
Obrovskobuněčná arteriitida manifestující se oboustrannou arteriitickou přední ischemickou neuropatií zrakového nervu (AION)
Giant cell arteritis manifested by bilateral arteritic Anterior Ischaemic Optic Neuropathy (AION)
Giant cell arteritis (GCA) is a systemic vasculitis of unknown etiology affecting medium and large calibre vessels by granulomatous panarteritis with the formation of giant multinucleate cell granulomas. Vision is affected in 25–50% of GCA patients. Affection of vision may be the first GCA symptom or a symptom which occurs weeks or months after the initial symptoms of the disease. Eye symptoms of the disease are mostly a manifestation of occlusion of ocular and orbital blood vessels. Permanent damage to the patient’s vision is a serious consequence of visual affection provoked by GCA. Arteritic Anterior Ischaemic Optic Neuropathy (AION) is the most frequent and most serious visual manifestation of GCA. It is manifested by partial or total loss of vision. Arteritic AION therapy in GCA uses high doses of glucocorticoids, but glucocorticoid therapy has a number of adverse effects. The proofs of the effect of the therapy on the improvement of the vision of patients with visual affection in GCA are not convincing. We report a case of a 76-year old man with biopsy-verified GCA whose primary manifestation was bilateral arteritic AION resulting in a complete loss of vision in one eye and dramatic worsening of visual acuity in the other eye. Glucocorticoid therapy only improved vision in one eye, and had adverse effects. Methotrexate was added to the therapy to achieve a glucocorticoid saving effect. Glucocorticoid therapy could be discontinued after 3 years. In the course of the therapy and for the subsequent 12 months after it was finished, there was no relapse of the underlying disease.
Key words:
Arteritic Anterior Ischaemic Optic Neuropathy – glucocorticoids – giant cell arteritis – therapy – vasculitis
Authors:
P. Němec 1; T. Jurečka 2; V. Žampachová 3; Z. Mašková 2; M. Souček 1
Authors‘ workplace:
II. interní klinika Lékařské fakulty MU a FN u sv. Anny Brno, přednosta doc. MUDr. Miroslav Souček, CSc.
1; Klinika nemocí očních a optometrie Lékařské fakulty MU a FN u sv. Anny Brno, přednosta doc. MUDr. Svatopluk Synek, CSc.
2; I. patologicko‑anatomický ústav Lékařské fakulty MU a FN u sv. Anny Brno, přednostka doc. MUDr. Markéta Hermanová, Ph. D.
3
Published in:
Vnitř Lék 2008; 54(12): 1195-1205
Category:
Case Report
Overview
Obrovskobuněčná arteriitida (OA) patří mezi systémové vaskulitidy neznámé etiologie postihující cévy středního a velkého kalibru granulomatózní panarteriitidou s tvorbou granulomů tvořených obrovskými mnohojadernými buňkami. U 25–50 % pacientů s OA se objevuje postižení zraku. To může být prvním projevem OA nebo se může objevit týdny až měsíce po iniciálních projevech onemocnění. Oční příznaky onemocnění jsou převážně projevem okluzivního postižení očních a orbitálních arterií. Závažným důsledkem očního postižení v rámci OA je trvalé poškození zraku. K nejčastěji se vyskytujícím a současně k nejzávažnějším očním projevům OA patří arteriitická přední ischemická neuropatie zrakového nervu (AION). Projevuje se částečnou nebo úplnou ztrátou zraku. V terapii arteriitické AION při OA se používají vysoké dávky glukokortikoidů. Terapie glukokortikoidy je však zatížena řadou nežádoucích účinků. Důkaz efektu této terapie na zlepšení vízu pacientů s očním postižením u OA není přesvědčivý. Předkládáme případ 76letého muže s biopticky ověřenou OA, jejíž primární manifestaci představovala oboustranná arteriitická AION vedoucí k úplné ztrátě vízu jednoho oka a závažnému poklesu zrakových funkcí druhého oka. Terapie glukokortikoidy u tohoto pacienta vedla ke zlepšení vízu pouze jednoho oka a byla zatížena výskytem nežádoucích účinků. Z důvodu glukokortikoidy šetřícího efektu byl do terapie přidán metotrexát. Terapie glukokortikoidy mohla být ukončena po 3 letech. V průběhu terapie a v průběhu následujících 12 měsíců po jejím ukončení nedošlo k relapsu základního onemocnění.
Klíčová slova:
arteriitická přední ischemická neuropatie optického nervu – glukokortikoidy – obrovskobuněčná arteriitida – terapie – vaskulitida
Úvod
Obrovskobuněčná arteriitida (OA) patří mezi systémové vaskulitidy neznámé etiologie postihující cévy středního a velkého kalibru. První popis tohoto onemocnění poskytl Jonathan Hutchinson v roce 1890, když popsal případ 80letého muže, který pro otok a bolestivost temporálních arterií nemohl nosit klobouk [1]. Teprve v roce 1932 Horton et al popsali typický histologický obraz arteriitidy z biopsie temporální arterie [2]. Pravděpodobně ale poprvé popsal toto onemocnění již v 10. století Tadkwat Ali Iba Isu. OA se vyskytuje celosvětově. V Evropě je pozorován nárůst incidence onemocnění zejména v severnějších zeměpisných šířkách [3]. OA postihuje starší osoby mezi 50. a 90. rokem života. Maximum výskytu je popisován u 70letých. Registr v Olmsted County v Minnesotě udává roční incidenci onemocnění u osob ve věku 50 a více let 17 na 100 000 obyvatel a prevalenci 223 na 100 000 obyvatel [4,5]. Onemocnění postihuje 2–3krát častěji ženy.
Etiologie onemocnění není doposud známá. OA postihuje zejména evropskou kavkazoidní populaci, což může svědčit pro určitou roli genetických faktorů v etiologii onemocnění. Prokázána byla asociace s geny hlavního histokompatibilního komplexu (HLA) II. třídy, zejména s alelami HLA‑DRB1*04 [6,7]. Mimo jiné byla prokázána asociace mezi těmito alelami a výskytem a závažností očních projevů onemocnění. Hazleman et al prokázali vyšší frekvenci alely HLA‑B8 u pacientů s OA (50 %) ve srovnání s pacienty s revmatoidní artritidou (27 %) [8]. Rovněž byla prokázána asociace mezi vnímavostí ke vzniku onemocnění a mikrosatelitním polymorfizmem v genu pro TNFα, a to zejména s alelou TNFα2 [9]. Na druhé straně se připouští existence zevního spouštěcího faktoru onemocnění, např. opakované infekce lidským virem parainfluenzy, adenovirem nebo parvovirem B19 [10–12].
V patogenezi onemocnění se uplatňují mechanizmy buněčné i humorální imunity. U pacientů s OA bývá prokazován pokles počtu CD8+ T‑lymfocytů a naopak zvýšený počet CD4+ T‑lymfocytů [13]. Současná představa o patogenezi tohoto onemocnění předpokládá vliv infračerveného a slunečního záření, které vede k poškození lamina elastica interna povrchově uložených arterií [14]. Toto poškození cévní stěny usnadní prezentaci neznámého (auto) antigenu (pravděpodobně virového proteinu) prostřednictvím antigen prezentující buňky (makrofág) ve vazbě na HLA molekulu II. třídy CD4+ T‑lymfocytům, které jsou následně aktivovány. Aktivované CD4+ T‑lymfocyty exprimují na svém povrchu receptory pro IL‑2. Aktivované makrofágy a CD4+ T‑lymfocyty následně produkují prozánětlivé cytokiny, které spouští zánětlivý proces cévní stěny. CD4+ T‑lymfocyty byly opakovaně prokázány v zánětlivých infiltrátech cévní stěny v bioptických vzorcích větví temporálních arterií pacientů s OA. U některých pacientů s OA byla detekována zvýšená sérová hladina IL‑6 [13]. Někteří autoři uvádějí zvýšení hladiny cirkulujících imunokomplexů, zvýšení koncentrace komplementu a IgG u pacientů s OA [15].
Morfologicky je onemocnění charakterizováno granulomatózní panarteriitidou postihující tepny středního a velkého kalibru [5]. Postižení cévní stěny je segmentární, mezi patologicky změněnými úseky cévní stěny se nacházejí segmenty bez patologických změn. Dochází k tvorbě granulomů tvořených obrovskými mnohojadernými buňkami. Je porušena lamina elastica interna. Může se vyvinout i nekróza hladké svaloviny cévní stěny. Lumen postižené cévy bývá výrazně zúženo nebo zcela obliterováno. Příčinou těchto změn je kromě proliferace intimy rovněž rozvoj intraarteriální trombózy v místech aktivního zánětu. V adventicii se nachází zejména mononukleární infiltrát. V médii bývají přítomny obrovské mnohojaderné buňky a převážně mononukleární infiltrát. Vaskulitida postihuje zejména hrudní aortu a její hlavní větve. Nejčastěji bývají postiženy temporální arterie, vertebrální a oftalmické arterie. Intrakraniální arterie bývají postiženy zřídka. Popsány byly i případy postižení koronárního řečiště a větví břišní aorty.
V úvodu onemocnění se u většiny pacientů vyskytují nespecifické celkové příznaky jako zvýšení tělesné teploty, únava, celková slabost, nechutenství, hubnutí a psychické poruchy. Bolesti hlavy bývají přítomny u 67 % pacientů s OA [5]. Bolest bývá intenzivní, někdy vystřelující, lokalizovaná nejčastěji do spánkové oblasti, ale může se objevovat i v parietální oblasti nebo v záhlaví. 45–50 % pacientů si stěžuje na citlivost kůže hlavy. Ve spánkové oblasti můžeme nalézt ztluštělé, palpačně bolestivé, někdy uzlovité větve temporální arterie s oslabenou nebo vymizelou pulzací. Kůže nad nimi bývá někdy zarudlá. Až 67 % pacientů si stěžuje na klaudikační bolesti žvýkacího svalstva [5]. Z dalších příznaků se může objevovat brnění jazyka, ztráta chuti a bolesti v ústech a hltanu. Postižení koronárního řečiště se může manifestovat rozvojem infarktu myokardu a kardiální insuficiencí. Postižení velkých tepen je popisováno u 10–15 % pacientů s OA. Může se manifestovat šelesty slyšitelnými nad velkými tepnami zejména horních končetin. Časným příznakem postižení karotického řečiště může být hyperreaktivita karotického sinu. Neurologické projevy zahrnující poruchy sluchu, hemiparézy, deprese či zmatenost se vyskytují u 20–30 % nemocných. Jsou popsány i případy postižení tepen dolních končetin. Postižení štítné žlázy bývá popisováno u méně než 10 % pacientů.
K častým projevům OA patří postižení zraku. Oční projevy se objevují u 25–50 % pacientů. Incidence trvalé ztráty zraku klesla na 6–10 % zejména z důvodu včasné diagnostiky a terapie. Postižení zraku může být prvním projevem OA nebo se může objevit týdny až měsíce po iniciálních projevech onemocnění [16]. Oční příznaky onemocnění jsou převážně projevem okluzivního postižení očních a orbitálních arterií. Jsou tak závažné, že pacienti často vyhledají nejdříve oftalmologa. Postižení zraku se vyvíjí náhle a je nebolestivé. Projevuje se zamlženým viděním, výpadky části zorného pole, případně úplnou slepotou. Následující tabulka uvádí přehled očního postižení v rámci OA (tab. 1).

K nejčastěji se vyskytujícím a nejzávažnějším očním projevům OA patří arteriitická přední ischemická neuropatie zrakového nervu (AION). Ve většině případů je provázena těžkou poruchou vízu až úplnou amaurózou. Toto postižení vzniká v důsledku zánětlivého postižení cévní stěny zadních krátkých ciliárních arterií, které zásobují prelaminární oblast zrakového nervu a oblast lamina cribriformis. V sérii 800 pacientů s AION tvořila arteriitická forma tohoto postižení 12 % případů [17]. V důsledku zánětlivých změn v cévní stěně dochází ke ztluštění intimy a často k tvorbě intraarteriální trombózy, která uzavře lumen tepny. Vyvíjí se ischemie optického nervu a edém s ischemickou nekrózou prelaminární, laminární a retrolaminární části zrakového nervu.
Laboratorně bývají u většiny pacientů s OA přítomny výrazně zvýšené hodnoty sedimentace erytrocytů (FW). Hodnoty FW jsou užitečným nástrojem k monitorování aktivity onemocnění a sledování efektu léčby. Průměrné hodnoty FW u biopticky potvrzené OA se pohybují okolo 84 mm/hod. Až u 20 % pacientů s potvrzenou diagnózou bývají prokazovány normální hodnoty FW [18]. Nález zvýšené hladiny C‑reaktivního proteinu (CRP) > 24,5 mg/l je považován za vysoce senzitivní pro diagnózu OA. Na druhou stranu stanovení hladin proteinů akutní fáze (α1-antitrypsin, orosomucoid, haptoglobin, CRP) nepřispívá dle některých autorů výrazněji k monitoraci aktivity onemocnění než hodnoty FW [19]. Podle Hayreha et al má průkaz arteriitické AION a současně nález FW > 47 mm/hod a CRP > 24,5 mg/l až 97% specificitu pro diagnózu OA [20]. Z dalších laboratorních parametrů bývá přítomna mírná hypochromní anémie. Normální bývá počet leukocytů včetně diferenciálního rozpočtu. Počet trombocytů bývá normální nebo zvýšený. Mohou být přítomny zvýšené hodnoty alkalické fosfatázy, zejména u pacientů se současnými projevy polymyalgia rheumatica. V elektroforéze bílkovin mohou být nalezeny zvýšené hladiny α2-globulinů, méně často α1‑globulinů a γ-globulinů. Někdy může býtpřítomna zvýšená hladina IgG a C3a C4 složek komplementu. Dalším nálezem bývá pozitivita antikardiolipinových autoprotilátek ve třídě IgG u aktivního onemocnění.
Diagnóza OA se stanovuje na základě přítomnosti klinických příznaků onemocnění. Bioptické ověření je doporučeno provést u všech pacientů, u kterých vznikne podezření na toto onemocnění. Materiál se odebírá obvykle z větve temporální arterie, případně z okcipitální nebo faciální arterie. Vzhledem k segmentárnímu postižení cévní stěny je doporučeno získat vzorek dlouhý 3–5 cm. Důležité je provést histologické vyšetření z více míst vzorku. V případě jednostranné biopsie větve temporální arterie je udávána falešná negativita přibližně ve 4 % případů [21]. Někteří autoři proto doporučují současně odebrat bioptické vzorky temporální arterie na obou stranách, což může zvýšit pravděpodobnost potvrzení diagnózy o 5 % [22]. Udává se, že systémová terapie glukokortikoidy neovlivňuje bioptický nález až 14 dní od jejího zahájení. Přesto je doporučeno provést biopsii nejlépe 24 hod od zahájení terapie glukokortikoidy. V případě postižení aorty a jejich větví je jako doplňkové vyšetření možné provést angiografii nebo vyšetření pomocí magnetické rezonance. V literatuře se rovněž objevují zprávy o použití 18F-FDG pozitronové emisní tomografie v diagnostice tohoto onemocnění [23,24].
Ke stanovení diagnózy OA se používá několika diagnostických kritérií. V současné době jsou nejužívanější diagnostická kritéria American College of Rheumatology z roku 1990 [25] (tab. 2).
![Diagnostická kritéria American College of Rheumatology pro obrovskobuněčnou arteriitidu [25].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/5c0b62de7375b8e724fd618cabc2bcdc.png)
Kazuistika
V květnu roku 2004 byl na ambulanci Kliniky nemocí očních a optometrie FN u sv. Anny Brno odeslán 76letý pacient k došetření oboustranného poklesu zrakové ostrosti. Subjektivně si stěžoval na přibližně 2 dny trvající progredující pokles vízu pravého oka spolu s mírnějším poklesem vízu vlevo. Pacient dále udával, že před týdnem podstoupil extrakci zubu s přechodnou cefaleou v parietální oblasti. V předchorobí byl údaj o traumatické amputaci levé dolní končetiny při autohavárii v roce 1975, dále údaj o léčené arteriální hypertenzi diagnostikované v roce 1989, o prostatektomii s orchiektomií provedené v roce 1985 pro karcinom prostaty a o totální náhradě levého kyčelního kloubu po úrazové fraktuře krčku stehenní kosti v roce 1994. Pacient byl doposud léčen antihypertenzivy (metoprolol, nitrendipin, amilorid, hydrochlorthiazid), dále užíval omeprazol, antitrombotikum sulodexid a kalcium. V rodinné anamnéze byl nález cerebrovaskulárního onemocnění u prvostupňových příbuzných.
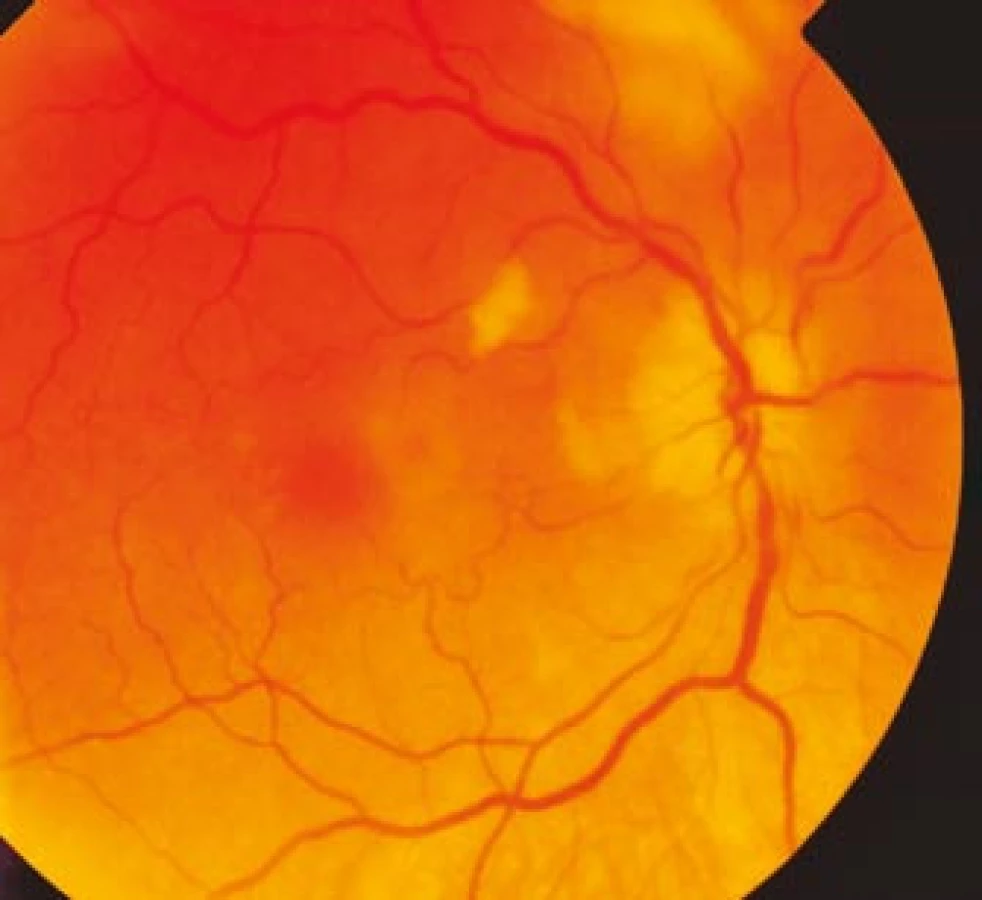
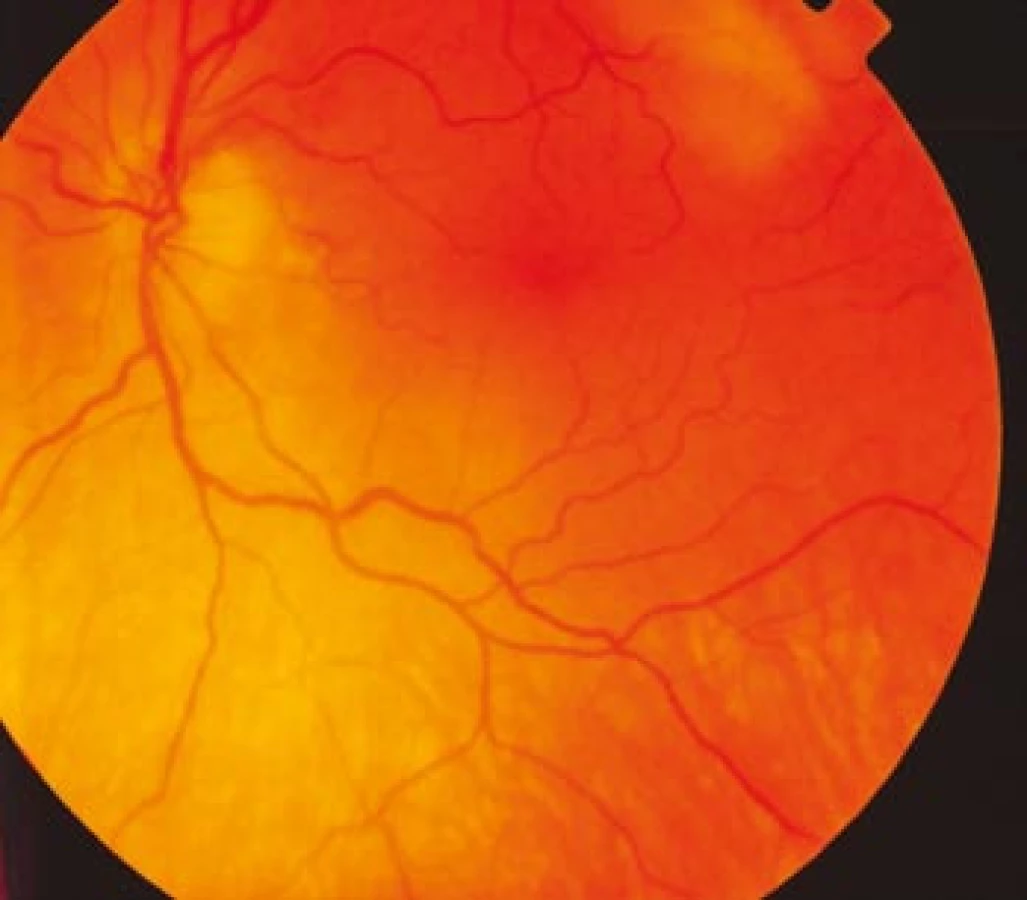
Zraková ostrost pravého oka při vstupním vyšetření byla na úrovni zbytků světlocitu s vadnou světelnou projekcí. Zraková ostrost levého oka byla 2/60 (0,03 při decimální notaci) se správnou světelnou projekcí. Nitrooční tlak byl 7/11 mm Hg. Nález na předním segmentu očním byl fyziologický, zornice pravého oka byla nepatrně širší (anizokorie). Přímá reakce na osvit byla vpravo nevýbavná, vlevo zachovaná. Nepřímá fotoreakce vpravo byla výbavná, vlevo chyběla, což svědčilo pro obraz amaurotické ztuhlosti zornice vpravo. Na očním pozadí byl vpravo terč zrakového nervu neostře ohraničen, bledý s ischemickým edémem, který zasahoval do okolní retiny. Peripapilárně se při horní temporální cévní arkádě nacházelo vatovité ložisko. Makulární krajina byla bez edému, sítnice byla přiložena do periferie. Žíly byly výrazně naplněny (obr. 1). Vlevo byla papila optického nervu temporálně nabledlá, lehce neostrých hranic, sítnice do periferie přiložena (obr. 2). Vyšetření zorného pole levého oka kinetickým perimetrem prokázalo centrální skotom (obr. 3). Evokované potenciály zrakové dráhy při zábleskové stimulaci byly na pravém oku nevýbavné, na levém oku byla hraničně prodloužená latence vlny P100. Bylo vysloveno podezření na pravostrannou arteriitickou AION se ztrátou zrakových funkcí a poklesem zrakové ostrosti levého oka.

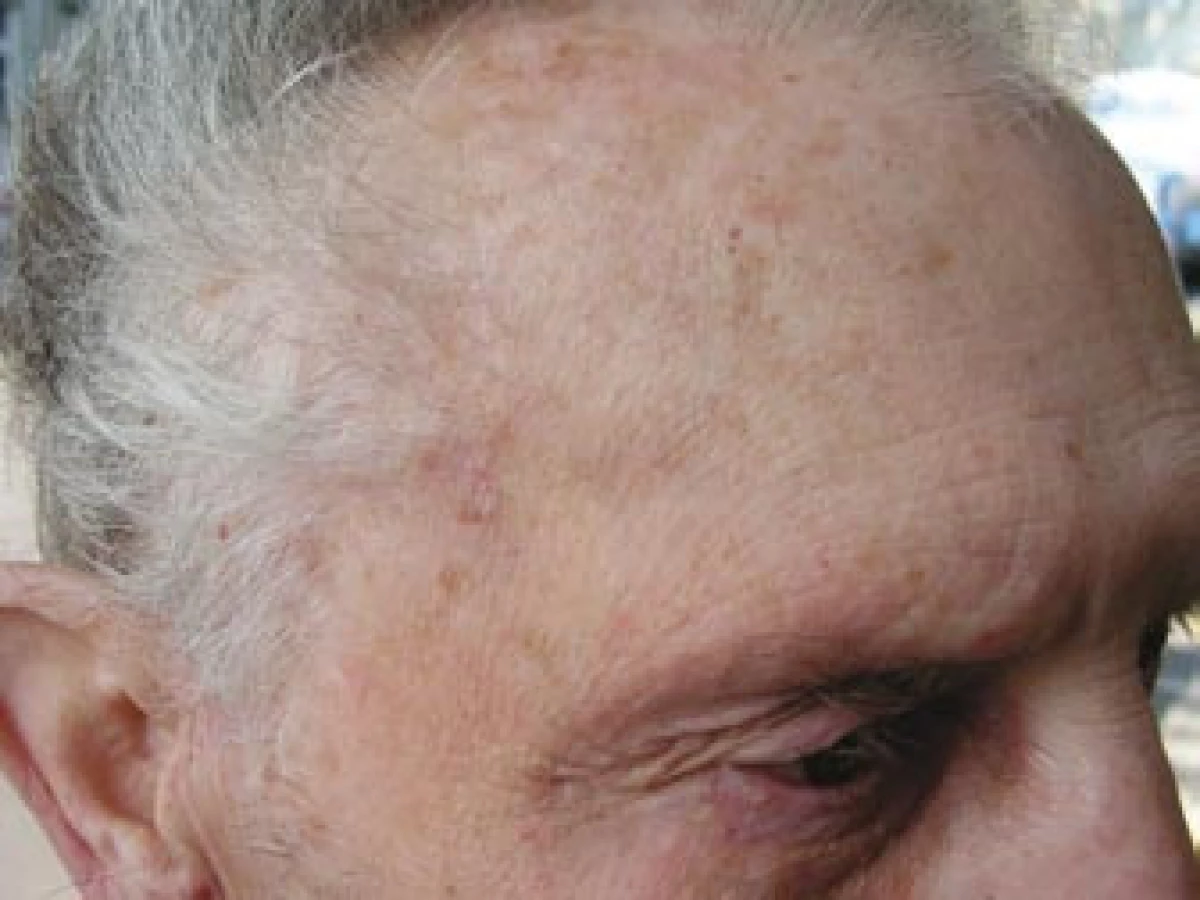
Pacient byl odeslán k vyšetření do revmatologické ambulance II. interní kliniky FN u sv. Anny Brno. Celkově se pacient cítil dobře, neudával zvýšení tělesné teploty, únavu, bolesti a ztuhlost svalů nebo svalovou slabost ani bolesti hlavy v posledním období. Na cílený dotaz udával přítomnost klaudikačních bolestí žvýkacího svalstva. Vyšetření hlavy prokázalo oboustranně klidnou spánkovou oblast, palpačně nebolestivou, se zachovanou pulzací větví temporální arterie, bez známek zánětu či zduření v jejím průběhu (obr. 4). Vyšetření hrudníku a břicha bylo bez pozoruhodností. Pulzace nad karotidami, tepnami horních končetin a pravé dolní končetiny byly hmatné bez slyšitelných šelestů. Krevní tlak byl 140/90 mm Hg, tepová frekvence byla pravidelná 64/min. Lymfatické uzliny nebyly zvětšené. Laboratorní vyšetření prokázalo zvýšenou hodnotu FW 70 mm/hod a středně zvýšenou hodnotu CRP 43,7 mg/l. V krevním obraze dominovala leukocytóza (12,8 × 109/l) s neutrofilií. Biochemické vyšetření prokázalo vyšší lačnou glykemii (7,9 mmol/l) a mírně zvýšenou hladinu kreatininu (119 μmol/l). Ostatní parametry byly v normě. V elektroforéze plazmatických bílkovin byly zvýšené hladiny α1-globulinů a α2-globulinů a snížená hladina γ-globulinů. Při imunologickém vyšetření byla prokázána mírně snížená hladina IgG 6,35 g/l (norma 7,00–16,00 g/l). Hladiny C3, C4 složek komplementu byly v normě, stejně jako hladiny cirkulujících imunokomplexů metodou PEGIKEM. Nebyla prokázána pozitivita autoprotilátek (ANA, anti ds-DNA, anti ENA, c-ANCA PR3+, p-ANCA MPO+, ACLA).Bylo vysloveno podezření na přítomnost OA s oboustrannou arteriitickou AION. Vzhledem k závažnému očnímu postižení byla neprodleně zahájena terapie 5 pulzy intravenózně podaného metylprednisolonu v dávce 250 mg. Následně byl pacient převeden na perorálně podávaný metylprednisolon v dávce 64 mg denně (1 mg/kg tělesné hmotnosti). Současně bylo zahájeno subkutánní podávání Fraxiparinu 0,8 ml denně.
V průběhu hospitalizace byla doplněna další vyšetření. RTG vyšetření orgánů dutiny hrudní neprokázalo patologii. Vyšetření mozku výpočetní tomografií prokázalo supratentoriálně v oblasti bazálních ganglií oboustranně starší ischemická ložiska o velikosti do 8 mm. Dalším nálezem bylo rozšíření Sylvické rýhy, až obraz arachnoidální cysty vlevo. Ultrasonografické vyšetření karotického povodí a vertebrálních arterií prokázalo pouze hemodynamicky nevýznamnou stenózu do 25 % a. carotis interna. Neurologické vyšetření konstatovalo přítomnost cévního onemocnění mozku a stav po opakovaných fokálních cerebrálních ischemických dysfunkcích v povodí a. carotis interna oboustranně.
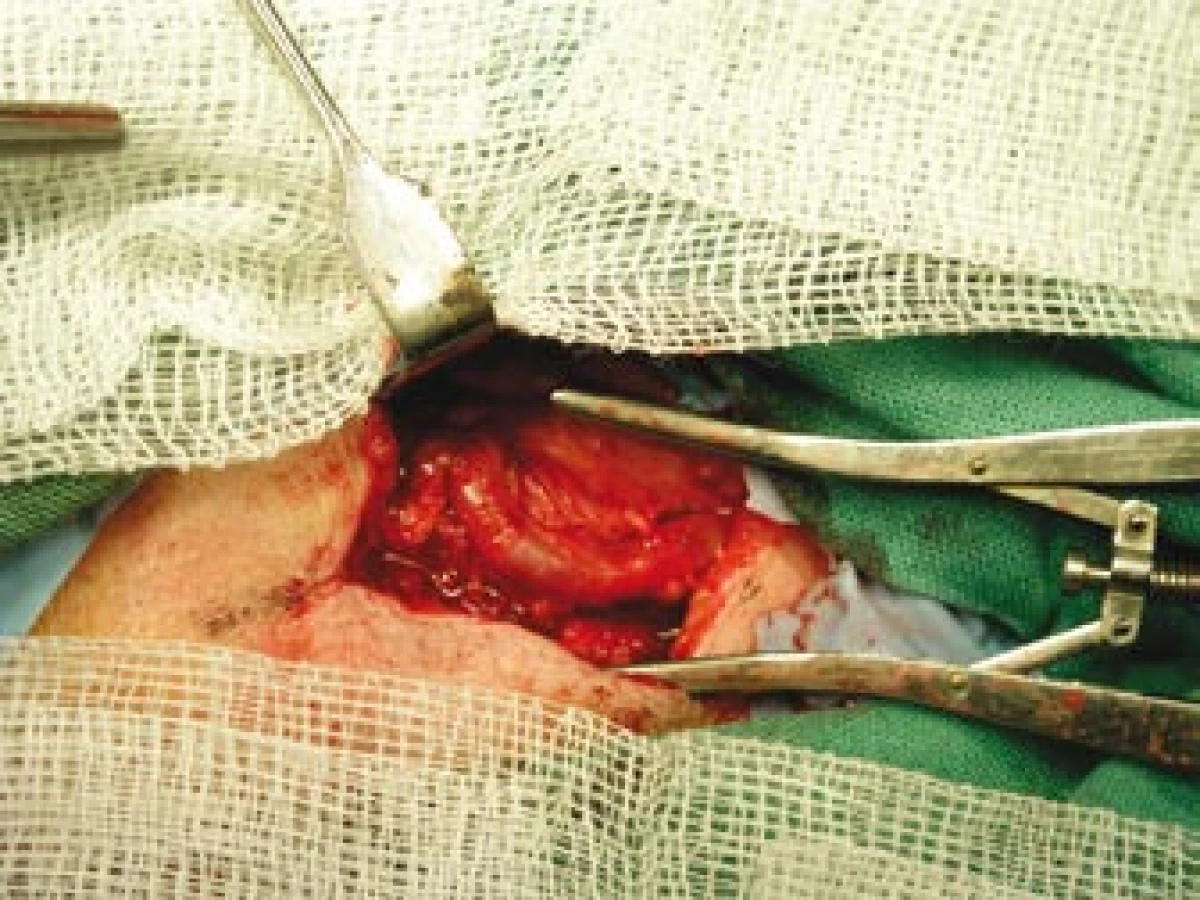
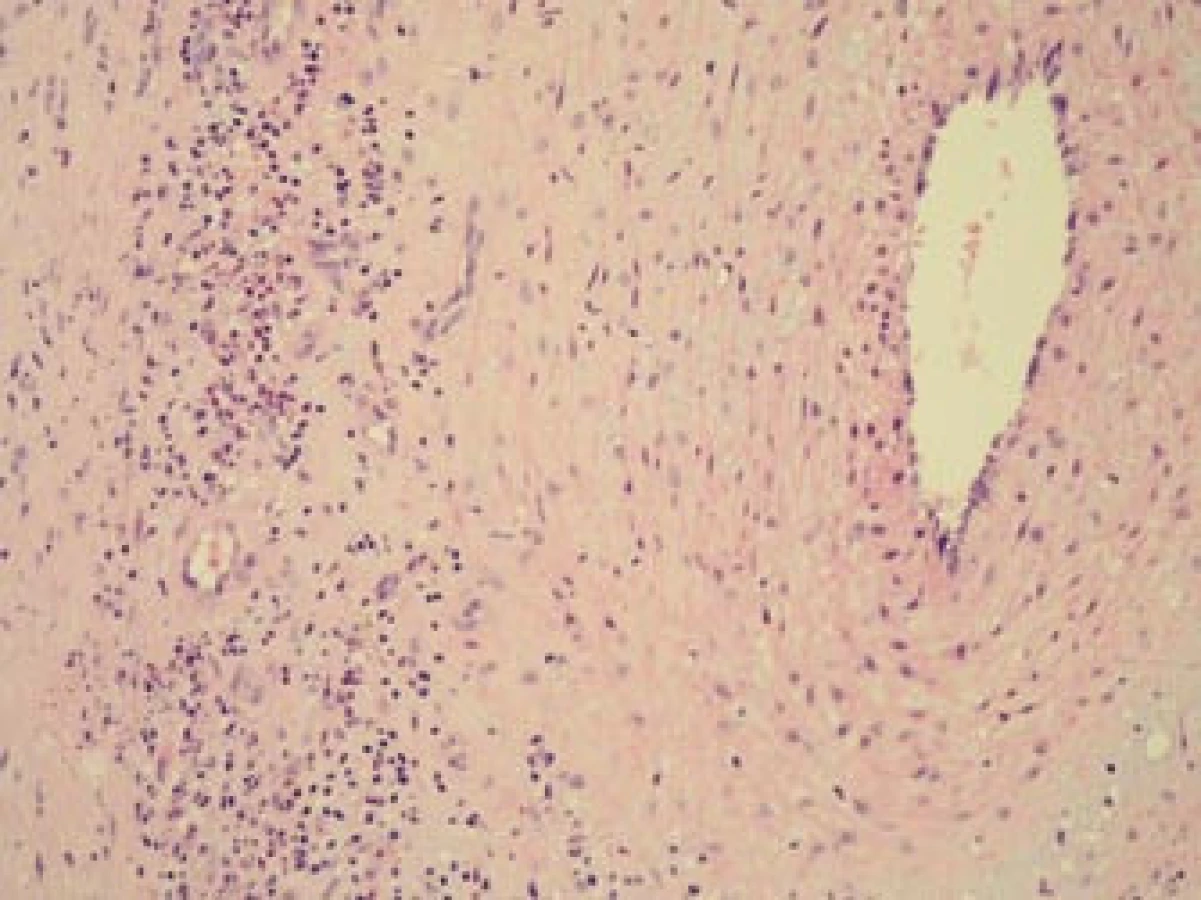
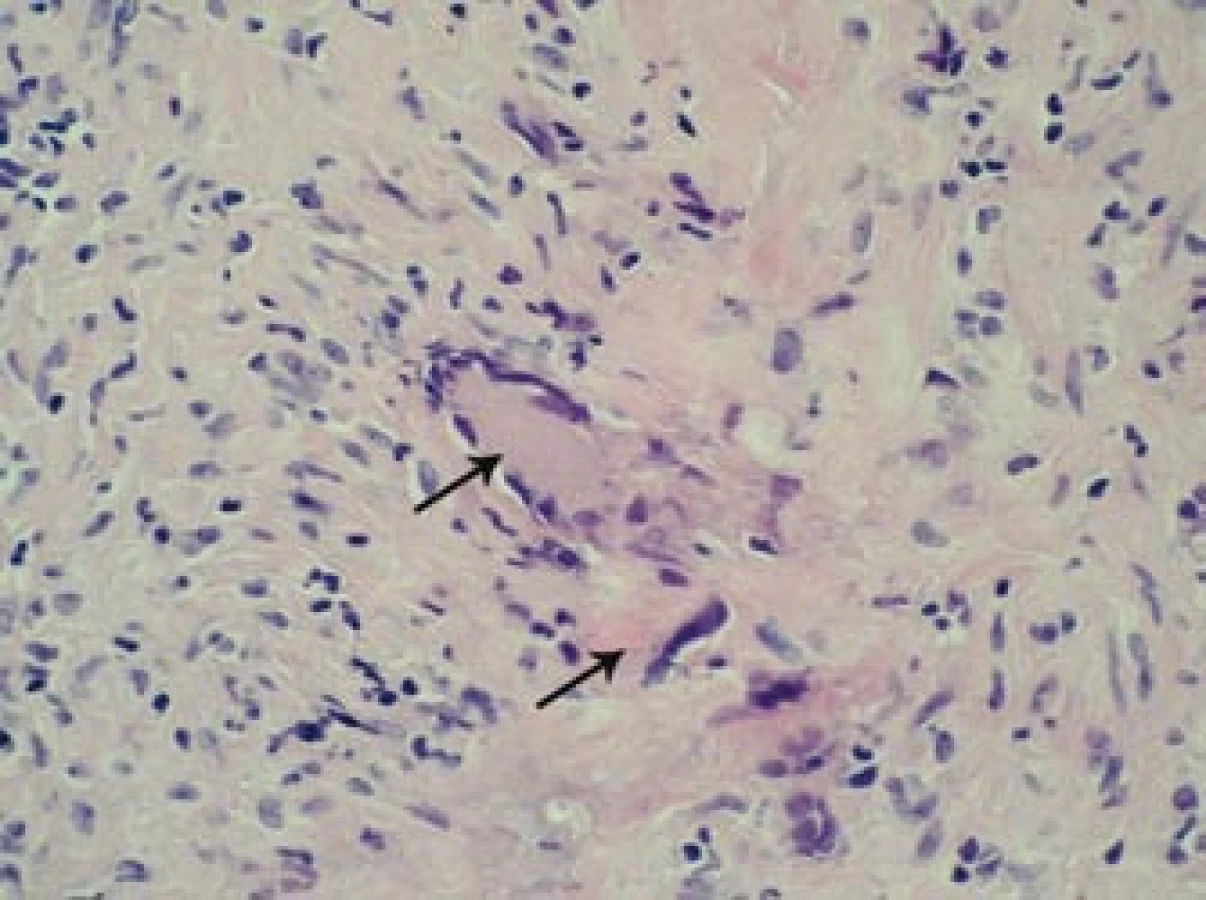
Šestý den od zahájení terapie byla provedena biopsie jedné z větví pravostranné temporální arterie v délce 4 cm (obr. 5, 6, 7). Histologické vyšetření prokázalo segmentární zánětlivé postižení cévní stěny s přítomností obrovských mnohojaderných buněk, s destrukcí lamina elastica interna a výraznou redukcí lumen cévy organizovanými tromby (obr. 8, 9, 10). Vyšetření potvrdilo diagnózu OA.



Od 2. dne hospitalizace na oční klinice zůstává pravé oko trvale amaurotické (bez světlocitu). Nejlépe korigovaná zraková ostrost (NKZO) levého oka 6/24, zaznamenaná 4. den hospitalizace, se mírně zlepšila 6. den hospitalizace na 6/12 a 12. den od zahájení terapie až na 6/9 parciálně. Pacient byl schopen s příslušnou addicí číst text Jäger č. 4.
Kontrolní laboratorní vyšetření, provedené 6. den po zahájení terapie, prokázalo normalizaci hladiny CRP (0,8 mg/l) a významný pokles FW (18 mm/hod). Pacient byl propuštěn do ambulantní péče.
Kontrolní vyšetření v revmatologické ambulanci v červnu roku 2004 prokázalo přetrvávající normalizaci laboratorních parametrů zánětu (CRP 0,9 mg/l, FW 14 mm/hod). Oční nález se významně nezměnil. Pokračovala terapie metylprednisolonem v dávce 64 mg/den. Do terapie byl přidán metotrexát v dávce 7,5 mg týdně se současnou substitucí kyseliny listové. Metotrexát byl do terapie přidán zejména z důvodu možnosti redukovat dávku glukokortikoidů a zabránit vývoji komplikací. V říjnu roku 2004 byla dávka metylprednisolonu postupně redukována na 32 mg denně a dávka metotrexátu zvýšena na 10 mg týdně. Rok po vzniku obtíží byl pacient léčen udržovací dávkou 8 mg metylprednisolonu denně a 10 mg metotrexátu týdně. Klinické ani laboratorní vyšetření neprokazovalo známky aktivity vaskulitidy. Denzitometrické vyšetření (DEXA) provedené v září roku 2004 prokázalo pokles T-skóre v oblasti bederní páteře (T-skóre –3,2) a v oblasti předloktí (T-skóre –4,2). Ke stávající terapii vápníkem a cholekalciferolem byl přidán risedronát v dávce 35 mg týdně. Při dlouhodobé terapii glukokortikoidy došlo k rozvoji cushingoidního syndromu a komplikované katarakty levého oka, která dále snížila vízus levého oka na 2/60. Proto bylo v září roku 2005 přistoupeno k fakoemulzifikaci katarakty s implantací umělé nitrooční čočky. Pooperační naturální zraková ostrost levého oka byla následně 6/9. Vízus 6/9 byl zaznamenán i při posledním očním vyšetření v dubnu roku 2008. Terapie metotrexátem byla ukončena v srpnu roku 2006 a terapie metylprednisolonem byla ukončena v květnu roku 2007. Doposud nejsou přítomny klinické ani laboratorní známky recidivy OA. Během období sledování byl zdravotní stav pacienta dále komplikován několika příhodami – v dubnu roku 2005 rozvojem levostranné bakteriální pleuropneumonie, v květnu roku 2006 rozvojem hluboké žilní trombózy pravé dolní končetiny, pro kterou bylo zahájeno podávání warfarinu. Striktura močové trubice byla řešena zavedením permanentního močového katétru.
Komentář
OA patří mezi systémové vaskulitidy neznámé etiologie postihující cévy středního a velkého kalibru granulomatózní panarteriitidou s tvorbou granulomů tvořených obrovskými mnohojadernými buňkami.
U 25–50 % pacientů s OA se objevuje postižení zraku. Může být prvním projevem OA nebo se může objevit týdny až měsíce po iniciálních projevech onemocnění [16]. Oční příznaky onemocnění jsou převážně projevem okluzivního postižení očních a orbitálních arterií. Oční postižení v rámci OA se projevuje zamlženým viděním, výpadky části zrakového pole, případně úplnou slepotou.
K nejčastěji se vyskytujícím a sou-časně k nejzávažnějším očním proje-vům OA patří arteriitická AION [26,27]. Projevuje se částečnou nebo úplnou ztrátou zraku. Vyvíjí se jako následek zánětlivého postižení cévní stěny zadních krátkých ciliárních arterií, které zásobují prelaminární oblast zrakového nervu a oblast lamina cribriformis. V důsledku zánětlivých změn v cévní stěně dochází ke ztluštění intimy a často k tvorbě intraarteriální trombózy, která uzavře lumen tepny. Dochází k rozvoji ischemie zrakového nervu, vývoji edému s ischemickou nekrózou prelaminární, laminární a retrolaminární částí zrakového nervu. V sérii 800 pacientů s AION Heyreha et al tvořila arteriitická AION 12 % případů [17]. Ostatní případy zahrnovaly tzv. nearteriitickou formu AION. Příčinou této formy AION je rovněž postižení cévního řečiště [28]. Nedostatečné prokrvení terče zrakového nervu, zhoršované strukturálním nakupením nervových vláken a podpůrných tkání na papile zrakového nervu může dosáhnout úrovně, kdy nedostatečná oxygenace vyústí v ischemii a otok papily zrakového nervu. Nearteriitická AION je často asociována s některými chorobami, které mohou vést k poklesu perfuzního tlaku nebo zvýšení rezistence krevního řečiště v rozsahu terče zrakového nervu, např. s arteriální hypertenzí, která bývá prokazována až u 47 % pacientů s nearteriitickou AION nebo s diabetes mellitus, který bývá přítomen až u 24 % pacientů. Prokazována je rovněž souvislost s přítomností kardiovaskulárních rizikových faktorů, např. s kouřením cigaret, hypercholesterolemií, dále s postižením tepen aterosklerózou nebo s přítomností ischemické choroby srdeční. K odlišení obou forem AION slouží především kombinace následujících parametrů: přítomnost systémových a očních příznaků, vysoké hodnoty FW a CRP, časná těžká ztráta zraku, bledý až křídově bílý otok terče zrakového nervu, asociace s okluzí cilioretinálních arterií, výrazně opožděné plnění až neperfuze cévnatky v průběhu fluorescenční angiografie fundu, biopsie větve temporální arterie [17]. V tab. 3 jsou uvedeny hlavní rozdíly mezi arteriitickou a nearteriitickou formou AION.

Základním lékem v terapii OA jsou glukokortikoidy [5]. Jejich podání vede k rychlému ústupu projevů onemocnění během několika dní. Glukokortikoidy snižují incidenci závažných komplikací onemocnění, např. očního postižení. Zlepšují kvalitu života pacientů s OA, ale chybí průkaz pro to, že by jejich podání vedlo ke zkrácení trvání choroby. V úvodu musí být podána dostatečná dávka glukokortikoidů, schopná potlačit aktivitu onemocnění. Obvykle doporučovanou dávkou na začátku onemocnění je 40–60 mg prednizonu denně. K monitorování účinku léčby je doporučeno sledovat hodnoty FW a CRP z důvodu rychlejší dynamiky změn [29,30]. Snižování dávek prednizonu by nemělo být rychlejší než 5 mg týdně a na konci 1. měsíce léčby by dávka neměla být nižší než 20 mg prednizonu denně. V případě nižších dávek se zvyšuje pravděpodobnost relapsu onemocnění a narůstá mortalita pacientů. Relaps onemocnění se objevuje nejčastěji v průběhu prvního roku a půl trvání choroby a v 1. roce po ukončení terapie glukokortikoidy. Po dosažení denní dávky 10 mg prednizonu by další redukce měla pokračovat pomaleji, přibližně o 1 mg každé 2–4 týdny. Po 2 letech léčby je možné u 1/3 až 1/2 pacientů ukončit terapii glukokortikoidy bez rizika relapsu onemocnění. Je proto doporučováno, aby terapie glukokortikoidy trvala minimálně 2 roky. Terapie glukokortikoidy je však spojena s výskytem nežádoucích účinků u 60–90 % pacientů. K nejčastějším patří nárůst tělesné hmotnosti (> 50 %) a glukokortikoidy indukovaná osteoporóza (> 40 %), která častěji postihuje ženy [31]. U pacientů rezistentních na terapii glukokortikoidy nebo při výskytu nežádoucích účinků je možné v terapii OA použít azathioprin, metotrexát, cyklosporin, dapson nebo hydroxychlorochin. Důkaz o jejich účinnosti je však založen pouze na jednotlivých kazuistických sděleních, případně na krátkodobých nekontrolovaných studiích s relativně malým počtem pacientů a s vysokým podílem pacientů s předčasně ukončenou terapií [32].
V případech OA s očním postižením, zejména při hrozící ischemii zrakového nervu, je doporučena vyšší dávka glukokortikoidů, přibližně 1 mg/kg prednizonu denně. Alternativou je pulzní parenterální podání vysokých dávek metylprednisolonu v intervalech 6–8 hod, následované perorálním prednizonem v dávce 1 mg/kg denně. Je‑li vážné klinické podezření na arteriitickou formu AION, měla by být terapie glukokortikoidy zahájena neprodleně bez ohledu na plánovanou biopsii temporální arterie. V posledních přibližně 50 letech bylo v odborné literatuře publikováno několik sdělení prokazujících částečný efekt terapie glukokortikody na zlepšení vízu u pacientů s poškozením zraku na podkladě arteriitické AION, případně s jiným typem ischemického očního postižení [33]. Podle principu medicíny založené na důkazech však tyto práce představují IV., případně III. kategorii evidence. Chybí tedy data z větších prospektivních kontrolovaných, ale i nekontrolovaných studií. V retrospektivní studii Hayera et al, zahrnující 84 pacientů (114 očí) s postižením zraku, zejména z důvodu arteriitické AION (91 %), došlo při terapii glukokortikoidy ke zlepšení vízu pouze u 4 % případů [33]. Podle autorů mohly být lepší výsledky popisované v předchozích studiích mimo jiné důsledkem nesprávného hodnocení vyšetření zorného pole kinetickým perimetrem. Autoři rovněž zdůrazňují nutnost velmi včasného zahájení terapie glukokortikoidy. Uvádějí, že možnost obnovení funkce zrakového nervu je závislá na tíži a délce trvání ischemie. Po překonání kritické periody trvání ischemie dochází k trvalému poškození nervových struktur, což jsme mohli pozorovat rovněž u našeho pacienta, u něhož zůstalo pravé oko i přes léčbu amaurotické, zatímco na levém oku došlo při léčbě k úpravě zrakových funkcí.
Další doporučovanou terapií arteriitické AION je podávání terapeutické dávky he-pa-rinu nebo nízkomolekulárního he-pa-rinu po dobu 5–7 dní s následnou terapií antiagregační dávkou kyseliny acetylsalicylové. Pro tuto terapii však rovněž neexistují kontrolované studie prokazující její efekt.
V odborné literatuře je možno nalézt rovněž několik kazuistických sdělení prokazujících efekt anti‑TNFα terapie (infliximab, adalimumab, etanercept) a anti CD20 monoklonální protilátky (rituximab) v terapii OA refrakterní na terapii glukokortikoidy [34–38]. V letošním roce byly publikovány výsledky prospektivní randomizované kontrolované studie, ve které bylo placebo nebo etanercept přidáno k redukované dávce glukokortikoidů u pacientů s OA s nežádoucími účinky terapie glukokortikoidy [39]. Primárním cílem studie byla možnost ukončit terapii glukokortikoidy a kontrola aktivity onemocnění během 1 roku sledování. Do studie bylo randomizováno celkem 8 pacientů léčených etanerceptem a 9 pacientů léčených placebem. Po 12 měsících sledování bylo po ukončení terapie glukokortikoidy onemocnění pod kontrolou u 50 % pacientů léčených etanerceptem oproti 22,2 % pacientů léčených placebem. Vzhledem k nízkému počtu zařazených pacientů nebyly výsledky statisticky významné. Kumulativní dávka glukokortikoidů byla nižší ve skupině léčené etanerceptem (p = 0,03). Autoři uzavírají, že terapie etanerceptem byla dobře tolerována, ale vzhledem k nízkému počtu pacientů je nutné tyto výsledky ověřit na větším souboru.
Arteriitická AION tedy představuje zá-važnou orgánovou komplikaci OA, která vede v mnohých případech k úplné ztrátě vízu s rizikem postižení obou očí. V terapii je doporučeno včasné zahájení podání vysokých dávek glukokortikoidů. Údaje o efektu glukokortikoidů na zmírnění poškození vízu jsou sporné. Navíc je terapie glukokortikoidy zatížena výskytem nežádoucích účinků. Neexistují jednotná doporučení pro dávku a délku trvání léčby glukokortikoidy. K terapii OA je tedy nutno přistupovat individuálně s ohledem na stav pacienta, rozsah postižení a riziko vývoje nežádoucích účinků léčby.
U našeho pacienta můžeme onemocnění označit jako tzv. okultní OA, která se projevuje jako náhlá ztráta zrakových funkcí s minimálními systémovými příznaky u přibližně 10–20 % pacientů s OA. Simultánní bilaterální postižení zrakového nervu je rovněž výjimečné. I při závažné prognóze a velmi nízké počáteční úrovni vízu obou očí se podařilo stanovením správné diagnózy a včasným zahájením imunosupresivní terapie uchovat dobré zrakové funkce alespoň jednoho oka, kde tkáňová ischemie nedosáhla kritické hranice pro nevratné poškození zrakového nervu.
MUDr. Petr Němec, Ph.D.
www.fnusa.cz
e‑mail: petr.nemec@fnusa.cz
Doručeno do redakce: 16. 6. 2008
Přijato po recenzi: 18. 7. 2008
Sources
1. Hutchinson J. A peculiar form of neurotic arthritis of the aged which is sometimes productive of gangrene. Arch Surg 1890; 1 : 323–327.
2. Horton BT, Magath TB, Brown GE. An underscribed form of arteritis of the temporal vessels. Mayo Clin Proc 1932; 7 : 700–701.
3. Watts RA, Lane S, Bentham G et al. Is there a latitudinal variation in the incidence of giant cell arthritis? Proceeding of the American College of Rheumatology, Philadelphia, October 2000. Arthritis Rheum 2000; 43 (Suppl): S137.
4. Machado EB, Michet CJ, Ballard DJ et al. Trends in incidence and clinical presentation of temporal arteritis in Olmsted County, Minnesota, 1950–1985. Arthritis Rheum 1988; 31 : 745–749.
5. Hazleman BL. Polymyalgia rheumatica and giant cell arthritis. In: Hoch-berg MC, Smolen JS, Winblatt ME et al. Rheumatology. 3rd ed. Edinburgh, London, New York, Oxford, Philadelphia, St. Louis, Sydney, Toronto: Mosby 2003 : 1623–1633.
6. González-Gay MA, Garcia-Porrua C, Rivas MJ et al. Epidemiology of biopsy proven giant cell arteritis in North Western Spain: trend over an 18 year period. Ann Rheum Dis 2001; 60 : 367–371.
7. Weyand CM, Hunder NN, Hicok KC et al. HLA‑DRB1 alleles in polymyalgia rheumatica, giant cell arthritis and rheumatoid arthritis. Arthritis Rheum 1994; 37 : 514–520.
8. Hazleman BL. Polymyalgia rheumatica and giant cell arthritis. In: Klippel K, Dieppe PA (eds). Rheumatology. St. Louis: Mosby 1994.
9. Mattey DL, Hajeer AH, Dababneh A et al. Association of giant cell arteritis and polymyalgia rheumatica with different tumor necrosis factor microsatellite polymorphisms. Arthritis Rheum 2000; 43 : 1749–1755.
10. Duhaut P, Bosshard S, Calvet A et al. Giant cell arteritis, polymyalgia rheumatica, and viral hypotheses: a multicenter, prospective case-control study. Groupe de Recherche sur l‘Artérite à Cellules Géantes. J Rheumatol 1999; 26 : 361–369.
11. Cimmino MA, Grazi G, Balistreri M et al. Increased prevalence of antibodies to adenovirus and respiratory syncytial virus in polymyalgia rheumatica. Clin Exp Rheumatol 1993; 11 : 309–313.
12. Salvarani C, Farnetti E, Casali B et al. Detection of parvovirus B19 DNA by polymerase chain reaction in giant cell arteritis: a case-control study. Arthritis Rheum 2002; 46 : 3099–3101.
13. Rovenský J, Tuchyňová A. Polymyalgia rheumatica a obrovskobuněčná (temporální) arteritida. In: Pavelka K, Rovenský J. Klinická revmatologie. 1. vyd. Praha: Galén 2003 : 317–323.
14. Cimmino MA. Genetic and environmental factors in polymyalgia rheumatica. Ann Rheum Dis 1997; 56 : 576–577.
15. Malmvall BE, Bengtsson BA, Nilsson LA et al. Immune complexes, rheumatoid factors, and cellular immunological parameters in patients with giant cell arteritis. Ann Rheum Dis 1981; 40 : 276–280.
16. Hayreh SS, Podhajsky PA, Zimmerman B. Ocular manifestations of giant cell arteritis. Am J Ophthalmol 1998; 125 : 509–520.
17. Hayreh SS. Anterior ischaemic optic neuropathy. Differentiation of arteritic from non‑arteritic type and its management. Eye 1990; 4 : 25–41.
18. Wise CM, Agudelo CA, Chmelewski WL et al. Temporal arteritis with low erythrocyte sedimentation rate: a review of five cases. Arthritis Rheum 1991; 34 : 1571–1574.
19. Kyle V, Cawston TE, Hazleman BL. Erythrocyte sedimentation rate and C reactive protein in the assessment of polymyalgia rheumatica/giant cell arteritis on presentation and during follow up. Ann Rheum Dis 1989; 48 : 667–671.
20. Hayreh SS, Podhajsky PA, Raman R et al. Giant cell arteritis: validity and reliability of various diagnostic criteria. Am J Ophthalmol 1997; 123 : 285–296.
21. Danesh-Meyer HV, Savino PJ, Eagle RC Jr et al. Low diagnostic yield with second biopsies in suspected giant cell arteritis. J Neuroophthalmol 2000; 20 : 213–215.
22. Pless M, Rizzo JF 3rd, Lamkin JC et al. Concordance of bilateral temporal artery biopsy in giant cell arteritis. J Neuroophthalmol 2000; 20 : 216–218.
23. Wenger M, Calamia KT, Salvarani C et al. Do we need 18F-FDG-positron emission tomography as a functional imaging technique for diagnosing large vessel arteritis? Clin Exp Rheumatol 2003; 21 (Suppl 32): S1–S2.
24. Blockmans D. The use of (18F)fluo-ro‑deoxyglucose positron emission tomography in the assessment of large vessel vasculitis. Clin Exp Rheumatol 2003; 21 (Suppl 32): S15–S22.
25. Hunder GG, Bloch DA, Michel BA et al. The American College of Rheumatology 1990 criteria for the classification of giant cell arteritis. Arthritis Rheum 1990; 33 : 1122–1128.
26. Otradovec J. Klinická neurooftalmologie. Praha: Grada 2003.
27. Jirásková N. Ischemická neuropatie optiku. In: Jirásková N. Neurooftalmologie, minimum pro praxi. Praha: Triton 2001 : 26–30.
28. Arnold CA. Ischemic Optic Neuropathies. In: Yanoff M, Duker JS (eds): Ophthalmology. 2nd ed. St. Louis: Mosby 2004 : 1268–1272.
29. Hayreh SS. Steroid therapy for visual loss in patients with giant-cell arteritis. Lancet 2000; 355 : 1572–1573.
30. Barrier JH, Chevalet P, Ponge T. Principles of acute treatment of Horton’s disease. Role of high‑dose corticosteroid therapy. Ann Med Interne 1998; 149 : 448–453.
31. Meola DC, Fierz A, Tschopp A et al. Corticosteroids in giant cell arteritis: primum nil nocere? Klin Monatsbl Augen-heilkd 2006; 223 : 379–381.
32. Pipitone N, Salvarani C. Improving therapeutic options for patients with giant cell arteritis. Curr Opin Rheumatol 2008; 20 : 17–22.
33. Hayreh SS, Zimmerman B, Kardon RH. Visual improvement with corticosteroid therapy in giant cell arteritis. Report of a large study and review of literature. Acta Ophthalmol Scand 2002; 80 : 355–367.
34. Benucci M, Manfredi M, Puce F et al. Improvement in visual acuity in a patient with ischaemic optic neuropathy (Horton arteritis) undergoing therapy with infliximab: a case report. Recenti Prog Med 2007; 98 : 624–626.
35. Pipitone N, Salvarani C. Improving therapeutic options for patients with giant cell arteritis. Curr Opin Rheumatol 2008; 20 : 17–22.
36. Ahmed MM, Mubashir E, Hayat S et al. Treatment of refractory temporal arteritis with adalimumab. Clin Rheumatol 2007; 26 : 1353–1355.
37. Bhatia A, Ell PJ, Edwards JC. Anti‑CD20 monoclonal antibody (rituximab) as an adjunct in the treatment of giant cell arteritis. Ann Rheum Dis 2005; 64 : 1099–1100.
38. Mayrbaeurl B, Hinterreiter M, Burgstaller S et al. The first case of a patient with neutropenia and giant-cell arteritis treated with rituximab. Clin Rheumatol 2007; 26 : 1597–1598.
39. Martínez-Taboada VM, Rodríguez-Valverde V, Carreño L et al. A double-blind placebo controlled trial of etanercept in patients with giant cell arteritis and corticosteroid side effects. Ann Rheum Dis 2008; 67 : 625–630.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2008 Issue 12
Most read in this issue
- Nová evidence‑based kritéria pro posouzení vhodnosti lékového režimu u seniorů. Kritéria STOPP (Screening Tool of Older Person’s Prescriptions) a START (Screening Tool to Alert doctors to Right Treatment)
- Schnitzlerův syndrom – popis čtrnáctiletého průběhu nemoci a přehled informací o této nemoci
- Obrovskobuněčná arteriitida manifestující se oboustrannou arteriitickou přední ischemickou neuropatií zrakového nervu (AION)
- Léčba invazivní kandidózy – doporučení odborných společností