
Pozdní komplikace chronických zánětů respiračního traktu u nemocných s běžnou variabilní imunodeficiencí
Late complications of chronic respiratory infections in patients with common variable immunodeficiency
Common variable immunodeficiency (CVID) is the most frequent serious humoral deficiency manifested in adulthood, and is due to acute and chronic respiratory infections which lead to respiratory failure. Retrospective analyses of 28 CVID patients were made. Mean age at time of diagnosis was 38.6 ± 18.6 years. The development of the first symptoms to the time of diagnosis was delayed 14.1 ± 10.2 years, three times longer than in other studies. Twenty-three (82.1 %) of the patients had respiratory complications. Chronic obstructive pulmonary diseases, n = 16 (57.1 %), and bronchiectasis, n = 10 (35.7 %), were the most frequent types of lung damage. In addition, two patients (7.1 %) had evidence of an interstitial lung process. The morbidity associated with CVID may be reduced by early diagnosis and adequate dosage of immunoglobulins to minimalize the occurrence and progression of lung damage.
Key words:
bronchiectasis - interstitial lung - fibrosis - antibody deficiency - common variable immunodeficiency - lung damage
Authors:
O. Kopecký 1; Š. Lukešová 1; V. Koblížek 3; V. Vroblová 1; J. Novotný 2
Authors‘ workplace:
Ústav klinické imunologie a alergologie Lékařské fakulty UK a FN Hradec Králové, přednosta MUDr. Otakar Kopecký, CSc.
1; Radiologická klinika Lékařské fakulty UK a FN Hradec Králové, přednosta prof. MUDr. Pavel Eliáš, CSc.
2; Plicní klinika Lékařské fakulty UK a FN Hradec Králové, přednosta doc. MUDr. František Salajka, CSc.
3
Published in:
Vnitř Lék 2006; 52(11): 1021-1029
Category:
Original Contributions
Overview
Běžná variabilní imunodeficience (Common Variable Immunodeficiency - CVID) je nejčastější závažnou imunodeficiencí manifestující se v dospělosti akutními a chronickými respiračními infekty, které vedou u většiny nemocných k respiračnímu selhání. Byla provedena retrospektivní analýza nálezů u 28 nemocných s CVID. Průměrný věk byl 38,6 ± 18,6 roků. Doba, která uplynula od prvních příznaků do stanovení diagnózy, byla 3krát delší. než je uváděna v podobných studiích, 14,1 ± 10,2 roků. U 23 (82,1 %) pacientů bylo diagnostikováno chronické plicní onemocnění, u 16 (57,1 %) nemocných byla diagnostikována chronická obstrukční plicní nemoc, 10 (35,7 %) nemocných mělo prokázáno bronchiektázie. U 2 nemocných (7,1 %) byla diagnostikována idiopatická intersticiální pneumonie. Studie řady autorů dokládají, že incidenci nevratných orgánových změn, které se rozvíjejí u nemocných s CVID, je možné snížit včasnou diagnózou, dostatečně dávkovanou substituční léčbou intravenózními gamaglobuliny a racionální antibiotickou léčbou.
Klíčová slova:
bronchiektázie - intersticiální plicní proces - protilátková deficience - běžná variabilní imunodeficience - poškození plic
Úvod
Běžná variabilní imunodeficience (CVID) je heterogenní chorobná jednotka neznámé etiologie. Podle údajů evropského a panamerického registru je prevalence této nejčastější, klinicky závažné imunodeficience, odhadována na 1 případ na 25 000-50 000 obyvatel. CVID je považována za vrozené onemocnění s pozdní klinickou manifestací, nejčastěji mezi 20. - 30. rokem života, vzácně před dovršením věku 15 let. Klinické i laboratorní nálezy u nemocných s CVID jsou pestré. Většina pacientů s CVID trpí na časté a závažné bakteriální, převážně respirační infekty. Nejčastějším vyvolavatelem bakteriálních infekcí horních i dolních dýchacích cest je Streptococcus pneumoniae, Haemophilus influenzae a Moraxella catarrhalis [12]. Pokud nejsou dominujícím symptomem závažné infekce, může být diagnóza protilátkové deficience nesnadná. CVID se může manifestovat jako autoimunitní cytopenie nebo revmatoidní artritida. Přibližně třetina nemocných trpí na chronické průjmy vyvolané Giardia lamblia. Není-li protilátková deficience provázena dysfunkcí T-lymfocytárních funkcí, je antivirová imunitní odezva zachována a u většiny nemocných s CVID není pozorována zvýšená náchylnost k virovým infekcím. Výjimku představují infekce vyvolané enteroviry, ECHO viry a cytomegalovirem [13,28]. V laboratorních nálezech je pozorován pokles sérových hladin IgG, IgA a nepravidelně IgM. Pacienti s CVID nejsou schopni odpovídat na antigenní podněty, chybí specifická protilátková odpověď na řadu bakteriálních antigenů. U části nemocných jsou prokazovány poruchy funkce T-lymfocytů [6,29]. Ačkoliv byl první případ hypogamaglobulinemie, která nebyla vázána na X-chromozom, popsána před více než 50 lety, její etiopatogeneze je objasněna pouze u malé části případů. U nemocných s CVID jsou popisovány poruchy funkce jak T - a B-lymfocytů, tak poruchy funkce monocytů, makrofágů [25]. Počty B-lymfocytů v periferní krvi jsou sníženy nepravidelně. U některých nemocných je pozorována porucha diferenciace B-lymfocytů po antigenní stimulaci, snížen je počet paměťových B-lymfocytů. Naivní zralé B-lymfocyty se diferencují v buňky nesoucí povrchovou molekulu CD27 (molekula náležící do rodiny receptorů pro TNF), ale proces je zablokován ve fázi izotypového přepnutí syntézy z IgM na IgG, IgA, IgE. B-lymfocyty takto postižených jedinců nesou fenotyp CD19+/CD27+/IgM+ (IgD+). Imunofenotypová analýza se stala základem nové klasifikace nemocných s CVID. Práce některých autorů poukazují na korelaci mezi Freiburgskou klasifikací (Warnatz) a klinickou manifestací CVID.
U nemocných s CVID byly popsány mutace 4 genů kódujících molekuly, které zajišťují přenos signálů mezi buňkami imunitního systému. Mutace genu kódujícího kostimulační molekulu ICOS byla popsána jako první. Molekula ICOS je vyjádřena na aktivovaných T-lymfocytech, které produkují IL-10. Ligandem je molekula ICOS-L na B-lymfocytu. Tato kostimulační vazba je nezbytným předpokladem diferenciace zralého B-lymfocytu v paměťovou buňku. ICOS molekula je kódována 3 geny nacházejícími se na 2. chromozomu. Vrozená homozygotní delece ICOS genu je příkladem jednobodové mutace zodpovědné za CVID. Mutace vede k zablokování pozdní diferenciace B-lymfocytů a poruše tvorby IL-10, který je důležitý pro izotypové přepnutí syntézy z IgM na IgA. Až dosud bylo popsáno 9 případů CVID s mutací genu ICOS [8,11].
Následně byly u pacientů s CVID popsány mutace dalších genů, TNFRSF13C kódující BAFF-R (B-cell activity factor of the TNF family-receptor) a TNFRSF13B kódující TACI (Transmembrane Activator Cyclophilin ligand Interactor). Zatímco BAFF-R je vazebným místem pouze pro BAFF, TACI molekula je ligandem jak pro BAFF, tak pro APRIL (A proliferation-inducing ligand). Uvedené molekuly náleží do rodiny molekul TNF (Tumor Necrosis Factor – tumor-nekrotizující faktor) a jejich receptorů [19,22]. Obě molekuly se nachází na membránách B-lymfocytů. Důsledkem mutace je zablokování vazby s ligandy a porucha funkce. Neprobíhá izotypové přepnutí z IgM syntézy na produkci IgG, IgA a IgE. Proces somatické hypermutace je nedostatečný, nebo neprobíhá vůbec. Zablokována je diferenciace naivních B-lymfocytů v paměťové B-lymfocyty. Až dosud bylo popsáno pouze několik desítek případů CVID s prokázanými mutacemi genů kódujícími BAFF-R a TACI [19,21,22]. Obě uvedené molekuly zajišťují mezibuněčné signály, které jsou společně s interakcí CD40 - CD154 nezbytné pro izotypové přepnutí.
Čtvrtou popsanou mutací u 3. nemocných s CVID je mutace CD19 molekuly. Tato molekula náleží do rodiny Ig molekul, nachází se na B-lymfocytech od časných vývojových stadií a její exprese zaniká s přechodem v plazmatickou buňku. Ve folikulární zóně germinálních center lymfatických uzlin je redukována proliferace a nedochází k somatické hypermutaci. Nejsou produkovány vysoce afinní protilátky [26,30].
Nemocní s opakovanými akutními a chronickými záněty dýchacích cest jsou vystaveni vysokému riziku vzniku a progrese nevratného poškození plic (bronchiektázie, CHOPN, intersticiální plicní procesy). U malé části nemocných s opakovanými záněty dýchacích cest k těmto změnám nedochází. Diskutovány jsou další faktory vedoucí k plicnímu poškození, jako např. kouření, expozice toxickým látkám, koincidence jiných nemocí. U více než 1/3 nově diagnostikovaných pacientů s CVID jsou přítomny známky poškození plic. Plicní změny jsou pozorovány i u nemocných, kteří netrpí na opakované respirační infekce. Nemocní s CVID umírají nejčastěji na respirační selhání na podkladě CHOPN. Druhou nejčastější příčinou úmrtí u nemocných s CVID jsou nádorová onemocnění.
Za rozhodující považuje většina autorů včasné stanovení diagnózy protilátkové imunodeficience a zahájení odpovídající léčby. Základem léčby nemocných s CVID je substituce intravenózními imunoglobuliny (IVIG) a antibiotická léčba v profylaktické nebo kauzální indikaci [17,23].
Cílem naší práce je upozornit na primární imunodeficienci CVID, která je pro pozdní manifestace a pestrost klinických projevů v řadě případů diagnostikována až při známkách orgánového poškození.
Soubor nemocných a metody
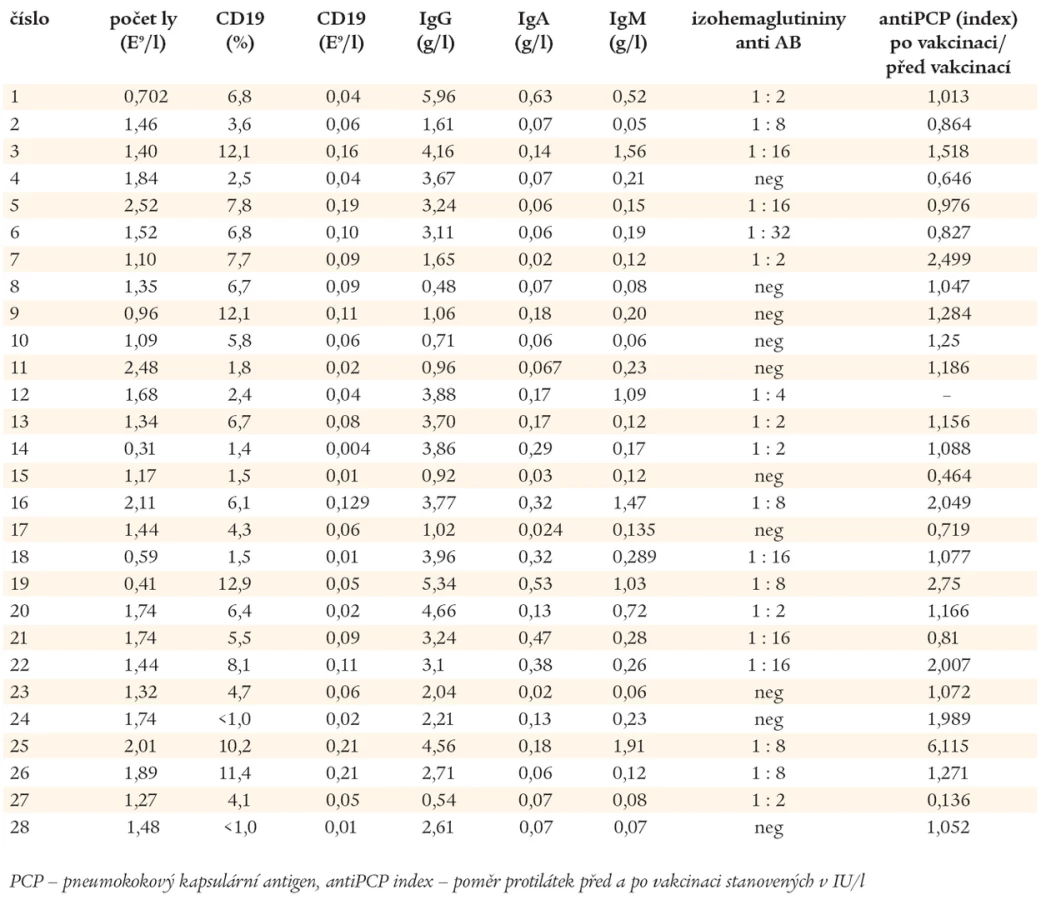
CVID byla diagnostikována u 28 nemocných, 16 žen a 12 mužů, léčených na našem pracovišti v letech 1993-2005. Porovnáním souboru žen a mužů nebyly zjištěny statisticky významné rozdíly ve věku výskytu CVID ani prodlevě stanovení diagnózy CVID od prvních příznaků. Za první klinický příznak protilátkové imunodeficience byl považován údaj o závažné infekci (pneumonii, chronické purulentní sinusitidě a otitidě v dospělém věku, bakteriální meningitidě, parazitární a bakteriální enteritidě a autoimunitní projevy). Všichni nemocní měli imunonefelometricky stanoveny hladiny sérových imunoglobulinů s využitím přístroje a diagnostik od firmy Beckman. Titry přirozených protilátek proti izohemaglutininům byly vyšetřeny před zahájením substituční léčby IVIG. Žádný z pacientů neměl krevní skupinu AB. Počty T a B-lymfocytů a jejich subpopulací byly stanoveny pomocí víceparametrové analýzy na průtokovém cytometru Cytomics 500FC (Beckman Coulter). V rámci první návštěvy na naší ambulanci bylo provedeno RTG plic a spirometrické vyšetření. Reverzibilita bronchiální obstrukce byla ověřena bronchodilatačním testem (400 µg salbutamolu přes spacer). Test byl považován za pozitivní při nárůstu FEV1 o 200 ml a více nebo zvýšení FEV1 o 12 % a více. V následujících 6 měsících od stanovení diagnózy CVID bylo provedeno HRCT plic (High Resolution Computed Tomography). U 8 nemocných, v období bez známek akutního infektu, s patologickým nálezem HRCT plic nebo závažnou ventilační poruchou, byla vyšetřena plicní difuze. Všichni nemocní splňovali diagnostická kritéria CVID Evropské společnosti pro imunodeficience - ESID (www.esid.com).
Výsledky
Laboratorní nálezy, o které se opírá diagnóza CVID, jsou uvedeny v tab. 1. U 26 nemocných s CVID dominovaly před stanovením diagnózy chronické a závažné infekce horních a dolních dýchacích cest. Mimoplicní manifestaci měli 4 nemocní (v 1 případě lambliová enteritida, v 1 případě kloubní záněty připomínající revmatoidní artritidu, v 1 případě purulentní meningitida a v 1 případě autoimunitní cytopenie). Pomocí imunofenotypové analýzy byly stanoveny počty nezralých CD19+/CD21-, zralých - naivních CD19+/CD27- a paměťových CD19+/CD27+ B-lymfocytů. Podle exprese IgM (IgD) byly paměťové B-lymfocyty rozděleny do 2 subpopulací, na paměťové B-lymfocyty, které neprošly fází izotypového přepnutí syntézy CD19+/CD27+/IgM+ (IgD+) a na B-lymfocyty CD19+/CD27+/IgM- (IgD-), které produkují imunoglobuliny ve třídách IgG, IgA a IgE. Na základě těchto stanovení byli pacienti zařazeni do 3 skupin, do skupiny Ia 8 nemocných, do skupiny Ib 10 nemocných a do skupiny II 10 nemocných (Freiburgská klasifikace) [27].
Výsledky spirometrického vyšetření (metodou křivka průtok - objem) jsou uvedeny v tab. 2. Na základě FVC (usilovné výdechové vitální kapacity) a FEV1 (usilovného výdechového objemu za 1 vteřinu) byli nemocní rozděleni na skupiny s obstrukční (O), restrikční (R) a smíšenou (S) ventilační poruchou. Závažnost patologického nálezu má stupně I - III v závislosti na poklesu sledovaných parametrů vztažených k náležitým hodnotám daných pohlavím, věkem a hmotností pacienta (graf 1).


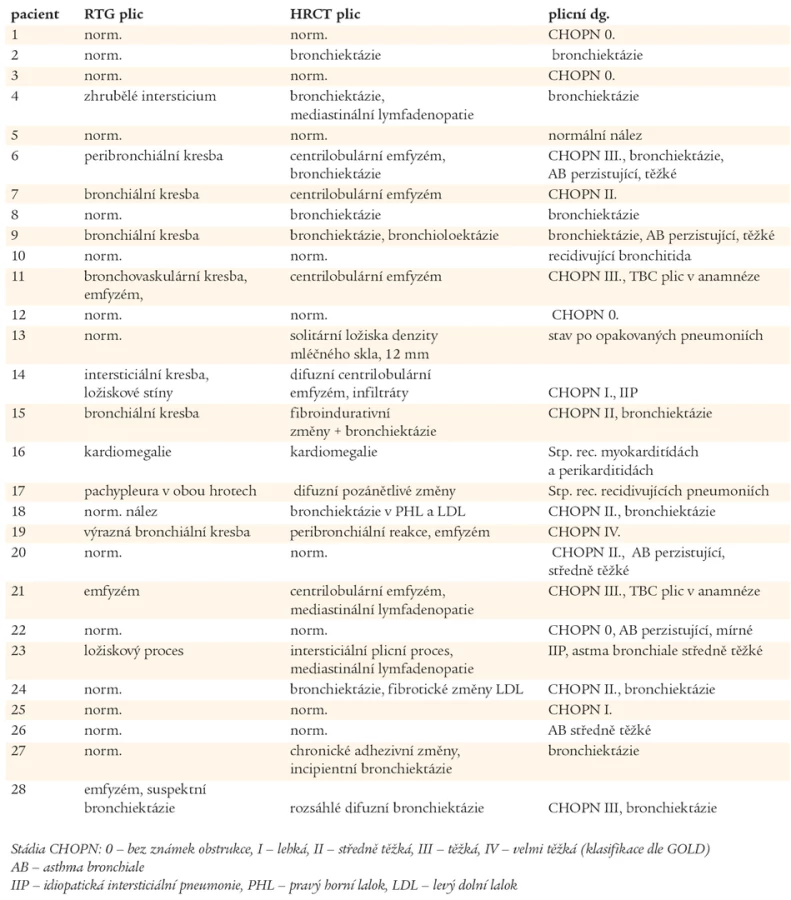
Výsledky zobrazovacích vyšetřovacích metod plic (RTG, HRCT) jsou uvedeny v tab. 3. U 23 nemocných s CVID bylo diagnostikováno chronické plicní onemocnění. CHOPN byla diagnostikována celkem u 16 nemocných, v 8 případech jako jediné plicní postižení. U 4 nemocných s CHOPN byly prokázány bronchiektázie. Ve 3 případech bylo po podání 400 µg salbutamolu dosaženo více než 12% zlepšení náležitých ventilačních hodnot, které však nedosáhlo fyziologického rozmezí hodnot náležitých, navíc, u jednoho z těchto pacientů byly HRCT vyšetřením plic odhaleny bronchiektázie. U jednoho nemocného s CHOPN byl diagnostikován intersticiální plicní proces. Bronchiektázie byly popsány u dalších 6 nemocných, 4krát u pacientů s normálními RTG nálezy plic a spirometrickými vyšetřeními. Kombinace patologických plicních nálezů jsou uvedeny v grafu 2. Z 8 vyšetření plicní difuze byla jednou zjištěna těžká (pacient č. 28) a jednou středně těžká porucha (pacient č. 14). U dvou nemocných s nálezy budícími podezření na intersticiální proces byla provedena transparietální biopsie s nálezem idiopatické intersticiální pneumonie (IIP - Idiopatic Intersticial Pneumonia), pacient č. 14 a 24. Pouze 5 nemocných mělo normální spirometrický nález a HRCT plic. Porovnáním plicích nálezů ve skupinách Ia, Ib a II nebyly zjištěny statisticky významné rozdíly.

Diskuse
Do klinického obrazu protilátkové imunodeficience náleží opakované a závažné infekce horních a dolních dýchacích cest, které ve svém důsledku vedou k nevratným plicním změnám. Chronické plicní změny jsou pozorovány i u nemocných bez známek akutních respiračních zánětů. U části nemocných jsou prvními příznaky autoimunitní projevy, např. idiopatická trombocytopenická purpura, hemolytická anémie nebo revmatoidní artritida. Poměrně časté jsou lambliové a bakteriální enteritidy, zvláště v oblastech s nižší hygienickou úrovní [13]. Svojí četností a spektrem byl výskyt mimoplicních projevů u našich nemocných s CVID podobný nálezům uváděným v evropských studiích [12,24,29].
Chronické plicní onemocnění bylo v našem souboru diagnostikováno u 23 z 28 (82,1 %) nemocných s CVID. V souboru 95 nově diagnostikovaných nemocných s CVID nalezla Kainulainen plicní komplikace pouze u 17 % nově diagnostikovaných pacientů. V této finské studii bylo stanovení diagnózy CVID opožděno v průměru o 8 roků od prvních příznaků, u našich nemocných to bylo o 14,1 roků. Nálezy jsou ale neporovnatelné, protože nemocní ve finské studii měli provedené pouze RTG plic a spirometrii [12]. Vyšetření RTG plic je pro malou citlivost nedostatečné, k odhalení počínajících intersticiálních změn a brochiektázií je nutné doplnit HRCT plic. Hodnota MMEF 75-25 je citlivým parametrem periferní bronchiální obstrukce a její pokles svědčí pro postižení bronchiolů. Thickett nalezl v souboru 47 nemocných s CVID pomocí vyšetření ventilačních plicních funkcí (FVC a FEV1) a HRCT plic ve 42 případech (89,4 %) chronické plicní změny [24]. Spektrem vyšetření a způsobem jejich hodnocení je tato studie podobná naší práci. Liší se však spektrem pacientů, protože do hodnoceného souboru byli zařazeni pouze nemocní, kteří trpěli na opakovanými respiračními infekty [24]. V našem souboru byli 2 nemocní s autoimunitními projevy jako dominujícími příznaky, 1 nemocný trpěl opakovanými průjmy vyvolanými Giardia lamblia a 1 nemocný prodělal bakteriální meningitidu.
U nemocných s CVID jsou častým nálezem difuzní intersticiální procesy plic označované jako idiopatické intersticiální pneumonie (IIP). V plicním intersticiu a oblasti malých bronchiolů se nachází jak zánětová reakce, tak zmnožení fibrotické tkáně různého stupně podle typu poruchy. Prognóza intersticiálních procesů je závažná, průměrná délka přežití je uváděna 3-6 roků od stanovení diagnózy. U nemocných s protilátkovou imunodeficiencí je nejčastěji diagnostikována idiopatická plicní fibróza (IPF) náležející do IIP. Do obrazu IIP je dále zahrnuta Lymphocytic Intersticial Pneumonia (LIP), obliterující bronchiolitida s pneumonickou reakcí (BOOP - Bronchiolitis Obliterans Organizing Pneumonia), nespecifická intersticiální pneumonie (NSIP - Nonspecific Interstitial Pneumonia) a autoimunitní intersticiální pneumonie (AIP - Autoimmune Interstitial Pneumonia) a „běžná“ intersticiální pneumonie (UIP - Usual Intersticial Pneumonitis). IIP byla v našem souboru diagnostikována u 2 nemocných, histologicky byly nálezy hodnoceny jako LIP. Kombinace kortikoidů a IVIG vedla v obou případech ke stabilizaci rentgenologických a spirometrických nálezů [2,10,20].
Přes závažnost klinických projevů se stanovení diagnózy CVID opožďuje vůči prvních příznakům až o několik roků. Údaje o zpoždění diagnózy se v jednotlivých studiích liší a pohybují se od 3,5 roků do 11 let. Za jednu z příčin opožděného stanovení diagnózy je považován všeobecně rozšířený omyl, že vrozené imunodeficience se manifestují v ranném dětství. V případě CVID a selektivní IgA deficience (sIgA) tato představa neplatí. CVID se manifestuje nejčastěji v období 2. a 3. decenia a sIgA je možné diagnostikovat až v období končící puberty [9]. Na diagnostické prodlevě se podílí i přístup specialisty, který bez hledání širších souvislostí řeší chronické záněty horních a dolních dýchacích cest, recidivující otitidy a průjmy opakovaným podáváním antibiotik. V neposlední řadě mají na prodlevu ve stanovení diagnózy CVID vliv úroveň a dostupnost zdravotní péče. Tyto rozdíly jsou patrny nejen mezi vyspělým a rozvojovým světem, ale i v rámci evropských zemích. Zavedení nových diagnostických a hlavně léčebných možností vedlo jak ke zlepšení kvality života, tak k prodloužení průměrné délky života nemocných s CVID. Ta byla uváděna ještě v 70. letech 20. století u mužů 28,8 roků, a u žen 55,4 roků [4,5,24]. V jiné studii byla CVID diagnostikována u mužů ve 33 letech a u žen ve 46,9 letech. Prodleva ve stanovení diagnózy u mužů činila 3 roky a u žen 7 roků. V tomto souboru 248 nemocných s CVID zemřelo v následujících 7 letech 57 nemocných, muži v průměrném věku 40 roků a ženy v průměrném věku 45,5 roků [6]. Další studie zabývající se otázkou věkových rozdílů v úmrtí a agresivitě onemocnění ve vztahu k pohlaví chybí. Za hlavní příčinu je považováno vyšší zastoupení kuřáků v populaci mužů a častější expozice toxickým látkám v pracovním prostředí u mužů a v neposledním řadě i méně zodpovědný přístup mužů ke zdravotním obtížím v porovnání s ženami.
Protilátková deficience je příčinou nedostatečné imunologické eliminace antigenních podnětů. Neúčinnou eliminací patogenních i podmíněně patogenních mikroorganizmů dochází k chronické stimulaci cytotoxických a fagocytujících buněk mikrobiálními antigeny a následnému uvolňování zánětových mediátorů. Doložena je zvýšená aktivita prozánětových mediátorů náležících do TNF rodiny [1]. Imunitní reakce má na sliznicích respiračního a gastrointestinálního traktu charakter nežádoucí poškozující reakce. V subepiteliální vrstvě dýchacích cest a v plicním intersticiu se hromadí lymfoidní buňky a makrofágy, prostřednictvím uvolňovaných cytokinů jsou aktivovány fibroblasty. Ve slizniční vrstvě gastrointestinálního traktu dochází ke zmnožení lymfatické tkáně pod obrazem nodulární lymfadenopatie. Dalším dokladem neúčinné eliminace patogenů a protrahované stimulace imunitního systému je nález splenomegalie, mediastinální a abdominální lymfadenopatie. Mediastinální lymfadenopatie se vyskytovala u naších 3 nemocných (pacienti č. 4, 21, 23) společně s abdominální lymfadenopatií a splenomegalií (nádorová a mykobakteriální etiologie nebyly prokázány). V průběhu léčby IVIG došlo k ústupu lymfadenopatie, přetrvávala splenomegalie.
Základem léčby je pravidelná substituce IVIG. Snížení frekvence akutních infekcí po substituční léčbě imunoglobuliny u nemocných s agamaglobulinémií popsal již Bruton. Přímý efekt IVIG je prokázán v řadě dalších studií. U nemocných s CVID vede podávání IVIG v dávce 205 - 372 mg/kg/měsíc k signifikantnímu snížení respiračních infektů, a to jak u nemocných s CHOPN, tak u nemocných bez CHOPN. K dosažení srovnatelného efektu nemocní s CHOPN vyžadují vyšší dávky IVIG [7]. Výsledky studie porovnávající účinnost dávek 200 mg/kg/měsíc a 600 mg/kg/měsíc ukázaly, že dávka 600 mg/kg/měsíc u imunodeficientních pacientů s CHOPN zpomaluje progresi poklesu ventilačních i respiračních parametrů, a to i přesto, že nedošlo k signifikantnímu snížení počtu akutních infekcí. Je prokázáno, že pravidelné podávání IVIG vede k poklesu spotřeby antibiotik a snižuje počet hospitalizačních dnů u nemocných s CVID [3]. Až u poloviny nemocných s bronchiektáziemi je však pozorována progrese změn bez závislosti na dávkování IVIG [14].
Z pestrosti klinických i laboratorních nálezů je zřejmé, že se v případě CVID jedná o několik chorobných jednotek, které jsou diagnostikovány až v okamžiku plně rozvinuté protilátkové imunodeficience. Poruchy v buněčných interakcích a cestách aktivace T - a B-lymfocytů a dendritických buněk, které mají základ v nově popsaných mutacích genů pro ICOS, BAFF, TACI a CD19 molekuly, jsou prokazovány u méně než 5 % nemocných s CVID. Nadto chybí reprezentativní počet nemocných ke zhodnocení souvislostí mezi popsanými mutacemi a klinickou manifestací. Neznáme ani odpověď na otázku, proč se vrozená porucha imunity typu CVID manifestuje převážně až v dospělosti, ojediněle i v seniu. V klinické manifestaci CVID se pravděpodobně uplatňuje více faktorů včetně zevních vlivů. Prognóza CVID je v jednotlivých případech obtížně odhadnutelná. Jsou nemocní s hodnotami sérového IgG pod 1,0 g/l bez klinických projevů imunodeficience. Naopak nemocní se sérovými hodnotami celkových Ig nad 5g/l, kteří mají poruchu tvorby specifických protilátek, mohou trpět závažnými, život ohrožujícími infekcemi [15]. Zařazení pacientů s CVID do skupin podle klasifikace založené na stanovení počtu naivních a paměťových B-lymfocytů v periferní krvi, vykazuje korelaci s některými klinickými nálezy, např. autoimunitními chorobami, splenomegalií, lymfadenopatií a granulomatózními procesy. Naopak u akutních infekčních projevů a jejich komplikací nebyly korelace prokázány. Dá se však předpokládat, že nemocní ze skupiny I jsou více ohroženi, protože diferenciace zralých B-lymfocytů v paměťové je u těchto nemocných zablokována. S touto poruchou je spojován závažnější defekt specifické protilátkové odezvy na proteinové i lipopolysacharidové antigeny, sérové hladiny IgG a IGA jsou u pacientů zařazených do I. skupiny nižší [15,16]. Dosud publikované výsledky bude nutné ještě ověřit na početnějších skupinách nemocných.
Závěr
Primární protilátková deficience typu CVID je onemocněním vyžadujícím celoživotní léčbu od stanovení diagnózy. Přes všechna nová poznání prognóza nemocných s CVID, tj. délka a kvalita života, závisí na včasné diagnóze, správné léčbě infekcí a oddálení pozdních komplikací. Proto je třeba věnovat pozornost nemocným s opakovanými respiračními záněty a na antibiotickou léčbu špatně reagujícími infekčními projevy. Neodpovídá-li klinický obraz laboratorním nálezům, je třeba spektrum testů rozšířit o vyšetření imunoglobulinových podtříd v séru, případně o průkaz tvorby specifických protilátek a s odstupem času vyšetření opakovat. Jsou známy případy selektivní IgA deficience nebo deficience IgG podtříd, které se vyvinuly v CVID [18,29].
Poděkování patří paní H. Kotlandové a J. Hrdinové za pomoc s grafickým zpracováním.
MUDr. Otakar Kopecký, CSc.
Ústav klinické imunologie a alergologie LF UK a FN Hradec Králové
www.lfhk.cuni.cz/UKIA
e-mail: kopecky.otakar@fnhk.cz
Doručeno do redakce: 28. 11. 2005
Přijato po recenzi: 29. 6. 2006
Sources
1. Aukrust P, Lien E, Kristoffersen AK et al. Persistent activation of the tumor necrosis factor system in a subgroup of patients with common variable immunodeficiency - possible immunologic and clinical consequences. Blood 1996; 87 : 674-681.
2. Bjoraker JA, Ryu JH, Edwin MK et al. Prognostic significance of histopathologic subsets in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 1998; 157 : 199-203.
3. Busse PJ, Razvi S, Cunningham-Rundles C. Efficacy of intravenous immunoglobulin in the prevention of pneumonia in patients with common variable immunodeficiency. J Allergy Clin Immunol 2002; 109 : 1001-1004.
4. Cadranel J, Bouvry D, Wislez M. Respiratory manifestations of common variable immunodeficiency in adults. Rev Mal Respir 2003; 20 : 126-133.
5. Cunningham-Rundles C. Clinical and immunologic analyses of 103 patients with common variable immune deficiency. J Clin Immunol 1989; 9 : 22-33.
6. Cunningham-Rundles C, Bodian C. Common variable immunodeficiency: clinical and immunological features of 248 patients. Clin Immun 1999; 92 : 34-48.
7. de Gracia J, Vendrell M, Álvarez A. Immunoglobulin therapy to control lung damage in patients with common variable immunodeficiency. International Immunopharmacol 2004; 4 : 745-753.
8. Grimbacher B, Hutloff A, Schlesier M et al. Homozygous loss of ICOS is associated with adult-onset common variable immunodeficiency. Nat Immunol 2003; 4 : 261-268
9. Hammarström L 2nd, Vorechovsky I, Webster D. Selective IgA deficiency (sIgAD) and common variable immunodeficiency (CVID). Clin Exp Immunol 2000; 120 : 225-231.
10. Homolka J. Idiopathic pulmonary fibrosis: adverse prognosis in artificial respiration patients. Pneumologie 2002; 56 : 161-162.
11. Hutloff A, Bücher K, Reiter K et al. Involvement of inducible costimulator in the exaggerated memory B cell and plasma cell generation in systemic lupus erythematosus. Arthritis Rheum 2004; 50 : 3211-3220.
12. Kainulainen L, Nikoskelainen J, Ruuskanen O. Diagnostic findings in 95 finnish patients with common variable immunodeficiency. J Clin Immunol 2001; 21 : 145-149.
13. Kainulainen L, Nikoskelainen J, Vuorinen T et al. Viruses and bacteria in bronchial samples from patients with primary hypogammaglobulinemia. Am J Respir Crit Care Med 1999; 159 : 1199-1204.
14. Kainulainen L, Varpula M, Liippo K et al. Pulmonary abnormalities in patients with primary hypogammaglobulinemia. J Allergy Clin Immunol 1999; 104 : 1031-1036.
15. Ko J, Radigan L, Cunningham-Rundles C. Switched memory B cells in common variable immunodeficiency. J Allergy Clin Immunol 2004; 113 (Suppl 1): S38.
16. Kokron CM, Errante PR, Barros MT et al. Clinical and laboratory aspects of common variable immunodeficiency. An Acad Bras Cienc 2004; 76 : 707-726.
17. Lamari F, Karamanos NK, Papadopoulou-Alataki E et al. Monitoring of two intravenous immunoglobulin preparations for immunoglobulin G subclasses and specific antibodies to bacterial surface antigens and relation with their levels in treated immunodeficient patients. J Pharmaceut Biomed Analysis 2000; 22 : 1029-1036.
18. Litzman J, Burianova M, Thon V et al. Progression of selective IgA deficiency to common variable immunodeficiency in a 16 year old boy. Allergol Immunopathol (Madr) 1996; 24 : 174-176.
19. Losi CG, Silini A, Fiorini C et al. Mutational analysis of human BAFF receptor TNFRSF13C (BAFF-R) in patients with common variable immunodeficiency. J Clin Immunol 2005; 25 : 496-502.
20. Popa V, Colby TV, Reich SB. Pulmonary interstitial disease in Ig deficiency. Chest 2002; 122 : 1594-1603.
21. Salzer U, Chapel HM, Webster ADB et al. Mutations in TNFRSF13B encoding TACI are associated with common variable immunodeficiency in humans. Nature Gen 2005; 37 : 820-828.
22. Salzer U, Grimbacher B. TACItly changing tunes: farewell to a yin and yang of BAFF receptor and TACI in humoral immunity? New genetic defects in common variable immunodeficiency. Curr Opin Allergy Clin Immunol 2005; 5 : 496-503.
23. Sewell WA, Buckland M, Jolles SR. Therapeutic strategies in common variable immunodeficiency. Drugs 2003; 63 : 1359-1371.
24. Thickett KM, Kumararatne DS, Banerjee AK et al. Common variable immune deficiency: respiratory manifestations, pulmonary function and high-resolution CT scan findings. QJM 2002; 95 : 655-662.
25. Thon V, Eggenbauer H, Wolf HM et al. Antigen presentation by common variable immunodeficiency (CVID) B cells and monocytes is unimpaired. Clin Exp Immunol 1997; 108 : 1-8.
26. Wang Y, Carter RH CD19 regulates B cell maturation, proliferation, and positive selection in the FDC zone of murine splenic germinal centers. Immunity 2005; 22 : 749-761.
27. Warnatz K, Denz A, Dräger R et al. Severe deficiency of switched memory B cells (CD27+IgM-IgD-) in subgroups of patients with common variable immunodeficiency: a new approach to classify a heterogeneous disease. Blood 2002; 99 : 1544-1551.
28. Webster AD Virus infections in primary immunodeficiency. J Clin Pathol 1994; 47 : 965-967.
29. Webster DB Common variable immunodeficiency. Immunol Allergy Clin North Am 2001; 21 : 1-22.
30. Weiler CR, Bankers-Fulbright JL Common variable immunodeficiency: test indications and interpretations. Mayo Clin Proc 2005; 80 : 1187-1200.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2006 Issue 11
Most read in this issue
- Mužská hormonální antikoncepce
- Transfúziou navodená imunomodulácia a infekčné komplikácie
- Mitrální regurgitace: umíme správně načasovat chirurgické řešení?
- Masivní plicní embolizace – pokus o embolektomii po selhání trombolytické léčby