
Rituximab (MabThera®) - nový biologický lék v terapii revmatoidní artritidy
Rituximab (MabThera®) – a new biological medicine in rheumatoid arthritis therapy
Rheumatoid arthritis (RA) is a serious, chronic, inflammatory disorder that damages the joints. The chronic destructive process causes pain to patients with RA and leads to the development of permanent disability. At present, great emphasis is placed on timely and effective therapy for RA, which is able to halt or slow the development of the disorder. At present we do not have any means of curing RA, the main objective for treatment is to induce remission of the disorder and prevent structural damage to the joints and the development of permanent disability. The relatively frequent failure of disease modifying medications (DMARDs) lead to efforts to find new resources for the treatment of RA. So called biological medicines were recently introduced into therapeutic use. These were mainly TNFα blockers. Experience has shown that approximately a third of patients with RA do not respond even to treatment with such medicines. Rituximab (MabThera®), a monoclonal antibody against CD20 positive B-lymphocytes, is a new biological medicine approved for RA therapy. It represents a new hope for patients with active RA, for whom earlier therapy with TNFα blockers has failed.
Key words:
rheumatoid arthritis – rituximab – therapies
Authors:
P. Němec
Authors‘ workplace:
Revmatologická ambulance II. interní kliniky Lékařské fakulty MU a FN u sv. Anny Brno, přednosta doc. MUDr. Miroslav Souček, CSc.
Published in:
Vnitř Lék 2007; 53(11): 1199-1210
Category:
Reviews
Overview
Revmatoidní artritida (RA) je závažné chronické zánětlivé onemocnění poškozující klouby. Chronický destruktivní proces přináší pacientům s RA bolest a vede k vývoji dlouhodobé disability. Velký důraz je v současné době kladen na včasnou a efektivní terapii RA, která by byla schopna zastavit, případně zpomalit vývoj onemocnění. V době, kdy nemáme k dispozici prostředky k vyléčení RA, je hlavním léčebným cílem navození remise choroby a zabránění strukturálnímu poškození kloubů a rozvoji dlouhodobé disability. Relativně časté selhání klasických chorobu modifikujících léků (DMARD) vedlo ke snaze najít nové prostředky k léčbě RA. Do terapie byly proto v nedávné době zavedeny tzv. biologické léky. Jednalo se zejména o preparáty blokující TNFα. Praxe ukázala, že přibližně jedna třetina pacientů s RA však neodpovídá ani na léčbu těmito léky. Rituximab (MabThera®), monoklonální protilátka proti CD20 pozitivním B-lymfocytům, je nový biologický lék schválený k terapii RA. Představuje novou naději pro pacienty s aktivní RA, u kterých selhala předchozí terapie léky blokujícími TNFα.
Klíčová slova:
revmatoidní artritida - rituximab - terapie
Úvod
Revmatoidní artritida (RA) je chronické zánětlivé onemocnění postihující přibližně 1 % dospělých jedinců s častějším výskytem u žen v poměru 2-4 : 1 [1]. Chronický zánět synoviální tkáně kloubního pouzdra vede k destrukci kloubní chrupavky, vzniku erozí subchondrální kosti a v konečné fázi k vývoji deformit postižených kloubů. Tento proces přináší pacientům s RA bolest a dříve či později vede k vývoji disability [2]. Disabilita bývá po 10 letech vývoje onemocnění přítomna přibližně u poloviny pacientů s RA [3]. Onemocnění rovněž zkracuje délku života pacientů s RA přibližně o 3-18 roků [4].
Etiologie RA není doposud známá. Předpokládá se však, že onemocnění vzniká u geneticky predisponovaných jedinců působením doposud neznámých faktorů (artritogenních agens), které vyvolají abnormální odpověď imunitního systému. Byla identifikována řada negativních prognostických faktorů vývoje RA. Patří k nim průkaz revmatoidního faktoru (RF) IgM a IgA [5-7], pozitivita anti-CCP protilátek [8,9], zvýšené hodnoty reaktantů akutní fáze [10,11], časný vývoj kloubních erozí a vyšší počet oteklých a bolestivých kloubů [6,11].
Velký důraz je v současné době kladen na včasnou a efektivní terapii RA, která by byla schopna zastavit, případně zpomalit vývoj onemocnění [3]. V době, kdy nemáme k dispozici prostředky k vyléčení RA, je hlavním léčebným cílem navození remise choroby a zabránění strukturálnímu poškození kloubů a rozvoji dlouhodobé disability [12,13]. American College of Rheumatology (ACR) i European League Against Rheumatism (EULAR) publikovali svá doporučení k terapii RA [13,14]. Česká doporučení k terapii RA, vytvořená Českou revmatologickou společností (ČRS), byla publikována v roce 1999 [15]. V roce 2002 byla doplněna o doporučení k terapii RA léky blokujícími TNFα [16]. V současné době jsou publikována nová doporučení ČRS týkající se účinnosti a monitorování bezpečnosti léčby RA.
Symptomatická terapie RA nesteroidními antiflogistiky umožňuje pouze kontrolu symptomů, ale nezabrání progresi onemocnění. Převážná většina pacientů s RA je proto léčena tzv. chorobu modifikujícími léky (disease modifying antirheumatic drugs - DMARDs), schopnými kontrolovat progresi onemocnění. Celosvětově nejužívanějším DMARDs je metotrexát. Metotrexát je často nasazován jako lék první volby zejména u pacientů s negativními prognostickými faktory vývoje onemocnění. Je lékem schopným zpomalit RTG progresi RA [17]. Ačkoliv monoterapie metotrexátem potlačuje aktivitu RA, kompletní remise choroby je dosaženo vzácně. Navíc může být terapie metotrexátem i jinými tzv. klasickými DMARDs spojena s výskytem nežádoucích příhod, které bývají často příčinou předčasného ukončení léčby [18]. Ostatní DMARDs, zejména leflunomid, sulfasalazin a antimalarika, jsou používána k terapii RA buď samostatně nebo v kombinaci s metotrexátem. To, že klasická DMARDs neúčinkují u všech pacientů, mají opožděný nástup účinku a jejich použití může být spojeno s výskytem nežádoucích příhod, vedlo ke snaze najít nové prostředky k terapii RA.
V posledních letech jsme proto svědky vývoje tzv. biologické léčby RA lépe cílené na patogenetický proces choroby. Ačkoliv v současné době neznáme příčinu vzniku RA, máme k dispozici řadu poznatků týkající se patogeneze choroby. Blokáda vzájemné interakce a aktivace imunokompetentních buněk, zejména T-lymfocytů a makrofágů, inhibice produkce cytokinů a inhibice aktivace různých efektorových buněk jsou hlavním cílem biologické terapie RA. Prvními biologickými léky schválenými k terapii RA byly látky blokující TNFα. Účinnost TNFα inhibitorů byla ověřena ve studiích na pacientech s RA nedostatečně odpovídajících na terapii klasickými DMARD, ale i na pacientech s časnou RA doposud neléčených [19-25]. Léčba TNFα inhibitory se ukázala být velmi účinná v potlačení symptomů, aktivity onemocnění a zpomalení a prevenci vzniku strukturálního postižení. Použití TNFα inhibitorů, většinou v kombinaci s metotrexátem, se stalo standardním postupem u pacientů, u kterých selhala předchozí terapie DMARDs. Přes vysokou účinnost těchto léků však zhruba třetina pacientů s RA na léčbu nereaguje. Nejméně 30 % pacientů nedosahuje alespoň 50% zlepšení skóre American College of Rheumatology (ACR) používaného v klinických studiích k hodnocení aktivity onemocnění [26]. Navíc se ukázalo, že léčba blokující TNFα může být spojena se zvýšeným výskytem závažných infekcí (zejména tuberkulózy), zhoršení příznaků chronického srdečního selhání a výskytem demyelinizačních onemocnění. Doposud nevyřešena zůstává otázka zvýšeného výskytu lymfomů a některých solidních tumorů (plíce, kůže, střevo) u pacientů s RA léčených TNFα inhibitory [27]. Dalšími biologickými léky schválenými k terapii RA, avšak doposud ne v České republice, jsou antagonista interleukinu-1 anakinra a abatacept, zasahující do vzájemné interakce T-lymfocytů a makrofágů [28-30].
Dlouho se předpokládalo, že v patogenezi RA mají výsadní postavení T-lymfocyty. Recentní výzkum však prokázal významnou roli B-lymfocytů. B-lymfocyty jsou totiž hojně přítomny v synoviální tekutině kloubů postižených RA. V patogenezi RA byly identifikovány 3 klíčové role B-lymfocytů: role antigen prezentující buňky, produkce autoprotilátek a produkce cytokinů. B-lymfocyty produkující RF jsou schopny vázat na svém povrchu různé antigeny, které jsou součástí imunitních komplexů a prezentovat je T-lymfocytům, a vést tak k jejich aktivaci. B-buňkami aktivované T-lymfocyty prostřednictvím prozánětlivých cytokinů, mimo jiné TNFα, aktivují makrofágy, které rovněž produkují prozánětlivé cytokiny. Tento proces vede k chronickému zánětu a následné destrukci kloubů. B-lymfocyty produkují autoprotilátky, např. RF. Imunitní komplexy, které se formují v synoviální tkáni, mohou aktivovat komplement, vázat se na makrofágy a aktivovat je. Aktivované B-lymfocyty jsou rovněž schopny produkovat prozánětlivé cytokiny, jako je TNFα, IL-6 a lymfotoxin α. Zejména produkce lymfotoxinu α vede k organizaci B-buněk a tvorbě lymfoidních struktur v synoviální tkáni. Vytváří se tak zárodečná centra, podobná zárodečným centrům B-lymfocytů v lymfatických uzlinách. V těchto centrech jsou produkovány četné autoprotilátky tvořící imunitní komplexy, které mohou výše uvedeným mechanizmem podporovat zánětlivý proces.
V roce 2007 byl v České republice schválen k terapii RA nový biologický lék rituximab (MabThera®). Jedná se o chimerickou monoklonální protilátku proti CD20 pozitivním B-lymfocytům. Rituximab byl poprvé schválen v roce 1997 v USA a celosvětově v roce 1998 k léčbě nonhodgkinských lymfomů (NHL). Doposud bylo tímto preparátem léčeno více než 730 000 pacientů s tímto onemocněním.
Struktura rituximabu
Rituximab je chimerická lidská/myší monoklonální protilátka specifická pro povrchový antigen CD20 B-lymfocytů vytvořená metodami genetického inženýrství [31]. Je tvořena humánní IgG1 konstantní oblastí a variabilní oblastí izolovanou s myších anti-CD20 protilátek (obr. 1). Molekula rituximabu se skládá ze 2 těžkých řetězců, každý je tvořen 451 aminokyselinami, a dvou lehkých řetězců s 231 aminokyselinami. Přibližná molekulová hmotnost je 145 kDa. Vazebná afinita k CD20 antigenu je přibližně 8,0 nM [31]. Antigenní specificita vůči CD20 antigenu je tedy vázána na myší komponentu, zatím co zbytek molekuly lidského původu zodpovídá za aktivaci komplementu a aktivaci imunokompetentních buněk. CD20 je povrchová molekula exprimovaná pouze na B-lymfocytech (zejména pre-B-lymfocytech a zralých B-lymfocytech) během procesu jejich dozrávání (obr. 2). CD20 není přítomna na kmenových buňkách ani na pro-B-lymfocytech a plazmatických buňkách, což znamená, že po podání rituximabu nedochází k poškození kmenových buněk a populace B-lymfocytů může být obnovována a rovněž nedochází k zástavě produkce sérových imunoglobulinů plazmatickými buňkami.

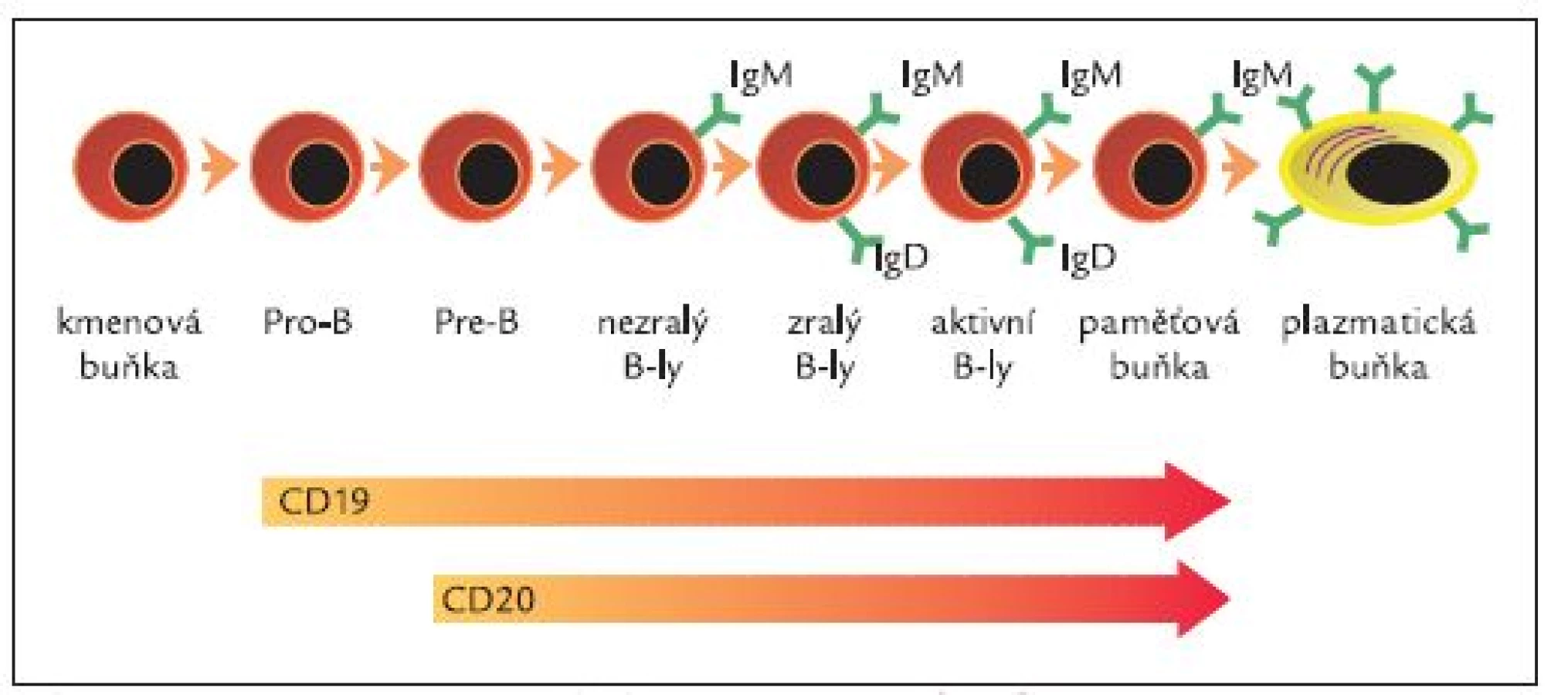
Mechanizmus účinku rituximabu
Rituximab se selektivně váže na povrchový antigen CD20 pouze těch B-lymfocytů, které tuto molekulu na svém povrchu exprimují. Neváže se tedy na kmenové buňky, pro-B-lymfocyty a plazmatické buňky. Vede k odstranění periferních B-lymfocytů z cirkulace 3 následujícími mechanizmy: komplementem zprostředkovanou lýzou B-lymfocytů, buňkami zprostředkovanou cytotoxicitou a indukcí apopotózy. Mechanizmus aktivace komplementu je umožněn vazbou C1q složky komplementu a jeho aktivací. Buňkami zprostředkována cytotoxicita je umožněna vazbou rituximabu na Fc receptory efektorových buněk, jako jsou makrofágy a NK-buňky, které spouštějí na protilátkách závislou buněčnou cytotoxicitu (ADCC) [32]. Bylo prokázáno, že rituximab je schopen spouštět programovanou buněčnou smrt B-lymfocytů in vitro [32].
Farmakokinetika rituximabu
Farmakokinetická data byla získána během klinického zkoušení fáze IIa, randomizované, dvojitě zaslepené studie se 161 pacienty s RA, kteří nedostatečně reagovali na terapii předešlými DMARD a u kterých bylo přítomno aktivní onemocnění navzdory pokračující terapii metotrexátem [33]. V této studii byl pacientům podáván buď pouze rituximab, nebo kombinace rituximabu s cyklofosfamidem, nebo rituximabu s metotrexátem a nebo metotrexát v monoterapii. Všem pacientům byl navíc před každou infuzí rituximabu aplikován intravenózně metylprednisolon a pacientům byl podáván prednison perorálně po dobu 2 týdnů. Vzorky séra k farmakokinetické analýze byly odebrány 1., 3., 15. a 17. den a v týdnech 4., 8., 16. a 24. po aplikaci rituximabu. Farmakokinetické parametry se významně nelišily, byl-li rituximab podáván monoterapeuticky nebo v kombinaci s cyklofosfamidem nebo metotrexátem. Ačkoliv clearence rituximabu a jeho distribuční objem nebyl ovlivněn konkomitantně podávanými DMARD, byly tyto parametry ovlivněny pohlavím a velikostí plochy tělesného povrchu. Muži měli rychlejší clearence a větší distribuční objem ve srovnání se ženami. Proporcionálně s rostoucí plochou tělesného povrchu narůstala clearence a distribuční objem rituximabu [34].
Farmakodynamika rituximabu
Během klinického zkoušení fáze IIa bylo po 2 aplikacích rituximabu 1 000 mg zaznamenáno rychlé a téměř úplné vymizení B-lymfocytů (měřeno úrovní povrchového antigenu CD19) z periferní krve [35]. U většiny pacientů docházelo k obnově B-buněčné populace po 6 a více měsících od podání rituximabu. U malého procenta pacientů byla zaznamenána protrahovaná deplece B-lymfocytů a nízký počet periferních B-lymfocytů byl přítomen i 2 roky po podání rituximabu [36]. Nebyla přítomna korelace mezi deplecí B-lymfocytů a klinickým efektem léčby ani korelace mezi obnovením B buněčné populace v cirkulaci a reaktivací RA. V dalším klinickém hodnocení fáze IIb (Dose-ranging Assessment: iNternational Clinical Evaluation of Rituximab in RA - DANCER) byl prokázán srovnatelný efekt dvou rozdílných dávkovacích schémat rituximabu (2krát 500 mg a 2krát 1 000 mg) na depleci periferních B-lymfocytů během 24 týdnů trvání studie [37,38]. Hladina imunoglobulinů zůstávala po celou dobu studie v normě. Podobná farmakodynamická data byla získána i ve studii fáze III (Randomised Evaluation oF Long-term Efficacy of rituXimab in rheumatoid arthritis - REFLEX) [39].
V Edwardsově studii byl rovněž sledován vliv podání rituximabu na produkci RF. Byl zaznamenán rychlý a výrazný pokles produkce RF, přetrvávající pod dobu 24 týdnů [35]. Naopak po podání metotrexátu byl pokles RF nevýrazný a pouze přechodný a mohl být způsoben současným podáním glukokortikoidů. Ve studii DANCER bylo podání 2 infuzí rituximabu v den 1 a 15 spojeno s poklesem zánětlivé aktivity měřené sérovou hladinou C-reaktivního proteinu (CRP). Pokles CRP oproti výchozím hodnotám byl výraznější po terapii rituximabem než po podání placeba (1,7 mg/dl vs 0,1 mg/dl) [37,38].
Účinnost rituximabu v terapii RA
První, rozsahem malé, otevřené studie hodnotící klinický efekt rituximabu v terapii RA, potvrdily hypotézu, že terapie cílená na B-buňky vede ke snížení aktivity choroby u pacientů s RA, u kterých selhala léčba několika DMARD [40-42]. Léčebné režimy v těchto studiích vycházely z léčebných režimů rituximab - CHOP (Cyklofosfamid, Doxorubicin, Vinkristin, Prednison) používaných v terapii NHL [43].
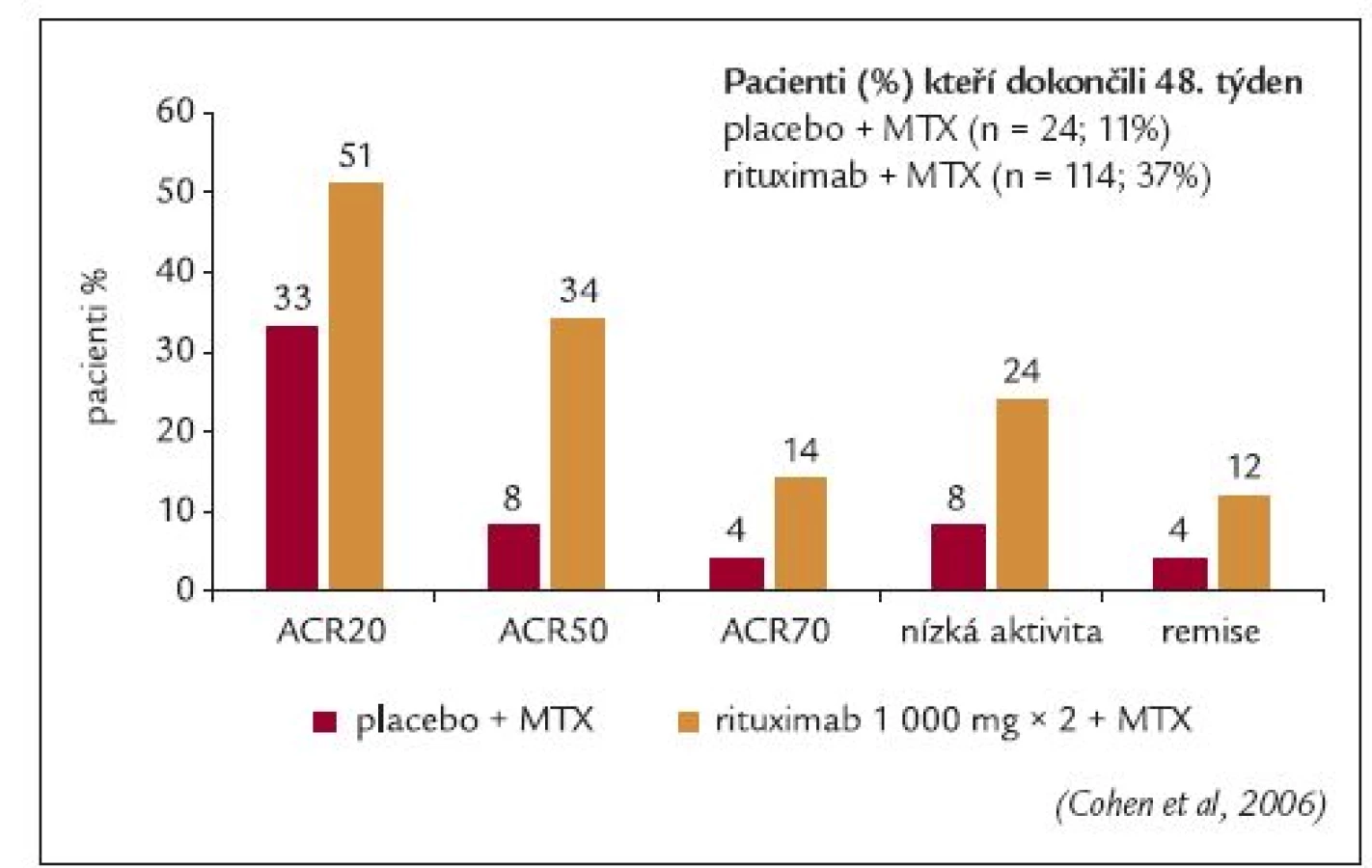
Edwardsovo klinické hodnocení fáze IIa bylo první velkou randomizovanou, dvojitě zaslepenou studií, do které bylo zařazeno 161 pacientů s aktivní RA navzdory terapii metotrexátem v dávce ≥ 10 mg/týden [35]. Všichni pacienti byli starší 21 roků a měli pozitivní RF. Základní studie trvala 24 týdnů. Pacienti, kteří dokončili tuto základní část, pokračovali v extenzi studie po dobu 104 týdnů od zahájení léčby. Pacienti byli rozdělení do 4 skupin, ve kterých byli léčeni buď pouze rituximabem (2 infuze 1 000 mg v den 1 a 15), nebo rituximabem a cyklofosfamidem, nebo rituximabem a metotrexátem a nebo metotrexátem samotným. Všem pacientům byl před každou infuzí rituximabu aplikován intravenózně metylprednisolon v dávce 100 mg. Pacientům byl podáván perorálně prednison po dobu 2 týdnů (prednison 60 mg 2. -7. den a prednison 30 mg v 8.-14. den). Primárním cílem studie bylo dosažení 50% zlepšení skóre aktivity ACR (ACR50). Sekundárními cíli bylo dosažení klinické odpovědi ACR20 a ACR70, odezva na léčbu dle kritérií EULAR, sledování sérové hladiny RF a monitorování bezpečnosti léčby.
Ve všech skupinách dokončilo 24. týden léčby 90-98 % pacientů. Avšak 48. týden léčby dokončilo větší procento pacientů léčených rituximabem než metotrexátem (80-95 % vs 65 %). Ve 24. týdnů léčby bylo primárního cíle dosaženo signifikantně častěji u pacientů léčených rituximabem v kombinaci s cyklofosfamidem nebo metotrexátem než u pacientů léčených metotrexátem v monoterapii (p = 0,005). Statisticky významný rozdíl mezi skupinami léčenými rituximabem a pouze metotrexátem byl rovněž zaznamenán v odezvě ACR20 (p = 0,025), obr. 3. Střední nebo dobré odezvy na terapii dle kritérií EULAR dosáhlo 83-85 % pacientů léčených rituximabem ve srovnání s 50 % pacientů léčených metotrexátem (p ≤ 0,004). Byl zaznamenám výraznější pokles skóre aktivity onemocnění DAS28 ve skupině léčené rituximabem (-2,2 až -2,6) v porovnání se skupinou léčenou pouze metotrexátem (-1,3) (p ≤ 0,002).

Rovněž po 48 týdnech léčby dosáhlo signifikantně vyšší procento pacientů léčených rituximabem ve srovnání s pacienty léčenými metotrexátem odpovědi ACR20, ACR50 a ACR70 (p ≤ 0,05). Dobrý efekt léčby dle kritérií EULAR přetrvával po 48 týdnech léčby u 23 % pacientů léčených rituximabem a metotrexátem oproti 0 % pacientů léčených metotrexátem samotným.
Po této fázi následovala 2letá extenze studie, ve které pacienti pokračovali v započatém léčebném režimu [44]. Nejčastější příčinou předčasného ukončení léčby v tomto období byla nutnost opakovat sérii infuzí (retreatment) rituximabu z důvodu relapsu RA. Výraznější léčebná odezva přetrvávala ve skupině pacientů léčených kombinací rituximabu a metotrexátu, v níž ve 104. týdnů dosáhlo odezvy ACR20 34 % a ACR50 20 % pacientů ve srovnání se 14 %, resp. 10 % pacientů léčených samotným metotrexátem. Klinická odezva ACR70 byla zaznamenána u 13 % pacientů léčených kombinaci rituximabu a metotrexátu, ale u žádného pacienta léčeného samotným metotrexátem.
Studie potvrdila, že jediná série 2 infuzí rituximabu představuje efektivní a dlouhodobou léčbu pacientů s RA, u kterých selhala předchozí terapie metotrexátem. Současně naznačila, že kombinace rituximabu a metotrexátu může znamenat větší přínos v terapii RA než použití rituximabu samotného. Účinnost této kombinační léčby byla následně ověřena ve 2 klinických studiích DANCER a REFLEX.
Studie DANCER byla randomizovaná, dvojitě zaslepená, placebem kontrolovaná studie fáze IIb, do které bylo zařazeno 465 pacientů s aktivní RA navzdory pokračující terapii metotrexátem, u kterých selhala předchozí léčba 1-5 DMARD, jinými než metotrexát, a/nebo biologickými léky [38]. Studie si kladla za cíl ověřit účinnost a bezpečnost 2 dávkovacích režimů rituximabu a 3 režimů glukokortikoidů v terapii RA. Pacienti dostávali buď 2krát 500 mg nebo 2krát 1 000 mg rituximabu v kombinaci s metotrexátem nebo dostávali metotrexát a placebo. Ve všech skupinách pacienti dále dostávali buď metylprednisolon intravenózně v den aplikace rituximabu, nebo intravenózně a následně prednison perorálně nebo glukokortikoidy nedostávali vůbec. Studie tedy zahrnoval celkem 9 léčebných skupin (obr. 4). Primárním cílem studie bylo dosažení ACR20 po 6 měsících léčby. Sekundárními cíli bylo dosažení ACR50 a ACR70, odpovědi EULAR, změna DAS28 a zlepšení kvality života.

Po 6 měsících léčby dosáhlo odpovědi ACR20, ACR50 a ACR70 signifikantně vyšší procento pacientů léčených oběma režimy rituximabu ve srovnání s pacienty, kteří užívali metotrexát a placebo. Ačkoliv odpovědi ACR20 a ACR50 dosáhlo podobné procento pacientů léčených oběma režimy rituximabu, odpovědi ACR70 bylo dosaženo mnohem častěji u pacientů léčených rituximabem v dávce 2krát 1 000 mg než v dávce 2krát 500 mg (20 % vs 13 %), obr. 5. Obdobné výsledky byly zaznamenány i při hodnocení odpovědi dle kritérií EULAR. Signifikantně větší procento pacientů léčených oběma režimy rituximabu ve srovnání s pacienty léčenými metotrexátem dosáhlo střední a dobré odezvy na léčbu dle těchto kritérií. 2krát více pacientů léčených rituximabem v dávce 2krát 1 000 mg, ve srovnání s pacienty léčenými rituximabem v dávce 2krát 500 mg, mělo dobrou odezvu na terapii dle kritérií EULAR (28 % vs 14 %; p < 0,0001), obr. 6. Studie tedy prokázala trend k nárůstu účinnosti vyšších dávek rituximabu. Byl rovněž sledován vliv terapie rituximabem na zlepšení kvality života pacientů. Klinicky významné zlepšení kvality života bylo zaznamenáno u 63 %, resp. 67 % pacientů léčených oběma režimy rituximabu ve srovnání s 34 % pacientů léčených metotrexátem a placebem. Studie dále prokázala, že srovnatelné léčebné odezvy u obou dávkovacích režimů rituximabu bylo dosaženo navzdory různým režimům glukokortikoidů. Glukokortikoidy však měly vliv na snížení výskytu akutních infuzních reakcí. Na základě těchto výsledků bylo doporučeno parenterální podávání glukokortikoidů před každou infuzí rituximabu.


Po 24 týdnech studie pokračovali pacienti, kteří nepotřebovali opakovaní série 2 infuzí rituximabu, ve sledování až do 24. měsíce. Pacienti, kteří vyžadovali podání další dávky rituximabu, byli dále sledování v otevřené extenzi studie. Pouze 27 % pacientů léčených metotrexátem a placebem a 48 % pacientů léčených 2krát 500 mg rituximabu a 49 % pacientů léčených 2krát 1 000 mg rituximabu dokončilo 48. týden sledování. Nejčastějším důvodem předčasného ukončení účasti ve studii byla ztráta odezvy na terapii a nutnost opakovat sérii infuzí rituximabu.
145 pacientům v této studii byla podána 2. série infuzí rituximabu. V době sdělení bylo 99 pacientů sledováno 24 týdnů po druhé infuzi. Byla zaznamenána srovnatelná nebo lepší klinická odpověď na terapii 2. sérií rituximabu ve srovnání se sérií 1. podle kritérií ACR, EULAR. Podání další série rituximabu tedy nejen přispívá k udržení klinického efektu léčby, ale může dále snížit aktivitu RA.
Studie DANCER prokázala, že 1 série 2 infuzí rituximabu poskytuje dlouhodobý efekt přetrvávající až 1 rok u pacientů s RA, u kterých selhala předchozí léčba DMARD včetně TNFα inhibitorů. Avšak velká část pacientů vyžadovala v období 6 a více měsíců od zahájení léčby opakované podání série infuzí rituximabu.
Studie rovněž prokázala bezpečnost terapie rituximabem. Jakákoliv nežádoucí příhoda byla zaznamenána u 70 % pacientů léčených samotným metotrexátem a u 81 % a 85 % pacientů léčených oběma dávkovacími režimy rituximabu. 82 % těchto nežádoucích příhod bylo mírné nebo střední intenzity (Common Toxicity Criteria – CTC - stupěň 1-2). Akutní infuzní reakce po 1. infuzi byla zaznamenána u 17 % pacientů léčených metotrexátem a u 23 % a 32 % pacientů léčených rituximabem. Parenterální podání glukokortikoidů snižovalo incidenci a závažnost infuzních reakcí. Výskyt akutních infuzních reakcí byl nižší po 2. infuzi rituximabu. Výskyt infekcí byl zaznamenán u 28 % pacientů léčených metotrexátem a u 35 % pacientů léčených rituximabem. Typy a závažnost infekcí se v obou skupinách významně nelišily. Závažné infekce se vyskytovaly v poměru 3,1/100 pacientských roků u pacientů léčených metotrexátem a v poměru 0 a 4,74/100 pacientských roků u pacientů léčených jednotlivými režimy rituximabu.
Reflex byla opět randomizovaná, dvojitě zaslepená, placebem kontrolovaná studie fáze III, do které byli zařazováni pacienti s RA, u kterých selhala předchozí léčba TNFα inhibitory [39]. Do studie bylo zařazeno celkem 520 pacientů. Pacienti dostávali rituximab (2krát 1 000 mg) nebo placebo v poměru 3 : 2. Všichni pacienti byli léčeni metotrexátem v dávce 10-25 mg týdně. Všichni pacienti dostávali intravenózně metylprednisolon 100 mg 30 min před každou infuzí a následně prednison perorálně po dobu 2 týdnů. Pacientům v metotrexátové skupině, kteří adekvátně nereagovali na léčbu, mohla být počínaje 16. týdnem aplikována série infuzí rituximabu jako záchranná medikace. Primárním cílem studie bylo opět dosažení klinické odpovědi ACR20 ve 24. týdnu léčby. Sekundárními cíli bylo dosažení ACR50 a ACR70, odpovědi EULAR, změna DAS28. Dále byly sledovány změny únavy (FACIT-F), kvalita života (SF-36) a RTG progrese RA (modifikované Sharp/Genant skóre).
51 % pacientů léčených rituximabem ve srovnání s 18 % pacientů léčených metotrexátem dosáhlo ve 24. týdnu klinické odpovědi ACR20 (p < 0,0001). Obdobné výsledky byly pozorovány i v kritériích ACR50 a ACR70 (obr. 7). Efekt léčby rituximabem byl zaznamenán nezávisle na pozitivitě či negativitě RF i počtu předchozích TNFα inhibitorů. Avšak odezva na terapii rituximabem byla výraznější u pacientů, kteří byli léčení pouze jedním předchozím TNFα inhibitorem ve srovnání s pacienty s předchozí neúspěšnou léčbou 2 a více TNFα inhibitory (ACR20 58 % vs 42 %), obr. 8 [46]. Průměrná změna skóre DAS28 byla -1,9 ve skupině rituximabové a -0,4 ve skupině s metotrexátem. Nízkou aktivitu onemocnění dle skóre DAS28 mělo ve 24. týdnu 15 % pacientů léčených rituximabem, ale pouze 2 % pacientů léčených metotrexátem. Klinicky významné zlepšení funkčního stavu, kvality života a únavy bylo zaznamenáno pouze u pacientů léčených rituximabem [45].


37 % pacientů léčených rituximabem a 11 % léčených metotrexátem pokračovalo ve sledování po dobu 48 týdnů. Ve 48. týdnu dosáhlo odezvy ACR20 51 % pacientů léčených rituximabem ve srovnání s 33 % pacientů léčených pouze metotrexátem. Rovněž procento pacientů s nízkou aktivitou a remisí onemocnění dle skóre DAS28 bylo vyšší v rituximabové větvi (obr. 9) [47]. Podobně jako v předchozí studii přetrvával efekt jedné série infuzí rituximabu po dobu 1 roku i déle, i když většina pacientů vyžadovala její opakování v období 6-12 měsíců od zahájení léčby.
Pacientům v metotrexátové skupině, kteří adekvátně nereagovali na léčbu, mohla být počínaje 16. týdnem podána série infuzí rituximabu jako záchranná medikace. V 56. týdnu pouze 19 % pacientů randomizovaných do skupiny metotrexát a placebo nevyžadovalo podání rituximabu. 81 % pacientům v této skupině byla tedy podána minimálně 1 série rituximabu (63 %; 17 %; < 1 % dostalo 1; 2, resp. 3 série rituximabu).
Celkem 279 pacientům v této studii byla podána druhá série infuzí rituximabu. V době sdělení bylo 155 pacientů sledováno 24 týdnů po 2. infuzi. Byla zaznamenána srovnatelná nebo lepší klinická odpověď na terapii 2. sérií rituximabu ve srovnání se sérií 1. 72 % pacientů po 2. sérii rituximabu ve srovnání s 65 % pacientů po 1. sérii dosáhlo odpovědi ACR20 [48]. Vyšší bylo i procento pacientů, kteří dosáhli po 2. sérii rituximabu odezvy ACR50, ACR70, nízké aktivity a remise choroby dle DAS28.
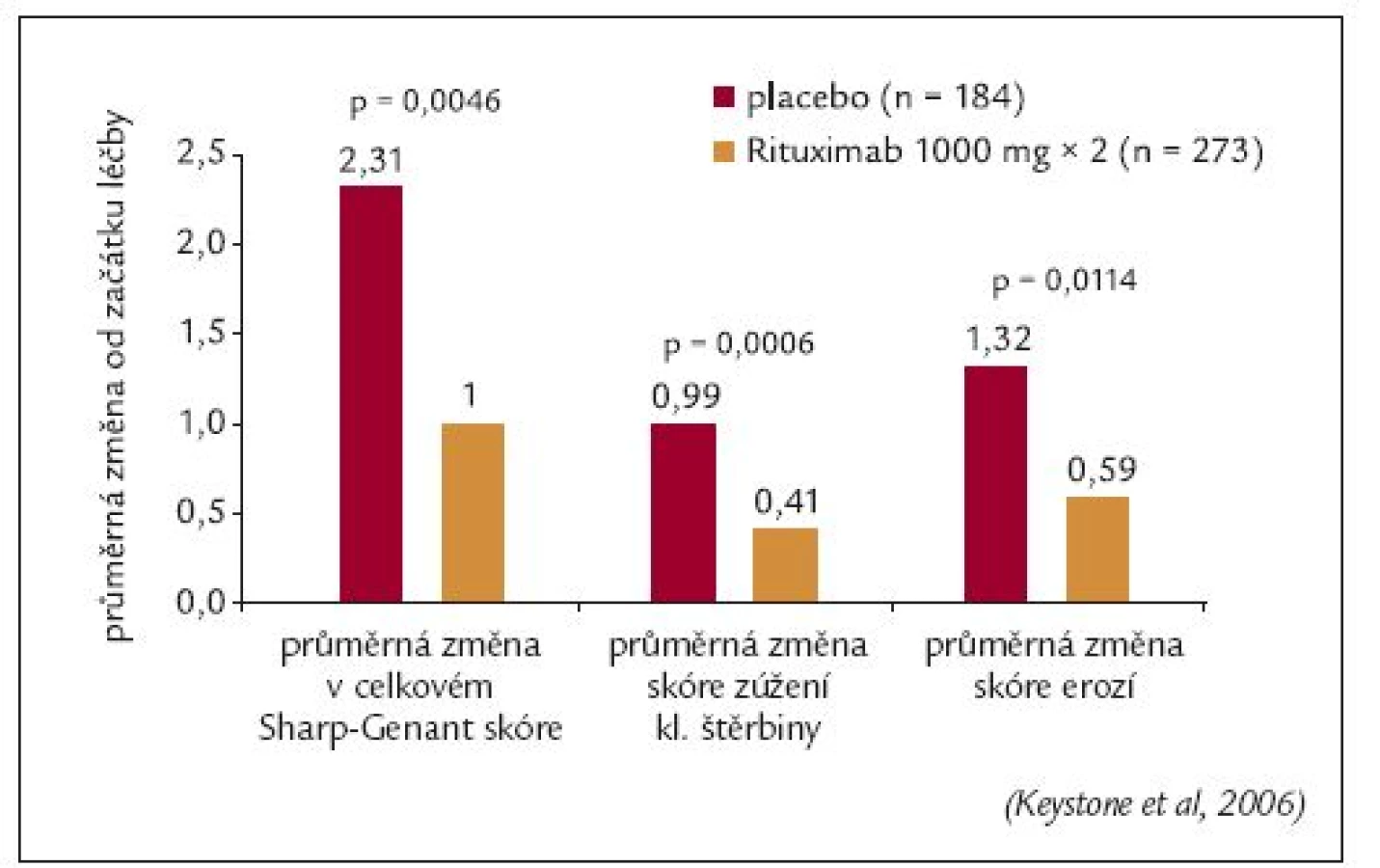
Všem pacientům ve studii byl proveden RTG snímek obou rukou a nohou před zahájením léčby a v týdnech 24. a 56. RTG progrese onemocnění byla hodnocena metodou podle Sharpa modifikovanou Genantem. Průměrná změna modifikovaného Sharpova skóre byla v 56. týdnu o 57 % nižší v rituximabové skupině ve srovnání s placebovou skupinou (1,00 vs 2,31; p = 0,0043), obr. 10. Signifikantní rozdíl mezi oběma skupinami byl rovněž zaznamenán v jednotlivých komponentách Sharpova skóre: počtu erozí a zúžení kloubních štěrbin. Navíc procento pacientů, u kterých nebyla zaznamenána žádná RTG progrese, bylo významně vyšší u rituximabové skupiny (61 % vs 52 %; p = 0,0445) [49]. Rituximab tedy zpomaloval RTG progresi u pacientů, u kterých selhala předchozí léčba TNFα inhibitory.
Výskyt nežádoucí příhod byl srovnatelný v obou skupinách (rituximab vs placebo; 85 % vs 88 %) a většina nežádoucích příhod byla mírné nebo střední intenzity. Hladina imunoglobulinů po jedné sérii rituximabu sice mírně klesala, ale zůstávala v normálním rozmezí po celou dobu sledování. Nebyl zaznamenán nárůst výskytu infekcí u pacientů léčených rituximabem a spektrum infekcí se nelišilo od běžné populace pacientů s RA. Nejčastěji byly zaznamenány infekce respiračního traktu, nazofaryngitidy, bronchitidy, sinusitidy a močové infekce. Závažné infekce se vyskytly v poměru 5,2/100 pacientských roků u pacientů léčených rituximabem a v poměru 3,7/100 pacientských roků u pacientů v placebové skupině. Akutní infuzní reakce byla pozorována po 1. infuzi u 23 % pacientů léčených rituximabem a u 18 % pacientů léčených placebem, po 2. infuzi u 8 %, resp. 11 % pacientů. Pouze u 2 pacientů léčených rituximabem byla infuzní reakce hodnocena jako závažná (anafylaktický šok, hypertenzní reakce).
Bezpečnost léčby rituximabem
Rituximab již prokázal příznivý bezpečností profil u více než 730 000 onkologických pacientů. Do listopadu roku 2005 bylo ve studiích fáze II a III léčeno jednou sérií rituximabu 1 039 pacientů s RA, 570 pacientů dostalo celkem 2 série a 191 a 40 pacientů dostalo 3 a 4 série rituximabu. Celková expozice představuje 1 669 pacientských roků [50,51]. Všichni pacienti dostávali jako základní terapii metotrexát. Výskyt nežádoucích příhod byl zaznamenán u 88 % pacientů po první sérii rituximabu. Většina z nich mírné nebo střední intenzity (CTC 1, 2). Závažné (CTC 3) a velmi závažné (CTC 4) nežádoucí příhody se vyskytly u 24 %, resp. 2 % pacientů. Incidence nežádoucích příhod klesala z hodnoty 939 příhod/100 pacientských roků během prvních 3 měsíců po podání rituximabu na hodnotu 399, resp. 212 příhod/100 pacientských roků během 4-6, resp. 10-12 měsíce po podání rituximabu. Vyšší výskyt nežádoucích příhod v období 3 měsíců po podání rituximabu souvisí zejména s výskytem akutních infuzních reakcí. Závažnost a výskyt nežádoucích příhod se snižoval s opakovaným podáním rituximabu (71 %; 64 % a 55 % po 2., 3. a 4. sérii rituximabu).
Nejčastější nežádoucí příhodou byla akutní infuzní reakce vyvíjející se během 24 hod po podání infuze rituximabu v souvislostí s vyplavením cytokinů následující po lýze B-lymfocytů. Výskyt symptomů jako je horečka, zimnice, třesavka, pruritus, vyrážka, angioneurotický edém, kašel, bronchospazmus, hypotenze nebo hypertenze byl zaznamenán u 15 % pacientů po podání rituximabu ve srovnání s 5 % pacientů po podání placeba. Většina reakcí byla mírné a střední intenzity. Incidence akutních infuzních reakcí se snižovala po opakovaném podání rituximabu i placeba (2 %). Závažné infuzní reakce se vykytovaly u méně než 1 % pacientů. Úpravu dávky (zpomalení, přerušení nebo ukončení infuze) vyžadovalo 10 % pacientů léčených rituximabem a 2 % pacientů léčených placebem. Premedikace intravenózně podaným metylprednisolonem snižovala incidenci a závažnost akutních infuzních reakcí [52].
Hladiny imunoglobulinů, zejména hladina IgM, sice mírně klesaly po 1 sérii rituximabu, ale zůstávaly v normálním rozmezí po celou dobu sledování (24 týdnů). Nebyl zaznamenán vztah mezi poklesem hladiny imunoglobulinů a zvýšeným výskytem infekcí.
Infekční komplikace ve studiích fáze II a III se vyskytly u 39 % pacientů léčených rituximabem a 34 % pacientů léčených placebem. Závažné infekce se vyskytly v těchto skupinách u 2 %, resp. 1 % pacientů. Nejčastěji byly pozorovány infekce horní části respiračního traktu, nazofaryngitidy, bronchitidy, sinusitidy a močové infekce. Nebyl zaznamenán žádný případ tuberkulózy ani oportunních infekcí. Při opakovaném podání rituximabu nebyl výskyt a závažnost infekcí ovlivněn protrahovanou deplecí B-lymfocytů a nelišil se od výskytu infekcí v běžné populaci pacientů s RA [50,53].
Byl rovněž sledován titr protilátek u pacientů očkovaných proti varicele, příušnicím, rubeole, tetanu, chřipce typu A a proti Streptococcus pneumonie a nebyl zaznamenán jejich pokles [50,53].
Rovněž nebyl zaznamenán nárůst výskytu malignit. Incidence malignit (1,6/100 pacientských roků) u pacientů s RA léčených rituxumabem se nelišila od očekávané incidence v běžné populaci obdobného věku a pohlaví. Nebyl zaznamenán ani jeden případ lymfoproliferativního onemocnění.
U 9,2 % pacientů byla alespoň jednou prokázána přítomnost HACA (Human Antichimeric antibody) autoprotilátek v souvislosti s léčbou rituximabem. Pozitivita HACA autoprotilátek v klinických studiích nebyla asociována se ztrátou účinnosti rituximabu ani s výskytem infuzních reakcí. Pouze u 1 pacienta byla v souvislosti s průkazem HACA autoprotilátek zaznamená ztráta účinnosti a zhoršení infuzních reakcí po podání dalších sérii rituximabu.
K dispozici jsou data od 78 pacientů, kteří byli po podání rituximabu následně léčeni TNFα inhibitorem [54]. Průměrná incidence nežádoucích příhod byla u těchto pacientů nízká a srovnatelná s očekávanou incidencí nežádoucích příhod u pacientů léčených TNFα inhibitory bez předchozí terapie rituximabem (7,62 vs 6,39/100 pacientských roků) [55].
Americká FDA (Food and Drug Administration) upozornila v prosinci roku 2006 na 2 fatální případy progresivní multifokální leukoencefalopatie (PML) u pacientů se systémovým lupus erythematodes (SLE) léčených rituximabem. Rituximab není v této indikaci schválen. PML je demyelinizační onemocnění způsobené reaktivací latentní infekce JC virem (typ lidského polyomaviru, původně nazývaný papovavirus), prokazatelné u 80 % dospělých. Doposud byl potvrzen výskyt 23 případů PML u pacientů s lymfoidními malignitami léčenými rituximabem. PML byla však popsána i u pacientů se SLE rituximabem neléčených.
V souvislosti s terapii rituximabem bylo dále popsáno 47 případů střevní obstrukce a 37 případů perforace zažívacího traktu u pacientů s NHL. Jednoznačná souvislost s terapií rituximabem nebyla potvrzena. Uvažuje se i vlivu konkomitantní léčby (chemoterapie, glukokortikoidy) a postižení zažívacího traktu v rámci základního onemocnění.
V Japonsku byl v posledních 2 letech popsán výskyt 18 případů, některých z nich fatálních, fulminantní hepatitidy u pacientů s NHL léčených rituximabem a současných nositelů viru hepatitidy B.
Závěrem lze říci, že rituximab je dobře tolerován. Nejčastější nežádoucí příhodou jsou akutní infuzní reakce mírné a střední intenzity, které se vyskytují zejména po první sérii rituximabu a jejichž incidence klesá po jeho opakovaném podání. Incidence závažných infekcí je nízká, i když mírně vyšší než po placebu, nenarůstá po opakovaném podání rituximabu a je srovnatelná s očekávaným výskytem závažných infekcí u běžné populace pacientů s RA.
Praktické použití rituximabu
Rituxumab je indikován v kombinaci s metotrexátem k terapii dospělých pacientů s těžkou aktivní RA (DAS28 > 5,1), kteří odpovídají nedostatečně nebo kteří netolerují jiné DMARD včetně jednoho a více TNFα blokujícího léku. Ve zvláštních případech, např. při současném onemocnění NHL, je možno rituximab podat jako lék první volby. Rituximab by neměl být použit u pacientů se známou přecitlivělostí vůči kterékoliv složce přípravku a vůči myším bílkovinám, dále u pacientů s aktivní těžkou infekcí, u pacientů s těžkou formou srdečního selhání (NYHA IV) a závažným nekontrolovaným srdečním onemocněním. Podání rituximabu se nedoporučuje těhotným a kojícím ženám a dále dětem, u nichž nebyla dosud účinnost a bezpečnost přípravku ověřena. V průběhu terapie rituximabem se nedoporučuje očkování živou vakcínou. Rituximab by měl být podáván pod dohledem zkušeného lékaře za dostupnosti pomůcek a léků nutných ke kardiopulmonální resuscitaci. Pacienti s historií kardiopulmonálního onemocnění by měli být pečlivě monitorováni. V České republice, v souladu s doporučeními ČRS, probíhá aplikace rituximabu ve vybraných centrech zařazených do projektu ATTRA. ATTRA je klinický registr, vedený od počátku roku 2002 pod odbornou garancí ČRS, určený k hodnocení průběhu a výsledků léčby RA TNF inhibitory a nově i rituximabem. Rituximab je podáván ve 2 intravenózních infuzích v dávce 1 000 mg podávaných odděleně s odstupem 2 týdnů (1. a 15. den). Zejména z důvodu snížení výskytu akutní infuzních reakcí je indikována premedikace metylprednisolonem v dávce 100 mg i.v. podaným 30 min před každou infuzí rituximabu. Součástí premedikace by mělo být podání paracetamolu v dávce 1 000 mg a antihistaminika (např. bisulepinu 2 mg) 60 min před každou infuzí rituximabu. K přípravě infuzního roztoku je doporučováno použít 250 ml fyziologického roztoku, ze kterého se odpustí 100 ml a přidají se 2 balení přípravku MabThera® 500 mg. Vznikne roztok o koncentraci 4 mg/ml. Doporučená počáteční rychlost infuze je 50 mg/hod a lze ji každých 30 min zvyšovat o 50 mg/hod do maximální rychlosti 400 mg/hod. V případě výskytu akutních infuzních reakcí mírné až střední intenzity je doporučeno snížení rychlosti infuze nebo její přerušení, podání antihistaminik (bisulepin nebo diphenhydramin) a případné podání glukokortikoidů. Po vymizení symptomů je možno v aplikaci infuze pokračovat poloviční nebo nižší rychlostí s možností postupného zvyšování rychlosti infuze. V případě výskytu závažné akutní infuzní reakce je nutné její okamžité přerušení a zahájení adekvátní agresivní léčby. Opakování léčby rituxumabem je poté nutné důkladně zvážit. 2. infuze rituximabu se aplikuje 15. den po podání 1. infuze za stejných podmínek. Hodnocení odpovědi na léčbu se provádí v 16. týdnu a je definováno jako pokles DAS28 > 1,2. Další sérii 2 infuzí rituximabu je možno podat nejdříve ve 24. týdnu léčby. Důvodem pro opakované podání rituximabu je nárůst aktivity onemocnění (zvýšení DAS28 > 0,6). Před zahájením a v průběhu léčby rituximabem je doporučováno provádět rutinní laboratorní biochemické a hematologické vyšetření, monitorovat hladinu imunoglobulinů a provádět screening hepatitidy B.
Závěr
RA je závažné chronické zánětlivé onemocnění. Chronický zánět vede k destrukci kloubní chrupavky, vzniku erozí subchondrální kosti a v konečné fázi k vývoji deformit postižených kloubů. Tento proces přináší pacientům s RA bolest a dříve či později vede k vývoji disability. Protože se předpokládá, že strukturální postižení kloubů u RA je nevratné, je v současné době kladen velký důraz na včasnou a efektivní terapii RA, která by byla schopna zastavit případně zpomalit vývoj onemocnění. V době, kdy nemáme k dispozici prostředky k vyléčení RA, je hlavním léčebným cílem navození remise choroby a zabránění strukturálnímu poškození kloubů a rozvoji dlouhodobé disability. Relativně časté selhání klasických DMARD vedlo ke snaze najít nové prostředky k léčbě RA. Do terapie byly proto v nedávné době zavedeny tzv. biologické léky. Jednalo se zejména a TNFα blokující preparáty. Praxe ukázala, že přibližně 1/3 pacientů s RA však neodpovídá ani na léčbu těmito preparáty. Rituximab (MabThera®), monoklonální protilátka proti CD20 pozitivním B-lymfocytům, je nový biologický lék schválený k terapii RA. Představuje novou naději pro pacienty s aktivní RA, u kterých selhala předchozí terapie TNFα blokujícími léky. Jedna série 2 infuzí rituximabu vede k dlouhodobému poklesu aktivity onemocnění s průměrným trváním 32-34 týdnů. Opakované podání série infuzí rituximabu (retreatment) je přinejmenším stejně účinné jako podání 1. série. Byl prokázán vliv rituximabu na zpomalení RTG progrese RA. Terapie rituximabem je bezpečná. Nejčastějšími nežádoucími příhodami jsou akutní infuzní reakce, jejichž incidence klesá při opakovaném podání rituximabu. Mírně zvýšena oproti placebu je incidence infekcí.
MUDr. Petr Němec, Ph.D.
Revmatologická ambulance II. Interní kliniky LF MU a FN u sv. Anny
Pekařská 53
656 91 Brno
e-mail: petr.nemec@fnusa.cz
Doručeno do redakce: 13. 5. 2007
Přijato po recenzi: 12. 6. 2007
Sources
1. Lee DM, Weinblatt ME. Rheumatoid arthritis. Lancet 2001; 358 : 903-911.
2. Lawrence RC, Helmick GG, Arnett FC et al. Estimates of the prevalence of arthritis and selected musculosceletal disorders in the United States. Arthritis Rheum 1998; 41 : 778-799.
3. Weinblatt ME. Rheumatoid arthritis: treat now, not later. Ann Intern Med 1996; 124 : 773-774.
4. Pincus T, Kavanaugh A, Sokka T. Benefit/risk of therapies for rheumatoid arthritis: underestimation of the “side effects” or risks of RA leads to underestimation of the benefit/risk of therapies. Clin Exp Rheumatol 2004; 22(Suppl 35): S2-S11.
5. Visser H, le Cessie S, Vos K et al. How to diagnose rheumatoid arthritis early: a prediction model for persistent (erosive) arthritis. Arthritis Rheum 2002; 46 : 357-365.
6. Brennan P, Harrison B, Barrett E et al. A simple algorithm to predict the development of radiological erosions in patients with early rheumatoid arthritis: prospective cohort study. BMJ 1996; 313 : 471-476.
7. Möttönen T, Paimela L, Leirisalo-Repo M et al. Only high disease activity and positive rheumatoid factor indicate poor prognosis in patients with early rheumatoid arthritis treated with “sawtooth” strategy. Ann Rheum Dis 1998; 57 : 533-539.
8. Goronzy JJ, Matteson EL, Fulbright JW et al. Prognostic markers of radiographic progression in early rheumatoid arthritis. Arthritis Rheum 2004; 50 : 43-54.
9. Vencovský J, Macháček S, Šedová L et al. Autoantibodies can be prognostic markers of an erosive disease in early rheumatoid arthritis. Ann Rheum Dis 2003; 62 : 427-430.
10. Combe B, Dougados M, Goupille P et al. Prognostic factors for radiographic damage in early rheumatoid arthritis: a multiparameter prospective study. Arthritis Rheum 2001; 44 : 1736-1743.
11. Dixey J, Solymossy C, Young A. Is it possible to predict radiological damage in early rheumatoid arthritis (RA)? A report on the occurrence, progression, and prognostic factors of radiological erosions over the first 3 years in 866 patients from the Early RA Study (ERAS). J Rheumatol Suppl 2004; 69 : 48-54.
12. Combe B, Landwe R, Lukas C et al. EULAR recommendations for management of early arthritis. Ann Rheum Dis 2006; 5 [Epub, v tisku].
13. American College of Rheumatology (ACR). American College of Rheumatology Subcommittee on Rheumatoid Arthritis Guidelines, Guidelines for the management of rheumatoid arthritis. Arthritis Rheum 2002; 46 : 328-346.
14. Combe B, Landewé R, Lukas C et al. EULAR recommendations for the management of early arthritis. Ann Rheum Dis 2007; 66 : 34-45.
15. Pavelka K, Bečvář R, Olejárová M et al. Standardní postupy v revmatologii. Revmatoidní artritida. Čes Revmatol 1999; 7(Suppl. 1): 4-8.
16. Vencovský J, Tegzová D, Pavelka K. Doplněk standardních postupů u revmatoidní artritidy. Čes Revmatol 2002; 10 : 31-40.
17. Strand V, Cohen S, Schiff M et al. Treatment of active rheumatoid arthritis with leflunomide compared with placebo and methotrexate. Leflunomide Rheumatoid Arthritis Investigators Group. Arch Intern Med 1999; 159 : 2542-2550.
18. Drosos AA, Tsifetaki N, Tsiakou EK et al. Influence of methotrexate on radiographic progression in rheumatoid arthritis: a sixty-month prospective study. Clin Exp Rheumatol 1997; 15 : 263-267.
19. Bathon JM, Martin RW, Fleischmann RM et al. A comparison of etanercept and methotrexate in patients with early rheumatoid arthritis. N Engl J Med 2000; 343 : 1586-1593.
20. Breedveld FC, Weisman MH, Kavanaugh AF et al. The PREMIER study: combination therapy with adalimumab plus methotrexate versus methotrexate alone or adalimumab alone in methotrexate-naive patients with early, aggressive rheumatoid arthritis. Arthritis Rheum 2006; 54 : 26-37.
21. Weinblatt ME, Keystone EC, Furst DE et al. Adalimumab, a fully human anti-tumor necrosis factor α monoclonal antibody, for the treatment of rheumatoid arthritis in patients taking concomitant methotrexate: the ARMADA trial. Arthritis Rheum 2003; 48 : 35-45.
22. Weinblatt ME, Kremer JM, Bankhurst AD et al. A trial of etanercept, a recombinant tumor necrosis factor receptor: Fc fusion protein, in patients with rheumatoid arthritis receiving methotrexate. N Engl J Med 1999; 340 : 253-259.
23. Klareskog L, van der Heijde O, de Jager JP et al. Therapeutic effect of the combination of etanercept and methotrexate compared with each treatment alone in patients with rheumatoid arthritis: double-blind randomised controlled trial. Lancet 2004; 363 : 675-681.
24. Lipsky PE, van der Heijde DM, St Clair EW et al. and the Anti-Tumor Necrosis Factor Trial in Rheumatoid Arthritis with Concomitant Therapy Study Group. Infiximab and methotrexate in the treatment of rheumatoid arthritis. N Engl J Med 2000; 343 : 1594-1602.
25. Maini R, St Clair EW, Breedveld F et al. Infliximab (chimeric antitumour necrosis factor α monoclonal antibody) versus placebo in rheumatoid arthritis patients receiving concomitant methotrexate: a randomised phase III trial. ATTRACT Study Group. Lancet 1999; 354 : 1932-1939.
26. Moreland L Unmet needs in rheumatoid arthritis. Arthritis Res Ther 2005; 7 (Suppl. 3): S2-S8.
27. Imperato AK, Bingham CO 3rd, Abramson SB Overview of benefit/risk of biological agents. Clin Exp Rheumatol 2004; 22(Suppl 35): S108-S114.
28. Cohen SB, Moreland LW, Cush JJ et al. A multicentre, double blind, randomised, placebo controlled trial of anakinra (Kineret), a recombinant interleukin-1 receptor antagonist, in patients with rheumatoid arthritis treated with background methotrexate. Ann Rheum Dis 2004; 63 : 1062-1068.
29. Genovese MC, Becker JC, Schiff M et al. Abatacept for rheumatoid arthritis refractory to tumor necrosis factor α inhibition. N Engl J Med 2005; 353 : 1114-1123.
30. Kremer JM, Weinblatt ME, Bankhurst AD et al. Treatment of rheumatoid arthritis by selective inhibition of T-cell activation with fusion protein CTLA41g. N Engl J Med 2003; 349 : 1907-1915.
31. Reff ME, Carner K, Chambers KS et al. Depletion of B-cells in vivo by a chimeric mouse human monoclonal antibody to C020. Blood 1994; 83 : 435-445.
32. Maloney DG, Smith B, Appelbaum FR. The antitumor effect of monoclonal anti-CD20 antibody (mAb) therapy includes direct anti-proliferative activity and induction of apoptosis in CD20 positive non-Hodgkin’s lymphoma (NHL) cell lines. Blood 1996; 88 (Suppl. 1): 637a.
33. Breedveld F, Agarwal S, Yin M et al. Relationship between clinical response, rituximab pharmacokinetics, and peripheral B cell levels in rheumatoid arthritis. Presented at ACR 2005 (Abstract 279).
34. Ng CM, Bruno R, Combs D et al. Population pharmacokinetics of rituximab (anti-CD20 monoclonal antibody) in rheumatoid arthritis patients during a Phase II clinical trial. J Clin Pharmacol 2005; 45 : 792-801.
35. Edwards JC, Szczepanski L, Szechinski J et al. Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N Engl J Med 2004; 350 : 2572-2581.
36. Emery P, Sheeran T, Lehane PB et al. Efficacy and safety of rituximab at 2 years following a single treatment in patients with active rheumatoid arthritis. Arthritis Rheum 2004; 50 (Suppl. 9): S659.
37. Emery P, Fillpowicz-Sosnowska A, Szczepanski L et al. Primary analysis of a double-blind, placebo-controlled, dose-ranging trial of rituximab, an anti-CD20 monoclonal antibody, in patients with rheumatoid arthritis receiving methotrexate (DANCER trial). Ann Rheum Dis 2005 : 64 : 58.
38. Emery P, Fleischmann R, Filipowicz-Sosnowska A et al. The efficacy and safety of rituximab in patients with active rheumatoid arthritis despite methotrexate treatment: results of a Phase IIb double-blind, placebo-controlled, dose-ranging trial (DANCER). Arthritis Rheum 2006; 54 : 1390-1400.
39. Cohen SB, Emery P, Greenwald M et al. Rituximab for rheumatoid arthritis refractory to anti-tumor necrosis factor therapy. Arthritis Rheum 2006; 54 : 2793-2806.
40. Protheroe A, Edwards JC, Simmons A et al. Remission of inflammatory arthropathy in association with anti-CD20 therapy for non-Hodgkin's lymphoma. Rheumatology (Oxford) 1999; 38 : 1150-1152.
41. Stewart M, Malkovska V, Krishnan J et al. Lymphoma in a patient with rheumatoid arthritis receiving methotrexate treatment: successful treatment with rituximab. Ann Rheum Dis 2001; 60 : 892-893.
42. Shaw T, Quan J, Totoritis MC B-cell therapy for rheumatoid arthritis: the rituximab (anti-CD20) experience. Ann Rheum Dis 2003; 62 (Suppl. 2): ii55-ii59.
43. Czuczman MS Combination chemotherapy and rituximab. Anticancer Drugs 2001; 12 (Suppl. 2): S15-S19.
44. Sheeran T, Emery P, Lehane PB et al. Duration of response to a single treatment course of rituximab (RTX) in active rheumatoid arthritis (RA): efficacy and safety data from a 2-year follow-up of a randomised trial. Rheumatology (Oxford) 2005; 44: i2.
45. Keystone EC, Burmester GR, Furie R et al. Improved healthrelated quality of life with rituximab pius methotrexate in patients with active rheumatoid arthritis who experienced inadequate response to one or more anti-TN F therapies. ACR 2005 (Abstract 287).
46. Kremer JM, Tony H, Tak PP et al. Efficacy of rituximab in active RA patients with an inadequate response to one or more TNF inhibitors. Ann Rheum Dis 2006; 65 (Suppl. 2): 326.
47. Cohen S, Emery P, Greenwald M et al. Prolonged efficacy of a single course of rituximab in rheumatoid arthritis patients with inadequate response to one or more TNF inhibitors: 1-year follow-up of a controlled trial (REFLEX study). Ann Rheum Dis 2006; 65 (Suppl. 2): 183.
48. Keystone EC, Fleischmann R, Emery P et al. Long term efficacy and safety of a repeat treatment course of rituximab in rheumatoid arthritis patients with an inadequate response to one or more TNF inhibitors. Ann Rheum Dis 2006; 65 (Suppl. 2): 323-324.
49. Keystone E, Emery P, Peterfy CD et al. Prevention of joint structural damage at 1 year with rituximab in rheumatoid arthritis patients with an inadequate response to one or more TNF inhibitors (REFLEX study). Ann Rheum Dis 2006; 65 (Suppl. 2): 58.
50. van Vollenhoven RF, Emery P, Bingham C et al. Safety of rituximab in rheumatoid arthritis: results of a pooled analysis. Ann Rheum Dis 2006 : 65 (Suppl. 2): 503.
51. van Vollenhoven RF, Emery P, Fleischmann RM et al. Safety and tolerability of rituximab in patients with moderate to severe rheumatoid arthritis (RA): results from the Dose-ranging Assessment iNternational Clinical Evaluation of Rituximab in RA (DANCER) study. Ann Rheum Dis 2005 : 64 : 432.
52. Fleischmann RM, Racewicz AJ, Schechtman J et al. Rituximab efficacy in rheumatoid arthritis is independent of coadministration of glucocorticoids: results from the Dose-ranging Assessment iNternational Clinical Evaluation of Rituximab in rheumatoid arthritis (DANCER) Study. Arthritis Rheum 2005; 52 (Abstract 263).
53. Doran MF, Crowson CS, Pond GR et al. Predictors of infection in rheumatoid arthritis. Arthritis Rheum 2002; 46 : 2294-2300.
54. Breedveld FC, Genovese M, Emery P et al. Safety of TNF inhibitors in rheumatoid arthritis patients previously treated with rituximab. Ann Rheum Dis 2006 : 65 (Suppl. 2): 178-179.
55. Dixon W, Watson K, Hyrich K et al. The incidence of serious infections is not increased in patients with rheumatoid arthritis treated with anti-TNF drugs compared to those treated with traditional DMARDs: results from a national prospective study. ACR 2005 (Abstract 1990).
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2007 Issue 11
Most read in this issue
- Syndrom horní duté žíly: definice, etiologie, fyziologie, symptomy, diagnostika a léčba
- Rituximab (MabThera®) - nový biologický lék v terapii revmatoidní artritidy
- Diagnostický prínos použitia implantovaného slučkového rekordéra (Reveal Plus) u pacientov so synkopou nejasnej etiológie
- Endoskopická diagnostika a léčba biliárních komplikací po laparoskopické cholecystektomii