
Poruchy metabolizmu železa I. Regulace homeostázy železa
Disorders of iron metabolism I. Regulation of iron homeostasis
Iron is an ubiquitously ocurring vital element which can be found in all living cell from bacterias and yeasts to mammals. The cells explore the redox potential of iron, which can be used in many essential and vital biochemical reactions. The iron homeostasis must be very finely tuned beacuse iron overload may lead to the generation of very toxic reactive oxid radicals, which may damage lipids, proteins and nucleic acids with possible severe cellular damage and death. Iron homeostasis is maintained on cellular and whole body levels and details of this fascinating network have been disclosed only very recently. The transferrin receptor (TfR) and ferritin play the main roles on cellular level of iron homeostasis maintaining. The expression of these proteins are under tight control of iron regulatory proteins (IRPs), which bind to specific parts of mRNA molecules, called iron regulatory elements (IREs). If the IRE is localized in 5’UTR (untranslated region) of mRNA molecule, the binding of IRP blocks translation of this mRNA. In case when the IRE lays in the 3’UTR, the binding of IRP stabilizes the mRNA and potentiates its translation. Excess of cellular iron causes high expression of ferritin genes and blocks the expression of TfR gene, in case of sideropenia the processes are reverse. The absorption and transport of iron to the blood by enterocytes and the retention or release of iron by macrophages represent the main regulatory processes of iron homeostasis on the whole body level. The enterocytes regulate the input of iron to the body by increasing or decreasing its absorption from the enteral lumen and its transport across the basolateral membrane to the blood. According to so called „preprogramming theory“ is the precursor cell of the enteral crypts preprogrammed by the body iron status to increased or decreased absorption and transport of iron. HFE protein, discovered by Feder et al. in 1996, plays very important role in these regulations. The C282Y mutation of HFE protein is the main cause of hereditary iron overload in causasoid populations worldwide as its homozygote status can be detected in more than 80 % of causasoid patients with hereditary hemochromatosis.
Key words:
iron – sideropenia – iron overload – hereditary hemochromatosis
Authors:
J. Novotný
Authors‘ workplace:
Oddělení klinické hematologie FN Brno, pracoviště Bohunice, přednosta prof. MUDr. Miroslav Penka, CSc., Transfuzní oddělení a krevní banka FN Brno, pracoviště Bohunice, přednosta prim. MUDr. Eva Tesařová
Published in:
Vnitř Lék 2005; 51(6): 681-689
Category:
Original Contributions
Overview
Železo je biogenní prvek vyskytující se ve všech živých buňkách od jednobuněčných organizmů (bakterie, kvasinky) až po savce. Atom železa je schopen velmi snadno vázat i uvolňovat elektron, čehož živé buňky využívají v celé řadě vitálních biochemických reakcí. Na druhé straně nadbytek železa ohrožuje organizmus generací toxických volných radikálů, které mohou oxidací poškozovat proteiny, lipidy i nukleové kyseliny, a způsobit tak závažné poškození až smrt buněk. Organizmy proto regulují homeostázu železa na buněčné i celotělové úrovni, jemnější detaily této integrované regulační sítě začínáme poznávat až v současnosti. Na buněčné úrovni hrají hlavní role v regulaci homeostázy železa transferinový receptor (TfR) a feritin, jejichž exprese je těsně kontrolována na úrovni translace regulačními proteiny (iron regulatory proteins – IRPs), které se vážou na specifické sekvence v mRNA (iron regulatory elements – IREs). Dle umístění IRE v molekule mRNA je po vazbě IRP její translace buď zablokována (lokalizace IRE v 5’untranslated region – 5’UTR), nebo potencována (lokalizace IRE v 3’UTR). Nadbytek železa v buňce tak způsobí zvýšenou expre si genů pro feritin a útlum exprese genu pro TfR, při buněčné sideropenii je tomu naopak. Na celotělové úrovni jsou klíčovými procesy regulace homeostázy železa absorpce železa enterocyty a retence nebo uvolňování železa z buněk monocyto–makrofágového s stému. Enterocyty regulují vstup železa do organizmu zvýšením nebo snížením jeho absorpce ze střeva a ovlivňováním jeho transportu přes bazolaterální membránu do krve. Na tomto procesu participuje řada proteinů, které jsou dle potřeb organizmu zvýšeně nebo sníženě exprimovány. Podle tzv. preprogramovací teorie („preprogramming theory“) je tak v závislosti na stavu zásob železa v organizmu prekurzorová buňka enterálních krypt, ze které se diferencuje zralý enterocyt, předprogramována k zvýšenému nebo sníženému transportu železa ze střevního lumen do krve. Velmi důležitým faktorem těchto regulací je tzv. HFE protein, objevený v roce 1996 Federem a spolupracovníky. Nejčastější příčinou vrozeného přetížení organizmu železem je bodová mutace C282Y v molekule HFE proteinu. Homozygotní stav pro tuto mutaci lze detekovat u více než 80 % pacientů kavkazské populace, trpících hereditární hemochromatózou.
Klíčová slova:
železo – sideropenie – přetížení železem – hereditární hemochromatóza
Úvod
Železo je biogenní prvek, který se vyskytuje prakticky ve všech živých organizmech. Najdeme je v jednobuněčných organizmech (kvasinky, bakterie), v buňkách rostlin a v tělech bezobratlých i obratlovců až po savce. Je tomu tak proto, že atom železa je schopen velmi snadno vázat i uvolňovat elektron, a tak měnit své mocenství z dvojmocné ferroformy na trojmocnou feriformu a naopak. Železo se ze všech biogenních kovů vyskytuje v organizmu v nejvyšším množství, což obnáší asi 35 mg/kg u žen a 45 mg/kg umužů. Největší podíl celkového množství železa v organismu je obsažen v hemoglobinu (60–70 %), asi 10 % je součástí myoglobinu, cytochromů a jiných enzymů, asi 20–30 % tvoří zásobní pool v podobě vazby na feritiny, méně než 1 % je obsaženo v cirkulujícím poolu v krvi. Nejdůležitější biosloučeniny železa a jejich funkce jsou uvedeny v tab. 1.

Vedle své nejznámější funkce transportéru kyslíku v podobě hemoglobinu plní železo i řadu dalších vitálních funkcí – je nutné pro syntézu nukleových kyselin (DNA i RNA), syntézu řady proteinů, účastní se řízení buněčné proliferace a diferenciace a apoptózy, je nutné pro syntézu myelinu a formování dendritů neuronů, což se obráží v ovlivňování pochodů učení a paměti.
Pozornost řady vědeckých týmů je upřena na úlohu železa v procesech stárnutí tkání, neurodegenerace, maligního bujení, aterosklerózy a role železa v imunitním systému.
Vlastnost atomu železa snadno vázat a uvolňovat elektron může však mít i negativní dopady na živé buňky generací toxických radikálů (zvláště hydroxyradikály OH – a superoxidové radikály O2–), které mohou oxidací poškozovat proteiny, lipidy i nukleové kyseliny. Je proto vitálně důležité, aby homeostáza železa byla citlivě regulována.
Homeostáza železa a její regulace
Organizmy jemně regulují homeostázu železa jak na buněčné, tak i na vyšších úrovních. Bez této regulace by v případě nekontrolovaného přísunu železa do buněk i celého organizmu docházelo k poškozování důležitých biosloučenin s následným poškozením až smrtí buněk, v případě chybějící odpovědi na nedostatek železa k rozvoji deficitu železa. Buněčnou a systémovou regulaci metabolizmu železa nelze od sebe navzájem oddělit, jelikož tvoří integrovaný celek vzájemně se ovlivňujících pochodů, které začínáme podrobněji poznávat teprve v posledních několika letech. Železo je ubikvitárním biogenním prvkem a v eukaryotických buňkách (od kvasinek po savčí buňky) byla prokázána výrazná konzervace a homologie genů, řídících metabolizmus železa, což umožňuje studovat aspekty metabolizmu železa na jednobuněčných i vícebuněčných objektech.
Z celkového pohledu je pro lidský organizmus nejdůležitější regulace vstřebávání železa enterocyty, největší podíl na exkreci železa má exfoliace epitelií trávícího traktu, u žen ve fertilním věku se přidávají ztráty menstruačním krvácením. Malé množství železa se z organizmu ztrácí odlučováním buněk jiných sliznic, kožního epitelu a kožních adnex a močí.
Železo je v krevním proudu vázáno na bílkoviny. Z největší části jde o transportní protein transferin. Molekula transferinu váže dva atomy trojmocného železa Fe+++. Denně dochází k obratu asi 25 mg železa v krvi. Za normálních okolností je transferin saturován pouze asi ze třetiny své vazebné kapacity. U stavů vrozeného a/nebo získaného přetížení organizmu železem stoupá saturace transferinu na 45 % a více.
Molekula transferinu, obsahující železo, má vysokou afinitu k specializovaným receptorům, lokalizovaným na buněčných membránách s nejhustší expresí na buňkách hemopoézy. Byly již popsány 2 typy transferinových receptorů (TfR) – TfR a TfR2. Po vazbě transferinu na TfR je komplex transferin/TfR internalizován do cytoplazmy v podobě endozomu. Uvnitř endozomu jsou výrazně kyselým pH atomy železa disociovány z vazby na transferin a zároveň dochází k jejich redukci na dvojmocné železo Fe++, které je pak dále exportováno do cytoplazmy. Transferin se váže hlavně na molekuly TfR, funkce TfR2 je méně jasná, nově byla popsána vrozená dispozice k přetížení organizmu železem, související smutací v genu pro TfR2. Po uvolnění železa jsou molekuly TfR recyklovány k expresi na povrch cytoplazmatické membrány, kde slouží opět jako receptory pro transferin. Celý proces nazýváme buněčná endocytóza transferinového železa.
Regulace buněčné homeostázy železa
Regulace metabolizmu železa na buněčné úrovni je řízena několika geny na transkripční, posttranskripční a posttranslační úrovni. Je nutno zdůraznit, že jisté aspekty celulární homeostázy železa jsou společné všem buňkám organizmu, jiné mechanizmy jsou tkáňově nebo orgánově specifické – například exprese divalent metal transporter 1 (DMT1) enterocyty, homeostáza železa v monocyto-makrofágovém systému a hematopoetických tkáních, akumulace zásobního železa v hepatocytech apod. Prvou úrovní regulace homeostázy Fe na buněčné úrovni je ovlivňování influxu tohoto prvku do buňky, což se děje prostřednictvím regulace exprese TfR a TfR2.
Volné atomární železo v cytoplazmě buněk tvoří tzv. labilní pool nitrobuněčného železa a je potencionálně toxické. Buňky mohou tuto toxicitu eliminovat v podstatě třemi způsoby [4].
- Volné železo je rozdělováno do různých kompartmentů, kde je dále využíváno k syntéze důležitých proteinů, ovlivňujících získávání energie, detoxikaci volných radikálů, proliferaci, diferenciaci a řadu dalších celulárních funkcí. Velmi důležitými kompartmenty utilizace nitrobuněčného železa jsou například mitochondrie, kde probíhají reakce Krebsova cyklu se syntézou ATP, dále buněčné jádro, lysozomy s železo obsahujícími enzymy aj.
- Volné cytoplazmatické Fe je exportováno z buňky. Tento pochod je relativně dobře prozkoumán u enterocytů, existence mechanizmů exportu Fe se však předpokládá ve všech ostatních buňkách.
- Nevyužité nebo nevyloučené Fe je uloženo do depozit.
Zásobní železo je v eukaryotických buňkách uloženo ve formě feritinu. U většiny obratlovců je feritin tvořen dvěma typy podjednotek, označovaných jako L - (Light, Liver) a H - (Heavy, Heart) feritin. Tyto podjednotky se organizují do schránek, z nichž každá je složena ze čtyřiadvaceti molekul L - nebo H-feritinu. Feritinové schránky jsou schopné každá pojmout několik tisíc atomů Fe+++. Jednotlivé feritiny se od sebe liší zastoupením L - a H - podjednotek a jsou označovány jako izoferitiny. H-feritin je feroxidáza a jeho aktivita usnadňuje rychlou inkorporaci atomu Fe do feritinových komplexů a na druhé straně je toto železo rychleji z feritinu uvolňováno. Izoferitiny, bohaté na L-podjednotky, přijímají Fe pomaleji a déle jej skladují. Jednotlivé tkáně a orgány se liší zastoupením exprese a translace mRNA pro L – a H – podjednotky feritinu.
Další formou existence zásobního železa v buňce je jeho vazba na hemosiderin. Jde o makromolekulární komplex feritinu s lipidovými strukturami, přesné chemické složení není zatím objasněno. Hemosiderin se fyziologicky vyskytuje především v Kuppferových buňkách, při chronickém přetížení železem ho nacházíme i v buňkách postižených orgánů.
Nově se ukazuje, že železo, vázané na feritin, může existovat v různých fyzikálně chemických formách (ferihydrid a goethit) a že tyto formy mohou vykazovat různý stupeň toxicity železa pro buňky [27].
TfR a feritin představují dva hlavní proteiny regulace buněčné homeostázy železa. Exprese obou proteinů je regulována na transkripční a hlavně na posttranskripční úrovni.
Savčí buňky se liší od rostlinných a nižších eukaryotických buněk i regulací metabolizmu železa. Zatímco rostlinné a nižší eukaryotické buňky ovlivňují expresi regulátorů homeostázy Fe převážně na transkripční úrovni, savčí buňky využívají vedle regulace transkripce hojně i posttranskripční mechanizmy. Messenger RNA mnoha ferotropních proteinů obsahuje ve své molekule specifické sekvence, schopné vázat regulační molekuly, které takto mohou modulovat translaci těchto mRNA. Tyto molekuly nazýváme IRPs – Iron Regulatory Proteins a jejich specifické vazebné sekvence v mRNA označujeme jako IREs – Iron Regulatory Elements. IRPs jsou považovány za centrální regulátory metabolizmu železa u savců. Po vazbě IRPs na IRE je translace mRNA buď zablokována nebo potencována, což je dáno lokalizací IRE v molekule mRNA. Vazba IRP na IRE v 5’UTR (untranslated region) molekuly mRNA zablokuje následnou translaci, zatímco vazba na IRE, lokalizované v 3’UTR stabilizuje mRNA a potencuje tak její translaci.
V tab. 2 jsou mRNA, jejichž translace je prokazatelně nebo velmi pravděpodobně regulována IRPs [4].
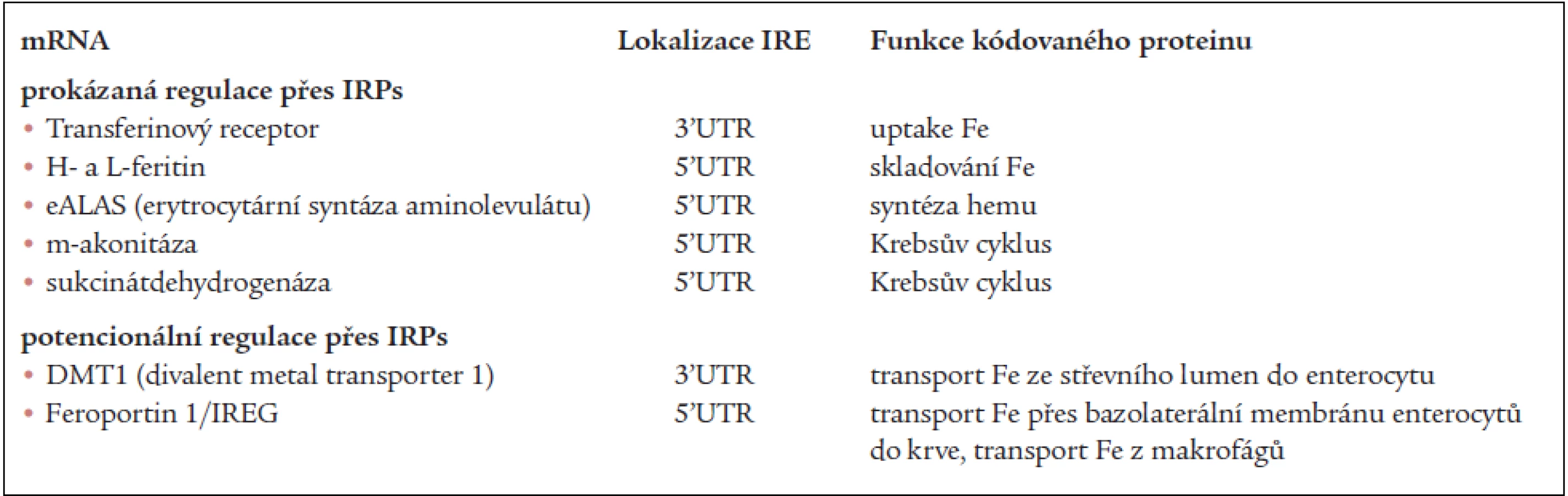
Přestože IRP regulují další osud mRNA, mohou faktory, ovlivňující transkripci genů regulovaného proteinu v mnoha případech převážit a konečná hladina produkce těchto proteinů je pak více výsledkem transkripce než posttranskripčních úprav (změna nebo „override“ IRP/IRE regulace).
Dobře prozkoumána je posttranskripční regulace exprese genů pro feritin a TfR. Molekula mRNA pro L - i H-feritin obsahuje 1 kopii specifické IRE sekvence v 5’UTR. Vazba IRP1 i IRP2 na IRE feritinové mRNA proto zablokuje její translaci, a tím snižuje konečnou expresi L - i H-řetězců feritinu. Na rozdíl od feritinu obsahuje molekula mRNA pro TfR pět kopií IRE, a to v 3’UTR. Vazba IRP1 a IPR2 na tyto IRE struktury snižuje degradaci cílové mRNA a nepřímo tak zvyšuje tak translaci [14]. Je zajímavé, že mRNA pro nově objevený TfR2 neobsahuje ve své molekule struktury blízké IRE a exprese genu pro TfR2 je tak regulována převážně na úrovni transkripce. Je zřejmé, že TfR a TfR2 hrají v organizmu rozdílné role v regulaci homeostázy železa.
Velmi zajímavý je mechanizmus regulace translace cílových mRNA přes IRP1 a IRP2 v souvislosti s koncentrací železa v labilním cytoplazmatickém poolu. IRP1 obsahuje ve své molekule úsek, kde je vázáno Fe a 4 atomy síry. Pokud jsou v této oblasti navázány tři atomy Fe („cluster“ 3Fe-4S), vykazuje molekula IRP1 vysokou afinitu k IRE. Při navázání dalšího atomu Fe (cluster 4Fe-4S) ztrácí IRP1 afinitu k IRE a nabývá aktivitu cytoplazmatické akonitázy (c–acon), která je schopna konvertovat citrát v izocitrát. Vidíme zde tedy změnu funkce IRP1 v závislosti na 3Fe-4Fe obsahu železa v Fe-S clusteru – jinými slovy modulace funkce molekuly tohoto regulačního proteinu ve smyslu IRP1 – c-akonitáza a obráceně [12]. Na rozdíl od IRP1, což je protein s dlouhým poločasem a jeho degradace a poločas nejsou příliš ovlivněny stavem intracelulárního železa, koncentrace Fe výrazně ovlivňuje stabilitu IRP2 [26]. IRP2 vykazuje asi 60% homologii s IRP1, tyto molekuly se výrazně liší unikátní sekvencí 73 aminokyselin, přítomnou v molekule IRP2. Tento motiv představuje vazebné místo pro Fe, které po navázání mění konformaci a/nebo oxidační stav molekuly IRP2 s její následnou urychlenou degradací [9]. Nadbytek železa tak způsobí rozdílnými mechanizmy nedostatek IRP1 i IRP2 pro vazbu na IRE s následnou zvýšenou transkripcí mRNA s IRE v 5’UTR (mRNA pro feritin) nebo degradací mRNA s IRE v 3’UTR (mRNA pro TfR). Vazebná afinita IRP1 a stabilita IRP2 je kromě koncentrace a buněčné lokalizace atomů Fe výrazně ovlivňována dalšími faktory – volnými radikály, NO, kyslíkem, peroxidy apod [20,21,30].
Enterocytové transportéry železa DMT1 (divalent metal transporter 1) a feroportin obsahují ve svých mRNA IRE, které jsou lokalizovány u DMT1 v 3’UTR a u feroportinu v 5’UTR. Při zvýšené koncentraci železa v labilním cytoplazmatickém poolu se proto translace DMT1 může snižovat a naopak translace feroportinu zvyšovat. V experimentech, zaměřených na regulaci exprese těchto transporterů, bylo však zjištěno, že vedle IRP je tato regulace nepochybně ovlivňována dalšími faktory – například železem ovlivňovanou transkripcí, transportem (traffic) nebo degradací těchto molekul [25].
Na posttranslační úrovni je funkce proteinů metabolizmu železa regulována např. fosforylací, změnou terciální struktury, ovlivněním jejich metabolizmu a/nebo lokalizace v buňce (cytoplazma, mitochondrie, endozomy, buněčná membrána aj). Pozoruhodným regulačním faktorem aktivity IRP je fosforylace těchto molekul. Fosforylace IRP například proteinkinázou C (PKC) nebo jinými kinázami obecně zvyšuje vazebnou aktivitu IRP k IREs. Fosforylace IRP1 navíc zvyšuje citlivost c–akonitázy k pertubacím v Fe–S clusteru, způsobeným kyslíkem, kyslíkovými radikály nebo NO [4].
Řada otázek a aspektů regulace homeostázy Fe není zatím zcela objasněna. Jde například o podíl IRP1 a IRP2 na posttranskripční regulaci exprese genů, mechanizmy transportu Fe mezi jednotlivými buněčnými kompartmenty, „crosstalk“ mezi jednotlivými regulačními systémy, export Fe z buněk apod. Lze shrnout, že řízení metabolizmu Fe na celulární i orgánové úrovni se uskutečňuje na mnoha stupních genové regulace, do níž zasahují vedle Fe i další faktory a celou tuto pestrou mozaiku teprve začínáme podrobněji poznávat.
Buněčnou a orgánovou regulaci homeostázy Fe nelze od sebe oddělit a je nutno je chápat jako celek.
Systémová regulace homeostázy železa
Na celotělové, systémové úrovni jsou citlivě regulovány vstřebávání, transport, utilizace a skladování železa. Jde o předávání signálů mezi hepatocyty, enterocyty, monocyto - makrofágovým systémem, erytronem, kosterními svaly i jinými kompartmenty, které má za následek jemné řízení homeostázy železa přesně dle potřeb tkání i celého organizmu. Klíčovými se v této souvislosti jeví regulace absorpce železa enterocyty a jeho transport přes bazolaterální membránu do krve a retence nebo uvolňování železa z elementů monocyto-makrofágového systému [19].
Vstřebávání železa
Zdrojem železa v potravě jsou hlavně živočišné bílkoviny, kde je nacházíme v podobě vázané na hem a jako nehemové železo. Hemové železo se vyskytuje v hemoglobinu a v myoglobinu svalstva. Vstřebávání hemového železa je pro organizmus nejsnadnější, řízení tohoto procesu však zůstává prozatím z velké části neobjasněno. Je prokázáno, že hem se z hemoglobinu a myoglobinu uvolňuje působení kyseliny solné a fermentů trávicích šťáv. Nehemové železo se v lumen gastrointestinálního traktu vyskytuje hlavně ve formě trojmocného Fe+++, na rozdíl od hemového železa je resorpce této formy železa ovlivněna přítomností řady faktorů, které mohou resorpci ovlivnit jak pozitivně, tak i negativně, takže tato může kolísat až 10násobně [10]. Hlavními faktory, usnadňujícími resorpci železa, jsou kyselina askorbová a bílkoviny masa, s resorpcí železa výrazněji interferují například vaječný bílek, kravské mléko a hlavně fytáty, obsažené v rostlinné dietě. Polyfenoly, obsažené v kávě, čaji a víně, rovněž inhibují vstřebávání železa [15].
Železo při své cestě ze střevního lumen do krve překračuje biomembrány enterocytu – apikální mebránu a bazolaterální membránu. Přes apikální membránu je regulován influx železa do cytoplazmy enterocytů, přes bazolaterální membránu je regulován vstup železa do krve.
Nejlépe je probádáno vstřebávání dvojmocného atomu železa, zprostředkované molekulou DMT1 (divalent metal transporter 1, dříve rovněž NRAMP2 nebo DCT1). Exprese DMT1 je regulována vlivem celotělových zásob železa, koncentrací železa v dietě nebo posttranslačně přes IRP1 a IRP2 [25]. Tento nosič ovšem vedle železa váže a transportuje přes buněčnou membránu enterocytu i další dvojmocné atomy kovů – například Zn++, Cu++, Mn++, Co++, Cd++, Ni++ a Pb++. Omechanizmech řízení absorpce těchto kovů a omožné interferenci se vstřebáváním železa například kompeticí je známo jen velmi málo.
DMT1 je transportní protein s 12 transmembránovými doménami a vykazuje homologii s Nramp (natural resistance-associated macrophage proteins) rodinou membránových proteinů. Vykazuje vysokou afinitu k dvojmocnému železu Fe++, nemá afinitu k Fe+++. Jde o ubikvitární protein se silnou expresí v apikálních membránách enterálních buněk kartáčového lemu duodena a v oblasti membrán časných endozomů, vytvářených při endocytóze transferinového železa. Lokalizace exprese DMT1 v membránách endozomů naznačuje jeho potenciální funkci liberátora Fe z endozomů do cytoplazmy. Funkce DMT1 byly zkoumány na zvířecích modelech i buněčných kulturách.
Transgenní exprese DMT1 v savčích buňkách vedla k akumulaci železa těmito buňkami, exprese mutované varianty Gly185Arg transport železa blokovala. Bodová mutace Gly185Arg je u myší a krys asociována s těžkou hypochromní mikrocytární anémií a zkrácenou dobou života postižených hlodavců. Podání železa parenterálně nevede k úpravě stavu, protože kromě těžké poruchy vstřebávání železa v duodenu vázne i transport Fe z endozomů do cytoplazmy erytroidních a pravděpodobně i nonerytroidních buněk. Je zajímavé, že existují dvě „splice“ varianty mRNA, jedna obsahuje IRE v 3’UTR, zatímco zkrácená forma DMT1 mRNA IRE neobsahuje, a proto její translace není ovlivňována přes IRPs. U pacientů s klasickou hereditární hemochromatózou byla popsána upregulace DMT1 mRNA, která obsahuje IRE [8].
Nově byl popsán protein, označovaný jako CYBRD1 nebo Dcytb, jde o ferireduktázu, exprimovanou na apikální membráně enterocytů (duodenální cytochrom b). Tento enzym redukuje trojmocné železo na dvojmocné, které se pak stává substrátem pro DMT1 [8,18].
Vstřebávání železa ve formě vázané v hemu představuje nejefektivnější cestu, mechanizmy tohoto procesu jsou však zatím neobjasněny.
Další možností je absorpce železa cestou mucin-mobilferin-integrin (tzv. paraferitinový komplex), tato problematika zatím není definitivněji dořešena [2].
K pochopení mechanizmů řízení absorpce železa je nutné zmínit aspekty proliferace a diferenciace enterocytů. Zralé enterocyty se vyskytují na povrchu klků enterální sliznice, diferencují se z prekurzorových buněk krypt, které jsou lokalizovány mezi klky. Při diferenciaci se buňky krypt postupně přesunují na vrcholy klků, kde nahrazují odlučované zralé enterocyty. Z pohledu regulace absorpce železa je důležité, že exprese několika genů, kódujících důležité proteiny systému vstřebávání a transportu železa, je odlišná u prekurzorové buňky a u zralého enterocytu. Labilní pool železa v enterocytech dále tuto expresi modifikuje. Prekurzorové buňky krypt exprimují molekuly TfR a mohou tak akumulovat železo z krve. V případě normální nebo zvýšené hladiny železa, vázaného na transferin, přechází do labilního cytoplazmatického poolu dostatečné množství železa a prekurzorové buňky jsou takto předprogramovány („primovány“) tak, aby vstřebávání a následný transport železa byly sníženy. Při sideropenii je tomu obráceně. V prekurzorových buňkách je rovněž exprimován tzv. HFE protein, který vazbou na TfR modifikuje transport transferinového železa do buňky. Přesná povaha této modifikace není jasná, porucha funkce HFE proteinu má však za následek dispozici k zvýšenému vstřebávání nehemového železa ze střevního lumen. Jelikož prekurzorové buňky krypt neexprimují molekuly DMT1, není množství železa v labilním poolu ovlivněno koncentrací nehemového železa v potravě a obráží tak převážně hladinu transferinového Fe. U zralých enterocytů dochází naproti tomu k útlumu exprese TfR a HFE proteinu a je zde dle potřeb organizmu řízeně zvyšována exprese genů pro DMT1, Dcytb, feroportin a hefestin s rezultující jemnou regulací vstřebávání železa a jeho transportu do krve.
Regulace influxu železa do organizmu
Bazolaterální membrána enterocytu představuje velmi pravděpodobně klíčovou strukturu regulace influxu železa do organizmu. Železo, které není přeneseno přes bazolaterální membránu, je vyloučeno exfoliací enterocytů.
Enterocyty hrají klíčovou roli v regulaci příjmu železa, a musí proto zpracovávat signály o aktuálním stavu zásob i potřeb tohoto biogenního prvku v jednotlivých tkáních i celotělově. Na hypotetické úrovni bylo postulováno několik regulátorů homeostázy železa, předávajících tyto signály enterocytům [25].
Regulátor zásob – „stores regulator“ monitoruje množství železa především v játrech, kosterních svalech a v krvi a při depleci tohoto prvku zvyšuje pomalou akumulaci nehemového železa, vstřebávání hemového železa není touto regulací výrazněji ovlivněno. Uvádí se, že stores regulator je schopen zvýšit akumulaci železa jen asi o 1 mg za den. Kandidátními molekulami signalizace stavu zásob železa jsou feritin, transferin a solubilní transferinové receptory.
Erytropoetický regulátor signalizuje zvýšenou erytropoetickou aktivitu a působí do jisté míry nezávisle na zásobním regulátoru, jelikož pacienti s anémií se zvýšenou erytropoetickou aktivitou mohou akumulovat železo i při normálních nebo dokonce zvýšených zásobách železa. Erytropoetický regulátor je výrazným faktorem řízení absorpce, neboť anemičtí pacienti mohou touto cestou zvýšit vstřebávání železa až na 40 mg za den.
Bylo prokázáno, že hypoxie rovněž zvyšuje akumulaci železa, není však jasné, zda působí nezávisle na erytropoetickém regulátoru. Zralé enterocyty exprimují v oblasti bazolaterální membrány proteiny feroportin a hefestin, které zde slouží transportu železa z enterocytů do krve.
Feroportin (Ireg1, MTP1)
Lokalizace exprese feroportinu svědčí pro jeho významnou roli v transportu železa přes biomembrány. Exprimují jej zralé enterocyty enterálních klků, a to v oblasti bazolaterální membrány, dále Kupfferovy buňky jaterních sinusoid, nižší exprese je prokázána v hepatocytech. Na bazolaterálních membránách placentálního trofoblastu byla rovněž detekována silná exprese Ireg1. Regulace exprese feroportinu probíhá jak na úrovni ovlivnění translace mRNA vazbou IRP1 a IRP2 na IRE v 5’UTR, tak i dalšími mechanizmy. Významnou se zdá regulace hladiny feroportinu erytropoetickým regulátorem, hladinou hemoglobinu a hypoxií. Další možností regulace funkce Ireg1 je ovlivnění cílové lokalizace exprese feroportinu, jelikož enterocyty myší s replecí železa vykazovaly vyšší podíl cytoplazmatické lokalizace exprese feroportinu než enterocyty zvířat s deplecí železa (převážně bazolaterální lokalizace exprese) [1]. Feroportin hraje rovněž důležitou roli v recyklaci železa, fagocytovaného v buňkách RES při odbourávání hemoglobinu ze zanikajících erytrocytů. Prvá popsaná mutace SLC11A3 genu, který kóduje feroportin, má za následek záměnu aminokyselin A77D s rezultující poruchou funkce feroportinu (nejpravděpodobněji „loss of function“) s fenotypickými projevy autozomálně dominantní non-HFE hemochromatózy s výrazným časným hromaděním železa v makrofázích. Jde velmi pravděpodobně o poruchu recyklace železa z makrofágů s následným sekundárním zvýšením jeho střevní rezorpce, pravděpodobně přes erytropoetický regulátor. Sideróza hepatocytů se rozvíjí na rozdíl od klasické HFE hemochromatózy až v pozdějších stadiích choroby. Další fenotypickou odchylku oproti klasické HFE hemochromatóze představuje snížená saturace transferinu železem a lehká hypochromní anémie [7,17]. Zajímavé a nevysvětlené zůstává, že přes poruchu funkce feroportinu je přenos železa přes bazolaterální membránu zvýšen.
Hefestin
Molekula hefestinu vykazuje signifikantní homologii s molekulou ceruloplazminu. Hefestin je exprimován zralými enterocyty, imunobarvením byla exprese hefestinu lokalizována perinukleárně a je postulována možnost recyklace hefestinu mezi cytoplazmatickým kompartmentem a bazolaterální membránou. Jelikož je hefestin exprimován vezikulárně, může hrát roli v blokování železa, určeného k extracelulárnímu transportu, od metabolizmu v cytoplazmě. Z hlediska enzymatické aktivity představuje hefestin feroxidázu, bohatou na atomy mědi. Hefestin v oblasti bazolaterální membrány oxiduje dvojmocné Fe++, transportované feroportinem, na Fe+++, které je pak schopno vazby na transferin. Deleční mutace několika exonů je odpovědná za fenotypické projevy u sla-myši.
Homozygotní samice a hemizygotní samci sla (sex linked anaemia) myši se rodí těžce anemičtí důsledkem defektního transplacentárního přenosu železa. Během života se anémie upravuje, i když postižení jedinci mají celoživotně velmi snížené zásoby železa. Soudí se, že postnatálně je funkci hefestinu schopen částečně zastoupit plazmatický protein ceruloplazmin, rovněž vykazující feroxidázovou aktivitu [25].
Ukazuje se, že měď hraje významnou roli jak v celotělovém, tak i v intracelulárním metabolizmu železa.
Hlavními orgány depozic zásobního železa jsou játra, slezina a kostní dřeň. O mechanizmech, jakými se železo dostává z buněk tkání a orgánů zpět do krve, je známo velmi málo. Tyto mechanizmy však zcela jistě existují, v opačném případě by byly buňky vystaveny neúměrně velkému riziku toxických účinků železa. Osudy železa se poněkud liší dle buněčného typu a tkáně. Nejdůležitějšími se jeví rozdíly mezi hepatocyty, erytroidními prekurzory a makrofágy. Pro eflux železa z hepatocytů je nutná přítomnost ceruloplazminu, hefestin zde nehraje důležitější roli. Aceruloplazminemie představuje vzácnou poruchu, vedoucí k akumulaci železa v hepatocytech, CNS a pankreatu, spolu s retencí železa v monocytomakrofágovém systému.Flebotomie je zde bez efektu pro poruchu efluxu železa z postižených buněk. Makrofágy jsou důležitým místem metabolizmu železa, které mohou získávat nejen v podobě endocytózy transferinu, ale hlavně fagocytózou erytrocytů. Makrofágy kostní dřeně, jater a sleziny představují stěžejní regulátory recyklace erytronového železa. Železo zde navíc slouží k obraně organizmu proti infekci. Odpovědí makrofágů a neutrofilů na stimulaci mikrobiálními produkty a nejrůznějšími cytokiny je tzv. oxidativní metabolické vzplanutí (oxidative burst) s produkcí řady toxických mikrobicidních radikálů. Export železa z monocyto–makrofágového systému je nutný k recyklaci železa pro další potřeby organizmu. Pro eflux železa z buněk RES je mimo jiné nutná neporušená funkce feroportinu. Nejčastější mutace v SLC11A3 genu 385–487del TTG má za následek deleci Val162del s fenotypickými projevy přetížení organizmu železem, které se nachází převážně v RES, parenchymatózní buňky jsou postihovány až v pozdějších stadiích tohoto typu hemochromatózy [22].
HFE protein
HFE gen byl popsán v roce 1996 Federem et al. Je lokalizován na 6. chromozomu v blízkosti genů hlavního histokompatibilního komplexu a kóduje transmembránový protein, složený ze 3 extracelulárních smyček (loops), transmembránové domény a krátké intracelulární domény, obsahující C-terminálový konec této bílkoviny. HFE protein je těsně asociován s molekulou β2-mikroglobulinu (β2m). Komplex HFE/β2m váže molekuly TfR na povrchu buněčných membrán a ovlivňuje tak influx transferinového železa do buněk. Mechanizmus modifikace endocytózy transferinového železa není jasný, mohlo by jít o vliv HFE na konformaci TfR s rezultující modifikovanou afinitou k ligandu [5], ale může se jednat i o ovlivnění recyklace TfR1 na povrch buněk a/nebo uvolňování atomů železa z transferinu [13,27]. V literatuře nacházíme zprávy postulující jak snížení, tak zvýšení endocytózy železa, vázaného na transferin. Zvýšená exprese normálního HFE proteinu v HeLa buňkách vedla ke snížení uptake transferinového železa, avšak v těchto experimentech byl HFE protein zvýšeně exprimován bez koexprese s β2-mikroglobulinem, což mohlo nefyziologicky modifikovat metabolizmus železa [11]. Waheed et al studovali ovlivnění influxu transferinového železa v CHO (Chinese hamster ovary) buňkách za podmínek koexprese HFE a β2-mikroglobulinu a zjistili zvýšení uptake železa, zatímco samostatná exprese pouze HFE vedla ke snížení vstřebávání transferinového železa do CHO buněk (!) [29].
Montosi et al nalezli u pacientů s HFE hemochromatózou snížený uptake transferinového železa makrofágy, stav se upravoval po transfekci buněk normálním HFE proteinem [52]. Ve světle těchto poznatků nabývá i „preprogramovací teorie“ prekurzorových buněk enterálních krypt logičtějších obrysů – porucha funkce HFE proteinu by pak vedla ke sníženému uptake transferinového železa prekurzorovými buňkami duodenálních krypt s rezultující sideropenií zralých enterocytů s následnou zvýšenou expresí transportního proteinu DMT1, vedoucí k zvýšenému vstřebávání železa z duodena. Zvýšená exprese DMT1 i feroportinu v duodenální sliznici pacientů s hereditární hemochromatózou byla opakovaně prokázána, duodenální tkáň HFE knock–out myší vykazuje snížený uptake transferinového železa a zvýšenou duodenální expresi DMT1 i feroportinu [23,28]. Je velmi zajímavé, že upregulace DMT1 i feroportinu je modifikována různě v různých genetických liniích myší, což jednak implikuje vliv dalších genů na fenotypické projevy hereditární hemochromatózy a dále částečně vysvětluje klinickou zkušenost o různé míře penetrace genotypu lidské hereditární hemochromatózy [3].
HFE protein velmi pravděpodobně není exprimován v hepatocytech, a proto uptake transferinového železa v těchto buňkách nepodléhá modifikaci tímto proteinem. Bylo prokázáno, že hepatocyty zvýšeně exprimují TfR2 a uptake železa by tak mohl probíhat prostřednictvím tohoto receptoru [27].
Nejčastější příčinou vrozeného přetížení organizmu železem je bodová mutace (single nucleotide polymorphism, SNP) G845A v HFE genu, zapříčiňující záměnu aminokyselin Cys282Tyr (C282Y) v molekule HFE proteinu (missense mutation). Tato záměna porušuje důležitou disulfidovou vazbu (disulphide bridge) v molekule HFE proteinu s následnou změnou konformace celé molekuly, zapříčiňující poruchu transportu mutovaného proteinu z endoplazmatického retikula a Golgiho komplexů s jeho následnou zvýšenou degradací a z toho rezultující sníženou expresí v cytoplazmatické membráně. Udává se i snížená asociace s β2-mikroglobulinem. Nejde však přitom o totální výpadek funkce HFE (null alela), ale o její snížení [6]. Kauzální roli mutací HFE genu v rozvoji HH prvého typu prokazují mimo jiné i pokusy na myších modelech. „HFE knockout“ myš a HFE C282Y myš vykazují zvýšenou koncentraci plazmatického železa, zvýšenou saturaci transferinu, vysokou hladinu sérového feritinu a rozvíjí se u nich obraz přetížení organizmu železem, který je velmi podobný manifestaci lidské hereditární hemochromatózy typu 1. U HFE „knockout“ myší je přitom obraz přetížení železem závažnější než u HFE C282Y zvířat, což dokazuje jistou reziduální funkci mutovaného C282Y HFE proteinu. Je rovněž velmi zajímavé, že u β2m „knockout“ myši byly prokázán zvýšený pool zásobního železa, implikující významnou roli asociace HFE proteinu s β2m pro normální funkci modifikace endocytózy transferinového železa [16,24,31].
Poruchy metabolizmu železa
Poruchy metabolizmu železa patří mezi nejčastější patologické stavy v klinické praxi. V zásadě je můžeme rozdělit na onemocnění, zapříčiněná absolutním nebo relativním nedostatkem tohoto prvku, na stavy související s přetížením organizmu železem a na poruchy utilizace železa. Jako platí v klinické medicíně obecně, mohou být tyto poruchy vrozené nebo získané, mnohdy však jde o kombinaci obou příčin. Tyto poruchy budou předmětem druhé části sdělení.
MUDr. Jan Novotný
www.fnbrno.cz
e-mail: novotnyj@fnbrno.cz
Doručeno do redakce: 27. 8. 2004
Přijato po recenzi: 26. 10. 2004
Sources
1. Abboud S, Haile DJ. A novel mammalian iron-regulated protein involved in intracellular iron metabolism. J Biol Chem 2000; 275 : 19906–19912.
2. Conrad ME, Umbreit JN. A concise review: iron absorption: the mucin-mobilferin - integrin pathway: a competitive pathway for metal absorption. Am J Hematol 1993; 42 : 67–73.
3. Dupic F, Fruchon S, Bensaid M et al. Inactivation of the hemochromatosis gene differentially regulates duodenal expression of iron-related mRNAs between mouse strains. Gastroenterology 2002; 122 : 745–751.
4. Eisenstein RS. Iron regulatory proteins and the molecular control of mammalian iron metabolism. Annu Rev Nutr 2000; 20 : 627–662.
5. Feder JN, Penny DM, Irrinki A et al. The hemochromatosis gene product complexes with the transferrin receptor and lowers its affinity for ligand binding. Proc Natl Acad Sci USA 1998; 95 : 1472–1477.
6. Feder JN, Tusuchihashi Z, Irrinki A et al. The hemochromatosis founder mutation in HLA-H disrupts beta2-microglobulin interaction and cell surface expression. J Biol Chem 1997; 272 : 14025–14028.
7. Fleming RE, Sly WS. Ferroportin mutation in autosomal dominant hemochromatosis: loss of function, gain in understanding. J Clin Invest 2001; 108 : 521–522.
8. Goswami T, Rolfs A, Hedinger MA. Iron transport: emerging roles in health and disease. Biochem Cell Biol 2002; 80 : 679–689.
9. Guo B, Phillips JD, Yu Y et al. Iron regulates the intracellular degradation of iron regulatory protein 2 by the proteasome. J Biol Chem 1995; 270 : 21645–21651.
10. Hallberg L, Rossander L. Absorption of iron from Western lunch and dinner meals. Am J Clin Nutr 1982; 35 : 502–509.
11. Harrison SA, Bacon BR. Hereditary hemochromatosis: update for 2003. J Hepatol 2003; 38(Suppl 1): S14–S23.
12. Hirling H, Henderson BR, Kuhn LC. Mutational analysis of the (4Fe–4S)–cluster converting iron regulatory factor from its RNA binding form to cytoplasmic aconitase. EMBO J 1994; 13 : 453–461.
13. Ikuta K, Fujimoto Y, Suzuki Y. et al. Overexpression of HFE alters transferrin recycling process in human hepatoma cells. Biochim Biophys Acta 2000; 1496 : 221–231.
14. Klausner RD, Rouault TA, Harford JB. Regulating the fate of mRNA: the control of cellular iron metabolism. Cell 1993; 72 : 19–28.
15. Lee GR, Herbert V. Nutritional factors in the production and function of erythrocytes. In: Wintrobe’s Clinical Hematology. 10th ed. Philadelphia: Lippincott, Williams and Wilkins 1999 : 228–266.
16. Levy JE, Montross LK, Cohen DE et al. The C282Ymutation causing hereditary hemochromatosis does not produce a null allele. Blood 1999; 94 : 9–11.
17. Montosi G, Donovan A, Totaro A et al. Autosomal dominant hemochromatosis is associated with a mutation in the ferroportin (SLC11A3) gene. J Clin Invest 2001; 108 : 619–623.
18. Muckenthaler M, Roy CN, Custodio AO et al. Regulatory defects in liver and intestine implicate abnormal hepcidin and Cybrd1 expression in mouse hermochromatosis. Nature Genetics 2003; 34 : 102–107.
19. Nicolas G, Viatte L, Lou DQ et al. Constitutive hepcidin expression prevents iron overload in a mouse model of hemochromatosis. Nature Genetics 2003; 34 : 97–101.
20. Pantopoulos K, Weiss G, Hentze M. Nitric oxide and oxidative stress (H2O2) control mammalian iron metabolism by different pathways. Mol Cell Biol 1996; 16 : 3781–3788.
21. Recalcati S, Taramelli D, Conte D et al. Nitric oxide–mediated induction of ferritin synthesis in J774 macrophages by inflammatory cytokines: role of selective iron regulatory protein-2 downregulation. Blood 1998; 91 : 1059–1066.
22. Roetto A, Merryweather–Clarke AT, Daraio F et al. A valine deletion of ferroportin 1: a common mutation in hemochromatosis type 4. Blood 2002; 100 : 733–734.
23. Rolfs A, Bonkovsky HL, Kohlroser JG et al. Intestinal expression of genes involved in iron absorption in humans. Am J Physiol 2002; 282: G598–G607.
24. Rothenberg BE, Voland JR. Β2m knockout mice develop parenchymal iron overload: a putative role of
class I genes of the major histocompatibility complex in iron metabolism. Proc Natl Acad Sci USA 1996; 93 : 1529–1534.
25. Roy CN, Enns CA. Iron homeostasis: new tales from the crypt. Blood 2000; 96 : 4020–4027.
26. Samamiengo F, Chin J, Uwai K et al. Molecular characterization of a second iron responsive element binding protein, iron regulatory protein 2. J Biol Chem 1994; 269 : 30904–30910.
27. Trinder D, Macey DJ, Olynyk JK. The new iron age (review). Int J Molec Med 2000; 6 : 607–612.
28. Trinder D, Olynyk JK, Sly WS et al. Iron uptake from plasma transferrin by the duodenum is impaired in the Hfe knockout mouse. Proc Natl Acad Sci USA 2002; 99 : 5622–5626.
29. Waheed A, Grubb JH, Zhou XY et al. Regulation of transferrin-mediated iron uptake by HFE, the protein
defective in hereditary hemochromatosis. Proc Natl Acad Sci USA 2002; 99 : 3117–3122.
30. Weiss G, Goossen B, Doppler W et al. Translational regulation via iron-responsive elements by the nitric oxide/NO-synthase pathway. EMBO J 1993; 12 : 3651–3657.
31. Zhou XY, Tomatsu S, Fleming RL et al. HFE gene knockout produces mouse model of hereditary hemochromatosis. Proc Natl Acad Sci USA 1998; 95 : 2492–2497.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2005 Issue 6
Most read in this issue
- Achalázia pažeráka
- Natriuretický peptid typu B (BNP) – použitelnost v diferenciální diagnostice dušnosti
- Doporučený postup diagnostiky a terapie esenciální trombocytemie a trombocytemie provázející jiné myeloproliferativní choroby
- Poruchy metabolizmu železa I. Regulace homeostázy železa