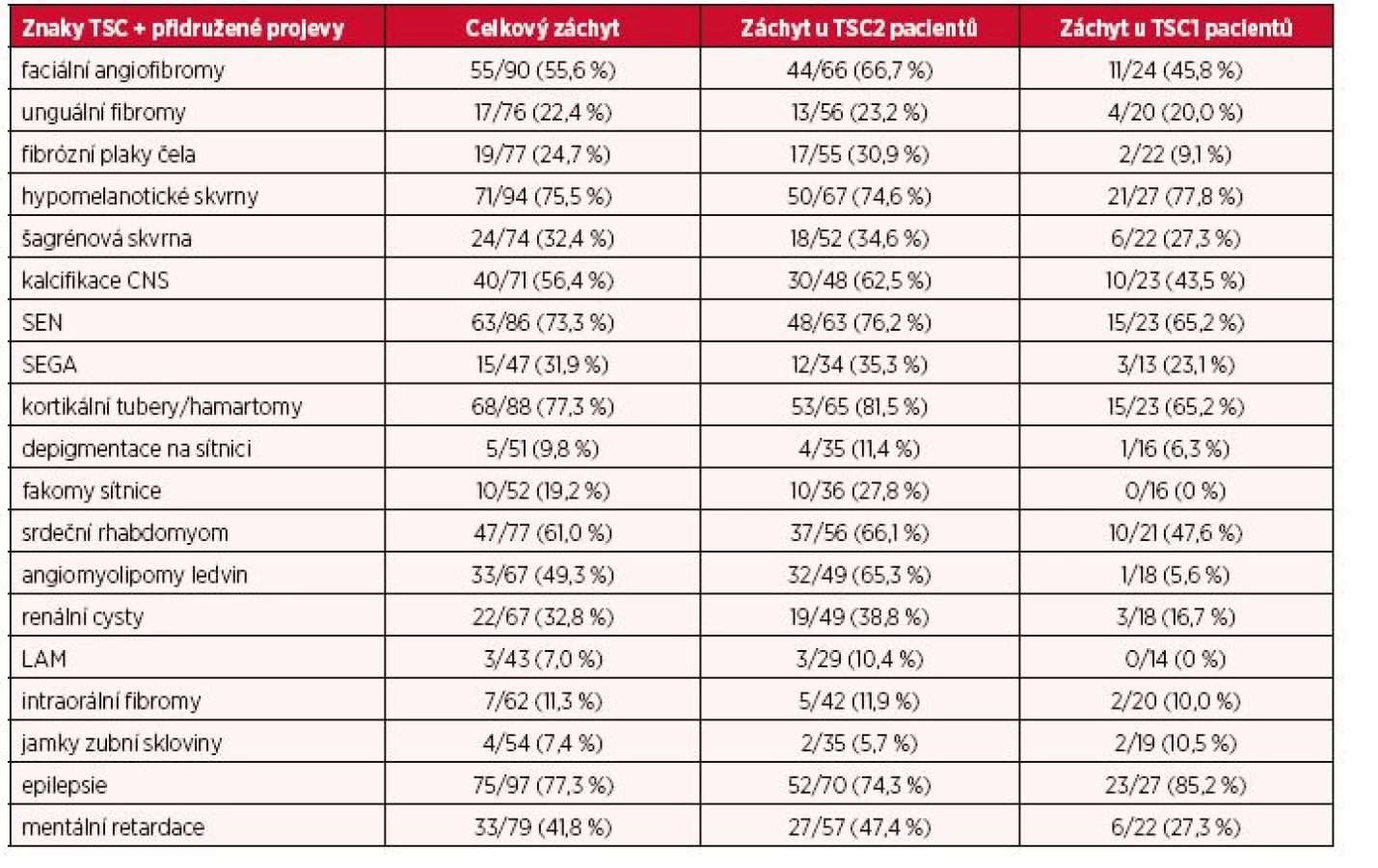
V rámci kožního postižení byl zaznamenán statisticky významný rozdíl u nálezu fibrózních plaků čela mezi jedinci s TSC2 mutacemi a osobami s TSC1 mutacemi. Navíc pacienti s TSC1 mutacemi málokdy vykazovali více než jeden TSC asociovaný znak na kůži než osoby s TSC2 mutacemi.
V případě CNS nebyl zjištěn statisticky významný rozdíl pro jednotlivé CNS znaky mezi TSC1 a TSC2 skupinou, nicméně pacienti s TSC2 mutacemi měli častěji dva a více typů postižení mozku oproti jedincům s TSC1 mutacemi (efekt na hraně významnosti). Rovněž efekt na hraně významnosti souvisel s porovnáním prevalence mentální retardace u TSC1 a TSC2 skupiny. Naopak statisticky nevýznamně se jevil rozdíl v záchytu epilepsie.
U očních TSC projevů bylo pozorováno, že celkové postižení sítnice (hamartomy a/či achromatickými skvrnami retiny) je méně častá u nositelů TSC1 mutací (efekt na hraně významnosti). Konkrétně pro fakomy sítnice byl tento efekt významný (p = 0,02).
Efekt na hraně významnosti byl stanoven v důsledku mírně zvýšeného výskytu rhabdomyomu v srdeční tkáni pacientů s TSC2 mutacemi oproti osobám se zasaženým TSC1 genem.
Jednoznačně závažnější postižení ledvin souviselo s TSC2 mutacemi, v případě angiomyolipomů byl efekt velký a statisticky významný (p = 0,00005).
Ohledně plicních a intraorálních TSC projevů bylo postižených v obou skupinách velmi málo, a tudíž sledování konkrétních trendů/efektů bylo statisticky zavádějící.
Mezi skupinou pacientů s TSC1 a TSC2 mutacemi nebyl odhalen statisticky významný rozdíl ve věku, formě výskytu TSC v rodině ani v počtu stanovených definitivních a možných TSC diagnóz.
Další fáze analýzy byla zaměřena na odhalení případné korelace mezi typem mutace a klinickým obrazem onemocnění. Na základě vlivu mutace na podobu proteinu byly nalezené mutace rozděleny do tří kategorií. Do první kategorie byly zařazeny mutace způsobující zkrácení proteinu (tzn. posunové mutace zapříčiněné inzercemi/delecemi malého rozsahu, nesmyslné mutace či rozsáhlé delece), druhou kategorii představovaly záměnové mutace a třetí skupinu tvořily sestřihové mutace. Rozdílné zastoupení těchto kategorií mutací ve vztahu k stanovené formě výskytu onemocnění je znázorněno v tabulce 2.
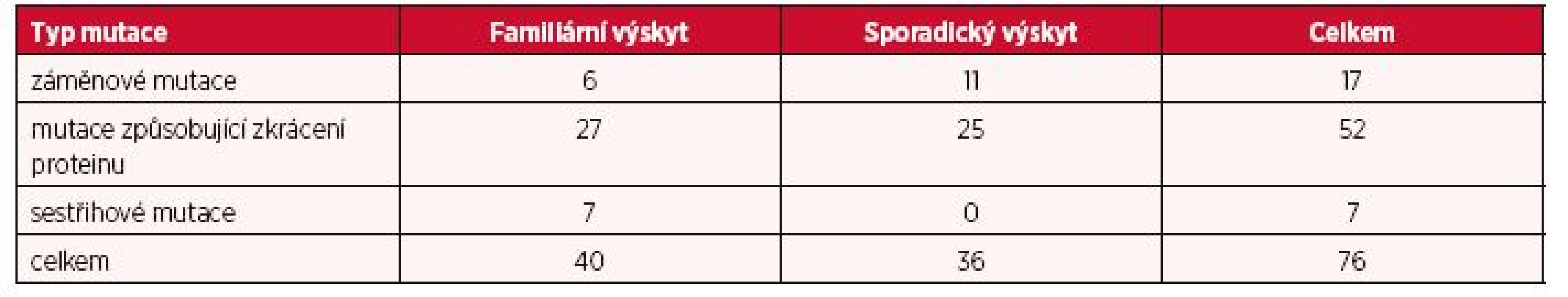
Jelikož všichni pacienti (kromě tří) s mutacemi v genu TSC1 nesli mutaci vedoucí ke zkrácení hamartinu, byla fenotyp/genotypová analýza omezena pouze na skupinu pacientů s TSC2 mutacemi, kde byly výše zmíněné tři kategorie mutací zastoupeny vyváženěji. Avšak, žádná statisticky významná korelace zde nebyla prokázána. Zajímavým poznatkem bylo zjištění, že jediní tři pacienti postižení LAM byli nositeli záměnových mutací TSC2 genu.
Dále bylo prostřednictvím neparametrické (Spearmanovy) korelace posuzován vzájemný vztah mezi konkrétními TSC znaky. Mezi statisticky významné korelace, které byly zjištěny analýzou souboru pacientů s TSC2 mutacemi, patřily např. korelace mezi celkovým postižením kůže a výskytem renálních angiomyolipomů. Konkrétně v případě postižení faciálními angiofibromy a vztahu k AML ledvin byl Spearmanův korelační koeficient 0,64. Další významná korelace byla mezi nálezem hypomelanotických skvrn a kortikálních tuberů (korelační koeficient 0,55), mezi hamartomy sítnice a srdečním rhabdomyomem (korelační koeficient 0,47) či mezi epilepsií a mentální retardací (korelační koeficient 0,58).
Zároveň bylo potvrzeno, že s narůstajícím věkem pacienta přibývají projevy onemocnění (kromě postižení srdce). Dokladem jsou např. významné korelace zaznamenané mezi věkem a výskytem angiomyolipomů ledvin (korelační koeficient 0,55) či postižením plic (korelační koeficient 0,53), záchytem unguálních fibromů (korelační koeficient 0,47) či faciálních angiofibromů (korelační koeficient 0,47). V případě srdečního rhabdomyomu byla v souvislosti se stoupajícím věkem prokázána významná antikorelace (korelační koeficient mínus 0,43).
Ve skupině osob s TSC1 mutacemi nebyla nalezena žádná výrazná korelace mezi konkrétními TSC znaky, pouze byla opět zjištěna významná korelace mezi věkem a postižením kůže a ledvin a silná antikorelace mezi věkem a postižením srdce.
DISKUZE
V souladu s výsledky zahraničních analýz TSC pacientů, bylo i v české TSC populaci prokázáno, že TSC2 mutace jsou příčinou závažnějšího fenotypu než TSC1 mutace. Rovněž poměr v nálezu mutací TSC2 : TSC1 a spektrum zachycených mutací v jednotlivých TSC genech bylo podobné. Dokladem je např. studie 362 pacientů, kdy poměr nalezených mutací TSC2 : TSC1 byl 3,4 : 1 a zastoupení jednotlivých typů mutací podpořilo tvrzení, že gen TSC1 je pouze vzácně zasažen rozsáhlými přestavbami, záměnovými mutacemi a in frame delecemi (7). V naší studii byl poměr TSC2 : TSC1 mutací v souboru probandů 3 : 1 a rozsáhlé a in frame delece jsme v genu TSC1 neprokázali žádné, záměnovou mutaci pouze jednu. Poměr sporadických/familiárních případů (40 : 17 ve skupině probandů, 40 : 36 v celém souboru) byl zkreslen tím, že u dalších 42 jedinců jsme neměli k dispozici DNA vzorky obou rodičů, a tudíž status výskytu jsme nemohli stanovit. Naše fenotyp/genotypová studie 118 jedinců v porovnání s analýzou rozsáhlejšího (224 osob) souboru TSC pacientů odhalila stejné zjištění, že angiomyolipomy a vícečetné renální cysty, fibrózní plaky čela a hamartomy sítnice jsou velmi vzácné u pacientů s TSC1 mutacemi (2). Přestože jsme prokázali, že všechny námi pozorované TSC znaky a přidružené projevy se s vyšší frekvencí vyskytovaly u nositelů TSC2 mutací, tak v případě epilepsie byla prevalence překvapivě mírně vyšší ve skupině TSC1 pacientů, nicméně se nejednalo o statisticky významný rozdíl. Statistické zpracování bylo problematické u plicních a intraorálních TSC projevů. Všichni tři pacienti postižení LAM měli detekované záměnové mutace TSC2 genu. Pro odlišení, zda se jedná o náhodu či trend, by v souboru muselo být pozorováno více postižených. Příčinou nízké prevalence TSC projevů v ústní dutině v obou TSC1/TSC2 skupinách mohla být skutečnost, že někteří pacienti v době vyšetření ještě/už neměli zuby, či vzhledem k možným opravám chrupu nebyly dentální jamky v zubní sklovině patrné.
Kombinovaný efekt (málo pacientů s mutacemi genu TSC1 a zároveň s mírnějším postižením) mohl být důvodem, proč ve skupině osob s TSC2 mutacemi byla nalezena celá řada významných korelací navzájem mezi určitými TSC znaky a ve skupině TSC1 osob žádná.
Klinická variabilita onemocnění byla mezi vyšetřovanými TSC pacienty značná, a to i v rámci rodin či mezi nepříbuznými jedinci se stejnou mutací. Porovnávání TSC fenotypu mezi pacienty bylo negativně ovlivněno chybějícími údaji o stavu TSC asociovaných orgánů, jelikož někteří pacienti nepodstoupili všechna doporučená vyšetření. Dalším problémem byl rozdílný věk porovnávaných osob. V případě jedné čtyřčlenné rodiny, u níž někteří příslušníci vykazovali pouze mírnou formu TSC projevů s pozdějším nástupem, byl nejmladší člen (10měsíční) v době probíhající analýzy dokonce bezpříznakový.
Možným vysvětlením značné variability onemocnění může být i vliv případných sekvenčních změn v dalších komponentách signálních drah.
Poděkování patří RNDr. Tomáši Fürstovi, Ph.D. za pomoc při statistickém zpracování dat a všem spolupracujícím indikujícím lékařům.
Střet zájmů: žádný.
ADRESA PRO KORESPONDENCI:
doc. RNDr. Radek Vrtěl, Ph.D.
Ústav lékařské genetiky FN a LF UP
I. P. Pavlova 6,
779 00 Olomouc
e-mail: vrtel@fnol.cz
Sources
1. Curatolo P, Moavero R, Roberto D, Graziola F. Genotype/phenotype correlations in tuberous sclerosis complex. Semin Pediatr Neurol 2015; 22 (4): 259–273.
2. Dabora SL, Jozwiak S, Franz DN, et al. Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. Am J Hum Genet 2001; 68(1): 64–80.
3. European Chromosome 16 Tuberous Sclerosis Consortium. Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell 199331; 75(7): 1305–1315.
4. Foster K, Acosta-Jaquez HA, Romeo Y, et al. Regulation of mTOR Complex 1 (mTORC1) by Raptor Ser863 and Multisite Phosphorylation. J Biol Chem 2010; 285(1): 80–94.
5. Gómez MR, Sampson JR, Whittemore VH. Tuberous sclerosis complex. Developmental perspectives in psychiatry. New York: Oxford University Press, 3rd ed., 1999 : 313–316; 11.
6. Northrup H, Krueger D. The international tuberous sclerosis complex consensus group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012. International tuberous sclerosis complex consensus conference. Pediatr Neurol 2013; 49 : 243–254.
7. Sancak O, Nellist M, Goedbloed M, et al. Mutational analysis of the TSC1 and TSC2 genes in a diagnostic setting: genotype-phenotype correlations and comparison of diagnostic DNA techniques in tuberous sclerosis complex. Eur J Hum Genet 2005; 13(6): 731–741.
8. van Slegtenhorst M, deHoogt R, Hermans C, et al. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science 1997; 277 : 805–808.
9. Vrtel R, Verhoef S, Bouman K, et al. Identification of a nonsense mutation at the 5‘ end of the TSC2 gene in a family with a presumptive diagnosis of tuberous sclerosis complex. J Med Genet 1996; 33(1): 47–51.
Labels
General practitioner for children and adolescents General practitioner for adultsArticle was published in
General Practitioner

2016 Issue 4
- Hope Awakens with Early Diagnosis of Parkinson's Disease Based on Skin Odor
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Advances in the Treatment of Myasthenia Gravis on the Horizon
- Memantine Eases Daily Life for Patients and Caregivers
- Current Insights into the Antispasmodic and Analgesic Effects of Metamizole on the Gastrointestinal Tract
Most read in this issue
- The purpose of MRI as a part of pre-surgery staging for breast cancer
- Genotype/phenotype correlation in Czech tuberous sclerosis patients
- Preview of physical activity in selected cancer patients
- Occupational diseases reported in the Czech Republic in 2015