
Schnitzlerův syndrom – popis čtrnáctiletého průběhu nemoci a přehled informací o této nemoci
Schnitzler syndrome – report on a fourteen-year course of the disease and an overview of information on the disease
Schnitzler syndrome is a rare disease characterised by chronic urticaria and the presence of monoclonal IgM immunoglobulin, and by other symptoms. We report our experience with 14-year treatment of a patient. The first medical examination in our workplace was at the beginning of 1995 and the patient was diagnosed with the disease in 1996 (at the age of 52). Antihistaminics, the first medication used to relieve the symptoms of urticaria, had no subjective or objective effect. After the detection of osteolytic-osteosclerotic changes in the pelvic region, in areas with intense pain, we started treatment with pamidronate (90 mg at 28-day intervals), and the pain disappeared completely within 3 months of application of the drug. When the bisphosphonate therapy was interrupted, the pain recurred and receded completely after renewal of bisphosphonate administration. After the diagnosis, we gave the patient high doses of dexametazone (40 mg day 1–4, 10–13 and 20–23, at 28-day cycles). However, the therapy suppressed urticaria only on the days dexametasone was administered and the effect did not last when the drug was discontinued. Therefore we moved to continuous daily doses of prednisone (10–30 mg, depending on the intensity of problems), which was the only therapy with a long-term effect which was relatively well tolerated at the same time. Based on the excellent effect of 2-chlordeoxyadenosine in Waldenström’s macroglobulinaemia, three cycles of this therapy were administered to the patient in 1996 (0.1 mg/kg/day, 7 days, at 28-day intervals). After the first infusion, urticarious lesions disappeared, but the positive effect on skin eruptions was limited in time and lasted only 14 days after the last infusion, i.e. the medication proved ineffective from a long-term point of view. The first improvement lasting for a longer period of time (partial remission) was achieved by regular application of interferon α (3 QU 3 times a week). However, adverse effects of interferon α prevailed after two years and the therapy was discontinued. Similarly phototherapy using the PUVA method resulted in partial regression of urticarious symptoms. Subsequently tested cyclosporine A (5 mg/kg/day) brought no benefit. Thalidomide (100 mg in the evening) administered on a continuous basis relieved pruritus and improved sleep disturbed by pruritus. However, adverse effects prevailed after 4 months and the therapy had to be discontinued, too. In 2005, we were hoping to achieve positive results with the most effective treatment for multiple myeloma of the time, a combination of bortezomib (1.3 mg/m2 i.v. on day 1, 4, 8 and 11, thalidomide 100 mg daily and dexametazon 20 mg p.o. on days 1–4 and 8–11 in 21-day cycles – VTD). A total of 4 complete cycles and 4 cycles with bortezomib reduced by 50% were applied. Urticarious eruptions were reduced by at least 50% in the course of the therapy, and also the concentration of monoclonal immunoglobin decreased temporarily by more than 50%. However, after the therapy was discontinued, the symptoms returned with their original intensity, which means that VTD regime did not provide a long-term therapeutic response. In 2007, we started the anakinra (Kineret) therapy. Skin symptoms disappeared after the first injection and a dose of 100 mg/day has kept the patient free of skin symptoms for 12 months by now. Also the CRP value which had been constantly high returned to normal, and haemoglobin values increased to achieve physiological range. In the course of 14 years, we confirmed partial therapeutic effect of glucocorticoids administered on a continuous basis, as well as a partial therapeutic effect of interferon α, thalidomide and PUVA, but all the therapies had to be discontinued due to adverse effects. A major turn, i.e. the complete disappearance of skin symptoms and normalisation of CRP and haemoglobin values, only came with anakinra which has become the drug of the first choice for the above syndrome.
Key words:
anakinra – bortezomib – bisphosphonates – cyclosporine A – interferon α – thalidomide – 2-chlordeoxyadenosin – Schnitzler syndrome – monoclonal gammopathy – Waldenström’s macroglobulinaemia – multiple myeloma
Authors:
Z. Adam 1; M. Krejčí 1; L. Pour 1; J. Neubauer 2; J. Prášek 3; R. Hájek 1
Authors‘ workplace:
Interní hematoonkologická klinika Lékařské fakulty MU a FN Brno, pracoviště Bohunice, přednosta prof. MUDr. Jiří Vorlíček, CSc.
1
Published in:
Vnitř Lék 2008; 54(12): 1140-1153
Category:
Reviews
Overview
Schnitzlerův syndrom je vzácná choroba, charakterizovaná chronickou kopřivkou a přítomností monoklonálního imunoglobulinu třídy IgM a dalšími znaky. V textu popisujeme 14leté zkušenosti s léčbou jednoho pacienta. První vyšetření bylo u nás počátkem roku 1995 a diagnóza byla stanovena v roce 1996 (ve věku 52 let). Antihistaminika, první léky, které tento nemocný užíval pro zmírnění kopřivkových potíží, byla zcela bez subjektivního i objektivního efektu. Po zjištění osteolyticko‑osteosklerotických změn v oblasti pánve, v místech s intenzivní bolestí, jsme začali nemocného léčit pamidronatem (90 mg ve 28denních intervalech) a po 3 měsících aplikace tohoto léku bolesti kostí zcela vymizely. Pokud se bisfosfonáty přerušily, bolesti se obnovily, po obnovení podávání bisfosfonátů bolesti opět zcela ustoupily. Po stanovení diagnózy jsme pacientovi podávali vysoké dávky dexametazonu (40 mg 1.–4., 10.–13 a 20.–23. den ve 28denních cyklech). Tato léčba odstranila kopřivkové problémy jen ve dnech podávání dexametazonu, po ukončení účinek dexametazonu nepřetrvával. Přešli jsme tedy na kontinuální denní dávky prednisonu (10–30 mg, dle intenzity potíží), které jediné byly dlouhodobě účinné a relativně dobře tolerované. Vzhledem k excelentnímu účinku 2-chlordeoxyadenosinu u Waldenströmovy makroglobulinemie byly v roce 1996 podány nemocnému 3 cykly této léčby (0,1 mg/kg/den 7 dní v 28denních cyklech). Po první infuzi sice vymizely kopřivkové morfy, příznivý vliv na kožní výsev byl časově omezený, přetrvával maximálně 14 dní od poslední infuze, takže z dlouhodobého pohledu byl tento lék nepřínosný. První dlouhodobější zlepšení (parciální remisi) přinesla pravidelná aplikace interferonu α (3 MU 3krát týdně). Po 2 letech léčby interferonem α převládly ale jeho nežádoucí účinky nad žádoucími, a proto byl vysazen. Podobně fototerapie metodou PUVA přinášela po 2 roky parciální regresi kopřivkových projevů. Následně testovaný cyklosporin A (5 mg/kg/den) byl bez jakéhokoliv přínosu. Thalidomid (100 mg večer) kontinuálně podávaný tlumil svědění a zlepšoval spánek, dříve rušený svěděním. Po 4. měsíci však převládly nežádoucí účinky nad žádoucími, a tak byl také vysazen. V roce 2005 jsme s nadějí zkoušeli v té době nejúčinnější léčbu u mnohočetného myelomu, kombinaci bortezomibu (1,3 mg/m2 i.v.1., 4., 8. a 11. den, thalidomidu 100 mg denně a dexametazonu 20 mg p.o. 1.–4. a 8.–11. den v 21denních cyklech – VTD). Celkem byly aplikovány 4 kompletní cykly a 4 cykly s 50% redukcí bortezomibu. Po dobu podávání této léčby se kopřivkové erupce nejméně o 50 % zmenšily a dočasně poklesla i koncentrace monoklonálního imunoglobulinu o více než 50 %, po ukončení této léčby se však projevy nemoci ihned navrátily v původní intenzitě, takže z dlouhodobého hlediska ani režim VTD nepřinesl léčebnou odpověď. V roce 2007 jsme zahájili léčbu preparátem anakinra (Kineret). Kožní projevy po první injekci ihned vymizely a při aplikaci 100 mg denně je pacient již 12. měsíc bez kožních projevů. Normalizovala se dříve trvale zvýšená hodnota CRP a hemoglobin se zvýšil na fyziologické rozmezí. V průběhu 14 let jsme potvrdili parciální léčebný efekt kontinuálního podávání glukokortikoidů, dále parciální léčebný efekt interferonu α, thalidomidu a PUVA, všechny postupy však musely být po čase ukončeny pro nežádoucí účinky. Zásadní zvrat, úplné vymizení kožních projevů a normalizaci hodnot CRP a hemoglobinu, přineslo až zahájení podávání preparátu anakinra, který se stává pro tento syndrom lékem volby.
Klíčová slova:
anakinra – bortezomib – bisfosfonáty – cyklosporin A – interferon α – thalidomid – 2‑chlordeoxyadenosin – Schnitzlerův syndrom – monoklonální gamapatie – Waldneströmova makroglobulinemie – mnohočetný myelom
Úvod
V roce 1974 popsal Schnitzler se spoluautory 5 pacientů s chronickou kopřivkou (urtikariální vaskulitidou), spojenou s kostními změnami (kombinace hyperostózy a osteolýzy), s lymfadenopatií a s přítomností monoklonálního imunoglobulinu třídy IgM [1,2]. V následujících letech se objevily další popisy této vzácné nozologické jednotky [3–9]. Její diagnostická kritéria a typické příznaky shrnuje tab. 1. Schnitzlerův syndrom je nutno mít na zřeteli vždy při diferenciální diagnostice chronických kopřivkových projevů a teploty nejasného původu [6,10–14].
![Diagnostická kritéria Schnitzlerova syndromu [12].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/9fdc2a141305e0e2ab203c63268211e3.png)
V následujícím textu popíšeme naše 14leté zkušenosti s léčením jednoho nemocného s touto vzácnou diagnózou a v diskuzi shrneme současné znalosti o této nemoci, její léčbě i naše zkušenosti.
Popis případu – kazuistika
Muž, narozený 1944, neměl do roku 1989 žádné vážnější onemocnění. Od roku 1989 se datují kožní problémy typu urtiky, pro něž navštívil četné dermatologické ambulance. Po vyšetřeních byl nález vždy shrnut do 3 slov: urtika nejasného původu. V roce 1995 byl odeslán k vyšetření na infekční kliniku s podezřením na kožní formu borreliózy. Borrelióza nebyla potvrzena, ale při vyšetření byla zjištěna přítomnost monoklonálního imunoglobulinu typu IgM, což vedlo k podezření na Waldenströmovu makroglobulinemii, a tak byl v roce 1995 odeslán na naše pracoviště k diferenciální diagnostice monoklonální gamapatie.
Při první návštěvě v dubnu roku 1995 si pacient vysloveně stěžoval na bolesti v oblasti bederní páteře a pánve a dále na velmi intenzivně svědící velkoplošné kopřivkové morfy. Svědění bylo tak intenzivní, že nemocného velmi trápilo nejen ve dne, ale neumožňovalo mu ani klidný odpočinek ve spánku.
Nemocný byl v roce 1995 vyšetřen v rozsahu, který byl v té době běžně prováděn u Waldenströmovy makroglobulinemie s následujícími výsledky:
- CT mediastina a CT břišní dutiny bez lymfadenopatie či jiného patologického nálezu
- trepanobiopsie lopaty kosti kyčelní: fibrózní kostní dřeň, bez patologické infiltrace
- cytologické hodnocení kostní dřeně: bez patologického nálezu, 26 % lymfocytů
- krevní obraz: leukocyty 9,78 × 109/l, erytrocyty 4,06 × 1012/l, hemoglobin128 g/l, trombocyty 278 × 109/l, neutrofily 6,3 ×109/l
- β2-mikroglobulin 0,90 g/l
- celková bílkovina v séru 85 g/l, bílkovina v moči 0,16 g/l
- imunofixace: v séru přítomen monoklonální imunoglobulinu IgM κ, v moči volné řetězce κ
- denzitometrie monoklonálního imunoglobulinu typu IgM κ: 17,4 g/l; v moči nekvantifikovatelné množství lehkých řetězců κ,
- polyklonální imunoglobuliny kvantitativně: IgG 16,6 g/l, IgA 2,30 g/l, polyklonální IgM nebyl pro přítomnost monoklonálního IgM stanoven
- C‑reaktivní protein zvýšen na 46 mg/l
- ostatní laboratorní hodnoty (urea, kreatinin, ionty, enzymy a další) byly zcela ve fyziologickém rozmezí
Při prvním kontaktu byl stav zhodnocen jako monoklonální gamapatie typu IgM nejasného významu (monoclonal gammopathy of unknown significance – MGUS), bez průkazu maligní choroby. Nemocný muž byl předán kožním specialistům, kteří otestovali dostupná antihistaminika, žádné z nich však nevedlo ke zmírnění utrpení, takže pacient sám hodnotil testovaná antihistaminika jako zcela neúčinná.
Jedině prednison v dávce kolem 20 mg denně (10–30 mg, dle intenzity potíží) dostatečně mírnil potíže nemocného, i když je neodstranil zcela. Tato dávka byla ponechána dlouhodobě, protože na rozdíl od antihistaminik pociťoval nemocný po prednisonu výraznou subjektivní úlevu a bylo zřetelné objektivní zlepšení kožních projevů. První pracovní diagnóza byla monoklonální gamapatie nejistého významu typu IgM (MGUS), provázená urtikou.
V červnu roku 1996 byla opakována zobrazovací vyšetření. Na kontrolním CT byly popsány strukturální změny v pravé lopatě kosti kyčelní, kombinace sklerotických změn a drobné ložiskové osteolýzy (obr. 1–3). Dále byla popsána mírná splenomegalie. Scintigrafie skeletu prokázala výrazně vyšší kumulaci radiofarmaka v pravé lopatě kosti kyčelní a v oblasti pravého sakroiliakálního skloubení, tedy ve stejných místech, v nichž CT potvrdilo zahuštění kostní tkáně (obr. 4). Histologie kůže popsala změny odpovídající urtice s perivaskulárními infiltráty, složenými z lymfocytů, histiocytů a neutrofilů, žádná klonální proliferace však nebyla v infiltrátech potvrzena, ani depozita monoklonálního imunoglobulinu typu IgM. Koncentrace monoklonálního imunoglobulinu IgM byla 22 g/l, takže jen nepatrně vyšší než při první návštěvě naší ambulance. Polyklonální imunoglobuliny byly v červnu roku 1996: IgG 11,47 g/l a IgA 1,32 g/l. Bylo tedy zaznamenáno jejich první snížení ve srovnání se vstupní hodnotou v roce 1995.

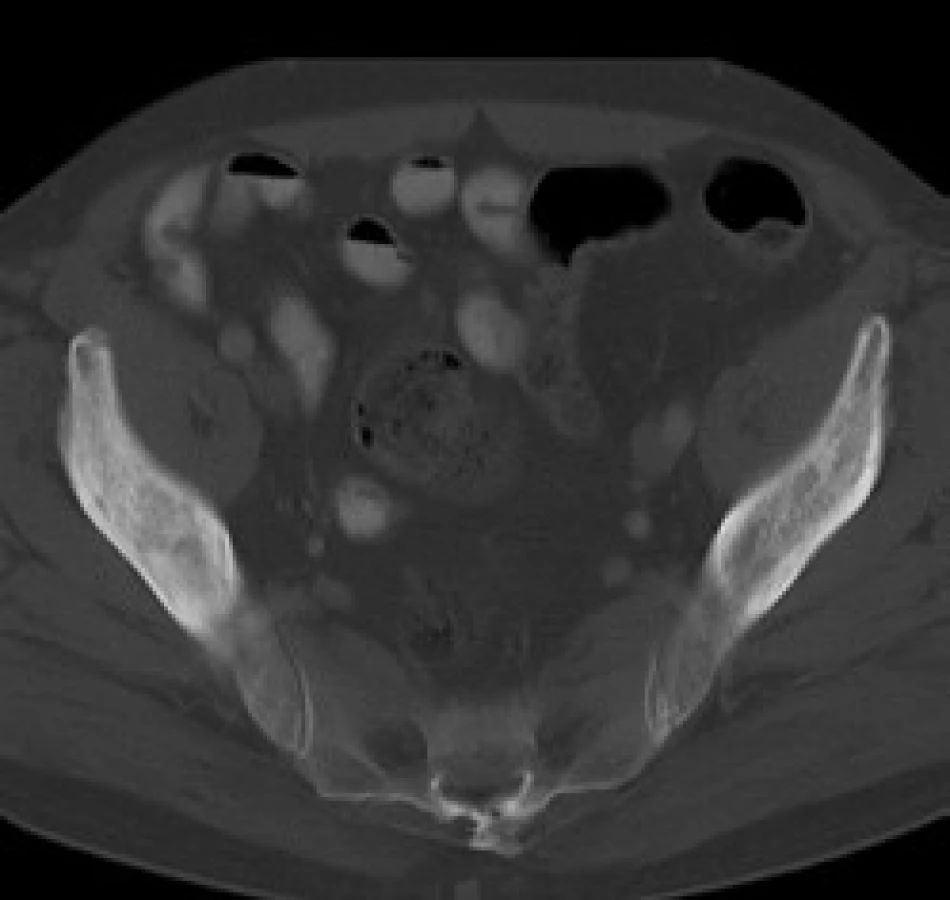
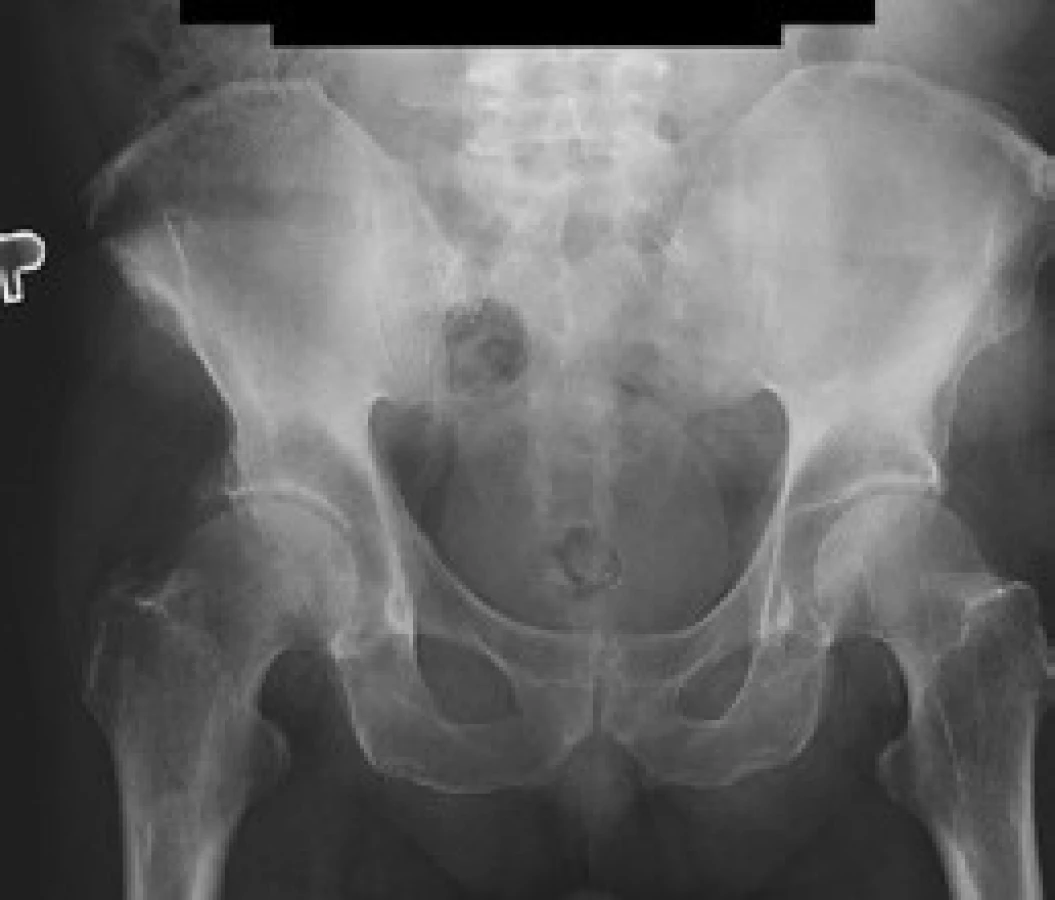
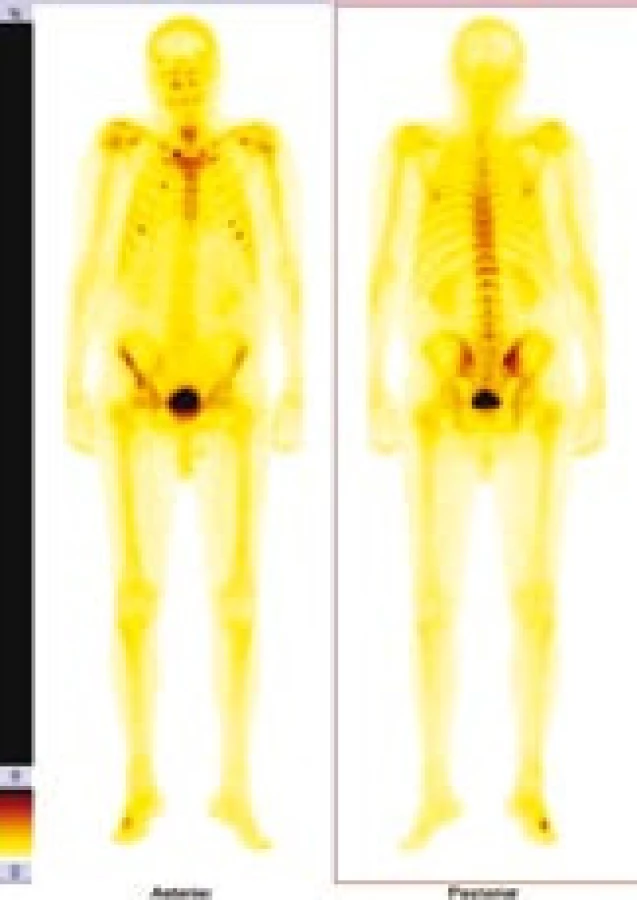
Sklerotické změny pánevních kostí se zvýšenou aktivitou na radioizotopovém vyšetření odpovídaly změnám popisovaným u Schnitzlerova syndromu, stejně jako nízká hodnota β2-mikroglobuinu (1,96 mg/l) a trvale zvýšená hodnota CRP kolísajícího při opakovaných kontrolách mezi 46 a 70 mg/l. Původní pracovní diagnózu MGUS jsme tedy změnili na Schnitzlerův syndrom.
Kostní hustota měřená metodou DEXA byla v roce 1999 –0,8 SD v oblasti L2–4, celková hodnota vyšla –0,6 SD, v roce 2008 se osteopenie v oblasti L2–4 prohloubila na –1,1 SD.
Bisfosfonáty
Pro bolesti kostí a známky kostní přestavby jsme ihned po stanovení diagnózy Schnitzlerův syndrom začali podávat pamidronat (90 mg ve 28denních intervalech). Po 3 měsících této léčby bolesti kostí zcela vymizely. Později jsme přešli na klodronat (900 mg i.v. infuze ve 14denních intervalech nebo 1 500 mg infuze ve 28denních intervalech). V průběhu 14 let jsme podávání bisfosfonátů 3krát přerušili a vždy došlo ke vzplanutí kostních bolestí, po obnovení nitrožilního podávání bisfosfonátu bolesti kostí opět ustaly.
Pulzní dávky dexametazonu
Po stanovení diagnózy Schnitzlerův syndrom jsme zkusili velmi nepříjemné urtikariální projevy zmírnit vysokými dávkami dexametazonu (dexametazon 40 mg p.o. 1.–4., 10.–13. a 20.–23. den v 28denních cyklech), celkem byly aplikovány 3 měsíční cykly. Subjektivní i objektivní zlepšení se dostavovalo pouze ve dnech dexametazonu, po jeho přerušení se opět subjektivní potíže i nález na kůži rychle vracely do původního rozsahu, takže po 3 cyklech (měsících) byla tato léčba ukončena a vrátili jsem se ke kontinuálnímu podávání středních dávek prednisonu (10–30 mg/den dle intenzity potíží).
2-chlordeoxyadenosin
Jedním z nejúčinnějších léků v monoterapii Waldenströmovy makroglobulinemie je 2‑chlordeoxyadenosin. Monoterapie 2‑chlordeoxyadenosinem (0,1 mg/kg/den infuze po dobu 7 dnů v 28denních cyklech) byla zahájena v říjnu roku 1996, celkem byly podány 3 cykly této léčby. Po dobu infuzí vymizela kopřivka a tento léčebný účinek přetrvával jen dalších 10–14 dní. Ve 4. týdnu léčebného cyklu, tedy týden před další aplikací, opět nabyla urtika i svědění své původní podoby a intenzity. Koncentrace monoklonálního imunoglobulinu zůstala po 3 cyklech prakticky stabilní. Podstatné je, že subjektivní potíže i objektivní nález na kůži nedoznal po 3 cyklech 2‑chlordeoxyadenosinu žádné podstatné změny, a tak jsme tuto léčbu ukončili a dále pokračovali v léčbě prednisonem a bisfosfonáty.
Neúspěšný pokus o sběr kmenových krvetvorných buněk
V červenci roku 1997 byl proveden pokus o sběr periferních kmenových buněk pro předchozím podání cyklofosfamidu a filgrastimu (Neupogenu) pro případnou pozdější vysokodávkovanou chemoterapii s autologní transplantací. U pacienta se však nepodařilo sebrat dostatečné množství kmenových buněk krvetvorby z periferní krve.
Interferon α
V roce 1998 jsme se rozhodli vyzkoušet vliv interferonu α ve stejné dávce, jakou jsme používali pro udržovací léčbu v remisi u nemocných s mnohočetným myelomem (3 miliony jednotek s.c. 3krát týdně). Interferon α má tlumivý vliv na svědění provázející myeloproliferativní choroby. Pacient si interferon α aplikoval od počátku roku 1998 do konce roku 1999. Dle subjektivního hodnocení interferon α částečně zmenšoval intenzitu svědění. V průběhu léčby interferonem α se zmenšil rozsah kopřivkových morf a četnost jejich výsevů. Takže počáteční léčebnou odpověď této nemoci na interferon α je možné zhodnotit jako parciální remisi, parciální léčebnou odpověď. Přesto byla koncem roku 1999 (tedy téměř po 2 letech) léčba interferonem α na žádost nemocného přerušena. Důvodem byly nepříjemné subjektivní příznaky po injekci interferonu α typu flu‑like syndromu a patologické chronické únavy (fatigue) i depresivních stavů, které dle subjektivního hodnocení pacientem postupně převážily nad přínosem interferonu α.
A tak v prosinci roku 1999 byla znovu obnovena symptomatická léčba prednisonem. V lednu roku 2000 byl zjištěn steroidní diabetes mellitus a zahájena léčba diabetu, prednison byl samozřejmě ponechán, protože nic účinnějšího na tlumení svědění nebylo k dispozici.
Léčba metodou PUVA
V květnu roku 2001 byla pro trvající svědění kůže celého těla zahájena léčba PUVA. Léčba metodou PUVA pak probíhala dlouhodobě. Po dobu léčby metodou PUVA se neobjevovaly velké kopřivkové plochy, ale jen malé tečkovité indurace, které však také silně svědily. Intermitentní léčba metodou PUVA trvala až do března roku 2003, kdy byla ukončena. Zajímavé je, že léčba metodou PUVA výrazně zmenšila kopřivkové projevy, zatímco subjektivní pocit svědění dle hodnocení nemocného zmírnila jen částečně, méně výrazně, než by odpovídalo minimalizaci kopřivkových morf. Po 2 letech intermitentní léčby metodou PUVA byla tato léčba na delší dobu přerušena a již jsme se k ní nevrátili. Důvodem k přerušení byla léčba recidivujícího svrabu, který se rozšířil na rodinu nemocného.
K léčbě metodou PUVA jsme se již znovu nevrátili, byť jsme konstatovali, že objektivní zmenšení kopřivkových výsevů bylo až překvapující, stejně jako byly překvapující stížnosti nemocného na svědění kůže i při vymizení velkých kopřivkových ploch. A tak jsme se vrátili k prednisonu a hledali jinou nadějnou alternativu.
Cyklosporin A
V červnu roku 2004 jsme se rozhodli otestovat léčbu cyklosporinem A v dávce 5 mg/kg, celkové dávce 300 mg. Cyklosporin A jsme podávali opět po dobu 3 měsíců, ale opět bez subjektivní i objektivní léčebné odpovědi. Nicméně ve 3. měsíci cyklosporinové léčby se zhoršily kožní projevy, byla provedena excise kůže a v ní byly zjištěny mykotické hyfy. Proto bylo podávání cyklosporinu ukončeno.
Thalidomid
V září roku 2004 na základě publikovaných pozitivních zpráv o účinku thalidomidu u mnohočetného myelomu a dalších krevních chorob, i u Schnitzlerova syndromu, jsme se rozhodli zahájit testování thalidomidu i u této diagnózy v dávce 100 mg večer. Po zahájení léčby udával pacient snížení svědění asi o 50 %. Thalidomid byl tedy po léčbě interferonem a fototerapií PUVA dalším lékem, který přinesl částečné zlepšení. V prosinci roku 2004, tedy po 4 měsících léčby, jsme s nemocným hodnotili přínos a nežádoucí účinky thalidomidu. Pacientovi výrazně vadila zácpa, celková ospalost a počáteční výrazné zmenšení intenzity svědění se při hodnocení aktuální situace pacientem již ztrácelo. A tak se pacient rozhodl dle svého subjektivního hodnocení přínosu a nežádoucích účinků v tomto léku již dále nepokračovat.
Bortezomib
Bortezomib se stal v roce 2005 zcela novým a velmi účinným lékem pro mnohočetný myelom. Proto jsme doufali, že by mohl pomoci i tomuto nemocnému. Dne 29. 12. 2005 jsme zahájili léčbu bortezomibem v rámci režimu VTD (bortezomib – Velcade 1,3 mg/m2 1., 4., 8. a 11. den cyklus, thalidomid – Myrin 100 mg denně a dexametazon 20 mg 1.–4. a 8.–11. den cyklu. Dexametazon byl redukován ze 40 na 20 mg pro diabetes mellitus a i přesto způsoboval polední a hlavně večerní hyperglykemie. Po ukončení IV. cyklu VTD v dubnu roku 2006 přetrvávala trombocytopenie, proto byla provedena modifikace a pacient dostával Velcade ve stejné dávce jen 1. a 4. den a dexametazon 1.–4. den a následně byla 14denní pauza. Takto redukované další 4 cykly snášel dobře, takže léčba byla ukončena po celkem 8 cyklech léčby v červnu roku 2006.
Intenzita kopřivky byla v průběhu léčby zmenšená, ale do měsíce od posledního cyklu se obnovily kopřivkové morfy v plné síle jako dříve. Léčba VTD vedla k signifikantnímu poklesu monoklonálního imunoglobulinu, při zahájení léčby byla koncentrace monoklonálního imunoglobulinu v séru 19 g/l, po ukončení 4. cyklu dosáhla 7,5 g/l, ale při redukované léčbě se opět koncentrace monoklonálního imunoglobulinu IgM vystoupila na 10,9 g/l. Stejně tak koncentrace volných lehkých řetězců v séru se v průběhu léčby nijak výrazně neměnila, koncentrace volných κ řetězců činila 56 mg/l na počátku léčby a 41 mg/l na konci, poměr κ/λ byl na počátku 5,14 a na konci 4,51. Hodnoty CRP v průběhu léčby kolísaly (nejnižší hodnota 10 a nejvyšší 79 mg/l) bez zřetelné souvislosti s vývojem koncentrace monoklonálního IgM a bez zjevných příčin pro toto kolísání.
Měsíc od ukončení léčby VTD již kopřivkové projevy nabyly své původní intenzity, takže bylo nutno opět navýšit dočasně sníženou dávku prednisonu (10 mg) na 20 mg denně. Dále tedy pokračovala symptomatické léčba prednisonem a bisfosfonáty.
Bortezomid v kombinaci s thalidomidem a dexametazonem nepřinesl žádné zlepšení, které by přetrvávalo po ukončení podávání léčby, pouze v době aplikací docházelo k dočasnému zmenšení kopřivkového výsevu a svědění.
Anakinra
V letech 2006 a 2007 se objevily zprávy o značném efektu léku anakinra u této nemoci. Tuto léčbu jsme zahájili 27. 12. 2007. Krevní obraz při zahájení léčby anakinrou byl: leukocyty 3,65 × 109/l, erytrocyty 4,62 × 1012/l , hemoglobin 138 g/l, trombocyty 92 × 109/l. Kopřivkový výsev před první injekcí anakinry ilustrují obr. 5–8. Pacient si aplikoval preparát anakinra (Kineret) v dávce 100 mg denně podkožně, a to dlouhodobě.




Po podání první injekce došlo ihned k výraznému zmenšení kopřivkového výsevu a po 3. injekci k vymizení kopřivkových morf i pocitu svědění (obr. 9–11). Injekce dosud snáší bez potíží. Pacient přestal užívat prednison, který doposud jako jediný ze všech symptomatických léků dlouhodobě a účinně tlumil svědění kůže. Tím pádem se výrazně zlepšila kompenzace diabetu a pacient je nyní bez aplikace inzulinu. V této léčbě pokračuje i nadále již 12. měsíc. Injekce si aplikuje sám a snáší je bez vedlejších nežádoucích subjektivních projevů.



V průběhu léčby preparátem anakinra kolísaly počty leukocytů, dne 5. 2. 2008 poklesl počet leukocytů na 2,89 × 109/l, neutrofily na 1,98 × 109/l, další hodnoty krevního obrazu byly: erytrocyty 4,44 × 1012/l, hemoglobin 133 g/l a trombocyty 100 × 109/l. Z obavy před dalším poklesem leukocytů jsme prodloužili intervaly aplikace anakinry na každý 2. den a při další kontrole již počet leukocytů vzestoupil na 9,60 × 109/l a počet neutrofilů na 9,07 × 109/l, takže se znovu přešlo na denní aplikace, které nyní pacient toleruje bez známek myelosuprese.
Urtika se při aplikaci ob den nepatrně, ale opravdu jen nepatrně, vrátila, nyní při aplikaci 1krát denně je pacient bez urtiky.
Vzhledem ke změnám na skeletu pacient pokračuje trvale v aplikaci bisfosfonátů v dávce 1krát měsíčně.
V současnosti nemá kostní bolesti, neužívá žádnou symptomatickou dávku prednisonu. Koncentrace monoklonálního imunoglobulinu se po dočasném poklesu při léčbě velcade, thalidomidem a dexametazonem opět vrátila k hodnotě kolem 17 g/l, kolem níž při kontrolách stále osciluje (14,8–18,4 g/l). Naopak hodnota CRP, která byla v prosinci roku 2007, před zahájením léčby, 74 mg/l, se v průběhu léčby anakinrou snížila na 10,8–2,5 mg/l.
V současnosti má pacient sice stabilní hodnotu monoklonálního imunoglobulinu IgM, která v roce 1995 byla 17,4 g/l a nyní v březnu roku 2008 je 21 g/l. Hodnoty polyklonálních imunoglobulinů však v průběhu let výrazně poklesly: IgG celkové z 16,6 g/l na 3,29 g/l; IgA z 2,30 na 0,12 g/l. Zvýšenou frekvenci infekcí však u pacienta nepozorujeme. Vyšetření komplementu, případně protilátky proti C1q jsme v průběhu nemoci neprováděli.
Diskuze a přehled publikovaných informací o Schnitzlerově syndromu
Schnitzlerův syndrom je vzácnou chorobou, která možná není často správně rozpoznávána. Počet popsaných případů v literatuře je nevelký, obsáhlý přehled literatury uvádí 94 případů, většina popisů je z Evropy. Průměrný věk nemocných je 60 let [14–19]. Klinické příznaky Schnitzlerova syndromu shrnuje tab. 2. Náš pacient měl mimo urtiky bolesti v oblasti LS páteře a pánve. Teprve později se přidaly bolesti v oblasti nosných kloubů. Febrilie nad 38 °C vyjma interkurentních infekcí neudával, ale subfebrilie v době intenzivnějších kožních projevů byly časté.
![Frekvence příznaků Schnitzlerova syndromu tak, jak je v přehledné analýze popsaných případů uvádí Lipsker [12].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/ea4f0db87ef6ec7859d6007135497944.png)
Vzhledem ke vzácnosti Schnitzlerova syndromu a nevelkém prostoru, který je této nemoci v českém písemnictví věnován, považujeme za vhodné uvést typické znaky Schnitzlerova syndromu tak, jak jsou v literatuře popsány, a zkonfrontovat je s příznaky a laboratorními a zobrazovacími nálezy u našeho nemocného. Následně připojíme přehled publikovaných zkušeností s léčbou této nemoci.
Etiopatogeneze Schnitzlerova syndromu a pro něj typických kopřivkových morf
Dominujícím příznakem jsou kopřivkové morfy. Histologické nálezy z kožních morf jsou v literatuře popisovány jako neutrofilní urtikarie (nejčastější), výjimečně pak jako spongiotická dermatitida anebo leukocytoklastická vaskulitida [7]. DeCastro histomorfologicky vyšetřil 25 kožních biopsií a ve většině případů odpovídaly nálezy neutrofilní urtikarii. Pouze ve 2 případech popsal vaskulitidu [20].
Při použití imunofluorescenčního vyšetření byla popsána imunoreaktivní depozita typu IgM v superficiálních dermálních cévách a v dermálním/epidermálním přechodu [21–25]. Bylo prokázáno, že v séru nemocných se Schnitzlerovým syndromem byly monoklonální protilátky IgM stejného izotypu, jako byla protilátka IgM nalezená v depozitech v dermálně/epidermálním spojení [26]. Je tedy možné, že tato depozita mohou spouštět lokální zánětlivou reakci, která pak způsobí kopřivkovou morfu. Tato hypotéza, dle níž je monoklonální imunoglobulin typu IgM etiologickým faktorem, se nám jeví jako nejpravděpodobnější, nebyla však potvrzena v žádném experimentu. Publikovány byly i jiné hypotézy, které však následně nebyly potvrzeny v dalších pozorováních. Jedna z nepotvrzených hypotéz uváděla souvislost s anti‑IL‑1 protilátkou, popsanou v několika případech tohoto syndromu [27,28].
U Schnitzlerova syndromu byla opakovaně zjištěna zvýšená plazmatická koncentrace interleukinu-6 (IL‑6), který je zásadní pro proliferaci plazmocytů a je také reaktantem akutní fáze. Proto se zvažovalo, zda přítomnost monoklonálního imunoglobulinu typu IgM není důsledkem zvýšených hladin interleukinu-6 a chronické antigenní stimulace, tedy sekundárním produktem stavu, anebo zda je primární příčinou. Popsaná depozita monoklonálního imunoglobulinu IgM v kůži svědčí spíše pro primární roli monoklonálního imunoglobulinu IgM v etiopatogenezi této nemoci. Nicméně zvýšená hodnota interleukinu-6 zde hraje asi důležitou roli, je dávána do souvislosti s chronicky zvýšenou hodnotou CRP a s anémií chronických chorob, která tento syndrom také provází.
Etiopatogeneze této nemoci zůstává v roce 2008 stále neobjasněna, monoklonální IgM má velmi pravděpodobně důležitou roli ve vzniku kožních morf. Je zřejmé, že u Schnitzlerova syndromu dále dochází k dysregulaci cytokinové sítě, což má za následek anémii chronických chorob a zřejmě i změny kostního metabolizmu [15–19,23,28].
Typické příznaky nemoci
Kožní projevy
Kopřivkové projevy jsou popisovány jako erytematózní makulopapulózní ložiska, až plaky červeného zbarvení, od 0,5 po 10 i více cm v průměru, někdy splývající. Všechna ložiska mají stejnou barvu i tvar. Denně se mohou objevovat nové výsevy, trvají 12–36 hod a potom pomalu mizí. Frekvence tvorby nových kopřivkových morf je v literatuře udávána s velkým rozptylem, u některého nemocného vznikají nové morfy denně, zatímco u jiných byl jejich výsev intermitentní s klidovými pauzami, trvajícími i několik týdnů. U většiny popsaných případů byly tyto změny přítomny kontinuálně. V jednom případně byla v kopřivkových morfách popsána purpura [29,30].
Morfy postihovaly v popsaných případech obvykle trup a končetiny, zatímco hlava a krk byly ušetřeny a stejně tak chodidla a ruce. Zpočátku mohou být kožní kopřivkové morfy bez svědění, to se přidává až v průběhu nemoci za více měsíců či roků [31].
Těmto popisům naprosto odpovídají i nálezy u našeho nemocného, velké kopřivkové morfy o průměru více než 10 cm, mapovitého vzhledu, pokrývaly trup, horní i dolní končeny, ne však dlaně a chodila. Kopřivkové morfy intenzivně svědily. Bez léčby byly přítomny trvale, staré postupně ustupovaly, zatímco nové vznikaly.
U některých pacientů byl popsán výsev nových morf v souvislosti s konzumací alkoholu, kořeněných jídel či stresem, tuto závislost jsme u našeho nemocného nepozorovali. U našeho nemocného nebyl přítomný angioedém, výjimečně popsaný Clavuelem a Sanchezem [32,33]. Při histologickém vyšetření biopsie kopřivkové morfy našeho nemocného byly popsány jako změny obvyklé u urtikarie.
Teplota
Intermitentní teplota je popisována jako kardinální symptom nemoci. U některých případů byla popsána horečka až 40 °C. Teplota je však dobře tolerována a nebývají přitom pocity zimnice. Teplota obvykle dobře reaguje na nesteroidní antiflogistika.
U našeho pacienta se vyskytovaly jenom subfebrilie, a to v závislosti na novém výsevu plošných kopřivkových morf. Subfebrilie byly nepříjemně vnímány, ale dobře reagovaly na paracetamol nebo na nesteroidní antiflogistika [34,35].
Příčina teploty není jasná, Lipskerpopisuje zvýšenou hladinu interleukinu-6 a interleukinu-2, zatímco tumor necrosis factor α (TNF‑α) a interleukin‑8 byl v normálním rozmezí [12].
Hladiny interleukinů (ani komplementu) jsme u našeho nemocného neanalyzovali, nicméně trvale zvýšenouhodnotu CRP bez dalšího zánětu a malignity si vysvětlujeme jako následek změny hladin cytokinů, a tedy zvýšené hodnoty IL‑6.
Muskuloskeletární postižení
Muskuloskeletární postižení je dalšímdůležitým znakem této nemoci, vyskytuje se asi u 80 % nemocných. Bolesti kostí jsou popisovány u 59 % nemocných a často jsou ještě doplňovány bolestmi kloubů (59 %), které však nevedou k jejich deformacím ani k destrukci.
Kostní bolesti jsou nejčastěji popisovány v oblasti pánve a v kosti holenní. Stehenní kosti, paže, předloktí a klíční kosti jsou postihovány méně často.
Bolesti kloubů bývají v kyčlích, kolenou, zápěstí či lokti, méně často v ostatních kloubech těla. V některých případech byly uvedeny i myalgie [14,15].
Náš pacient uváděl v době stanovení diagnózy bolesti v oblasti sakroiliakální a v oblasti kyčelních kloubů. Vzhledem k lokalizaci nešlo zcela odlišit, zda jde o bolest kosti (kyčelní kosti, kde byly změny kostní struktury), či zda jde o bolest kloubní. Stížnosti nemocného jsme chápali jako kombinované bolesti kostní i kloubní. V následujících letech se objevily u našeho pacienta bolesti v ramenních kloubech.
Palpačně zvětšené lymfatické uzliny a hepatosplenomegalie
Palpačně zvětšené uzliny byly popsány u 50 % nemocných a hepatosplenomegalie u 33 % nemocných. Při bioptickém vyšetření těchto uzlin byly u těchto nemocných popsány jen nespecifické zánětlivé změny [12]. V našem případě nebyla přítomna lymfadenopatie ani hepatomegalie, na CT byla popsána jen mírná splenomegalie.
Laboratorní a zobrazovací nálezy
Monoklonální imunoglobulin typu IgM
Monoklonální imunoglobulin třídy IgM patří do definice této nemoci. U 89 % nemocných se jedná o κ IgM. Koncentrace monoklonálního imunoglobulinu typu IgM je obvykle při stanovení diagnózy nízká, u 67 % popsaných případů byla pod 10 g/l. Koncentrace monoklonálního imunoglobulinu zůstává stabilní nebo se pozvolna v průběhu času zvyšuje, obvykle o 0,5–1,0 g/l za rok [12].
Vyšší hodnoty vyvolávají podezření na transformaci ve Waldenströmovu makroglobulinemii.
Nashan v roce 1995 první popsal případ urtiky horečky a atralgií s přítomným monoklonálním IgG imunoglobulinem jako variantní typ Schnitzlerova syndromu a posléze přibyly další popisy těchto variantních případů [36,37]. Lipsker dále popsal případ urtikariárních kožních změn, které souvisely s myelomovým monoklonálním imunoglobulinem IgA, u něhož ale chyběly další typické změny pro Schnitzlerův syndrom [17]. Je otázka, zda lze připustit Schnitzlerův syndrom bez monoklonálního imunoglobulinu [9,38].
Bence-Jonesova proteinurie byla popsána ve 44 % případů [12]. Snížení koncentrací IgG a IgA imunoglobulinů popisuje Lipsker ve 26 % případů.
Biochemické nálezy spadají tedy u našeho nemocného do standardního popisu. V průběhu let je zřetelný výrazný pokles polyklonálních imunoglobulinů zbývajících tříd (IgG a IgA). Jakou měrou se na snížení IgG a IgA podílí léčba anebo vlastní onemocnění, nelze odlišit.
Další laboratorní změny provázející Schnitzlerův syndrom
Trvale zvýšená hodnota sedimentace erytrocytů je charakteristickým, u všech nemocných popisovaným laboratorním nálezem, stejně tak zvýšená hodnota CRP. Trvale zvýšené hodnoty CRP měl i náš nemocný, míra zvýšení CRP odpovídala tíži nemoci. Teprve při zahájení léčby anakinrou se hodnoty CRP i hemoglobinu upravily na normu.
Hodnoty komplementu se popisují jako normální nebo zvýšené. Pokud by hodnoty komplementu byly snížené, je nutné pomýšlet na jinou diagnózu, u 2 nemocných s podobnými potížemi byl popsán deficit C4 [39]. Náš pacient měl vstupní hodnoty komplementu v normálním rozmezí a další analýzy komplementu jsme neprováděli.
Trombocytóza a anémie, odpovídající anémii chronických chorob, je popisována u 10 % nemocných. U 2 nemocných byla anémie chronických chorob tak závažná, že vyvolávala symptomy [21]. Náš pacient měl anémii odpovídající anémii chronických chorob [40]. Hodnoty hemoglobinu měly v průběhu 13 let pozorování vlnitý charakter, nejnižší hodnota byla 86 g/l. Nejvyšší hodnoty trombocytů jsme zachytili v roce 1996 – 359 × 109/l, od té doby se počet trombocytů pohyboval spíše při dolní hranici fyziologického rozmezí a v posledních letech kolísají kolem hodnoty 100 × 109/l.
U většiny nemocných byla pozorována trvalá leukocytóza nad 10 × 109/l, a to bez jakékoliv léčebné intervence, a stejně tomu bylo i u našeho nemocného, kdy v roce 1995 v době stanovení diagnózy se počet leukocytů pohyboval mezi 10 a 16 × 109/l a kolísání jejich počtu zpočátku odpovídalo aktivitě nemoci, později bylo také ovlivněno léčbou.
Změny skeletu při Schnitzlerově syndromu
Schnitzlerův syndrom způsobuje i změny kostního metabolizmu. Zvýšení kostní denzity je nejčastějším radiologickým nálezem. V oblastech zvýšené kostní denzity jsou často pociťovány bolesti [41–44]. Osteolytická ložiska byla popsána u 2 nemocných [12,45] a periostální apozice byla popsána u dalších 2 [41,46,47]. Podobné změny může způsobit mastocytóza, POEMS syndrom a Erdheimova-Chesterova choroba. Scintigrafie skeletu pomocí technecium-pyrofosfátu odhalí ložiska se zvýšenou kostní přestavbou [41].
Magnetická rezonance byla provedena u 3 nemocných, prokázala změny signálu oproti signálu fyziologické kostní dřeně [35,46,48].
V našem případně byly při CT vyšetření popsány smíšené osteolyticko‑osteosklerotické změny.
Scintigrafie skeletu u našeho pacienta opakovaně zobrazila ložiska přestavby (obr. 4). Podobné změny jsou popsány i v literatuře [49,50].
Biopsie kosti a hodnocení kostní tkáně není běžným vyšetřením, v literatuře jsme našli informace o nálezech u 9 nemocných. Ve 3 případech byl popsán normální nález [1,51,52], v 5 případech byly popsány nespecifické zánětlivé změny někdy se zřetelnou zvýšenou osteoblastickou aktivitou [32,53–56]. Pouze u 1 nemocného byla histologicky popsána přítomnost osteosklerózy [6].
Pro diferenciální diagnózu IgM gamapatií je zásadní biopsie kostní dřeně [57], v době stanovení diagnózy mělo 80 % vyšetřených normální nález v kostní dřeni, u zbývajících 20 % byly nalezeny nespecifické polyklonální lymfocytární nebo plazmocytární infiltráty [12].
V našem případě byla biopsie kostní dřeně bez jednoznačné patologické odchylky, biopsii a histologické hodnocení kosti nebylo u našeho pacienta provedeno. Na CT snímcích byly popsány projevy osteosklerózy (obr. 1–3) a na scintigrafii skeletu byla ložiska přestavby (obr. 4).
Diferenciální diagnóza
Nemoci, které se také mohou projevovat erytémem a urtikou, uvádí tab. 3. Podobné kožní projevy jako Schnitzlerův syndrom může způsobit Stillova nemoc vznikající v dospělosti, nicméně v prvním případě dominuje monoklonální imunoglobulin IgM a v druhém vysoké hodnoty feritinu [29]. Kožní urtika může být také projevem kryoglobulinemie, hypokomplementární kopřivkové vaskulitidy, získaného deficitu C1 inhibitoru, hyper‑IgD syndromu.

Přidruženými nemocemi může být pseudoxanthoma elasticum [55,58], periferní neuropatie s přítomností IgM anti‑MAG (myelin asociovaný glykoprotein) [59–61], deficit C4 komplementu [34,62], antifosfolipidový syndrom [63], nodulární hyperplazie v játrech [64].
Léčba Schnitzlerova syndromu
Léčba Schnitzlerova syndromu byla až do příchodu preparátu anakinra velmi neuspokojivá a deprimující. Žádný z popsaných léčebných postupů nevedl u všech případů k žádoucí léčebné odpovědi. Ibuprofen v jednom případě příznivě ovlivnil kožní projevy [65], ale dle dalších zkušeností nemají blokátory cyklooxygenázy žádný vliv na kožní projevy [23–25,41,66–68]. Blokátory cyklooxygenázy mají však pochopitelně účinek na teplotu, je‑li přítomna, a na bolesti kostí a kloubů.
Bolesti kostí však výrazně mírní či zcela odstraňují bisfosfonáty [35,69].
Antihistaminika nemají u Schnitzlerova syndromu schopnosti mírnit kožní projevy [4,21,25,41,66–68] a také u našeho nemocného byly veškeré léky ze skupiny antihistaminik zcela neúčinné.
Léky, které inhibují migraci neutrofilů, jako je kolchicin či dapson, byly také testovány, ale jejich přínos nebyl nikterak přesvědčivý [22,48,55,66].
Hydroxychlorochin a chlorochin nebyly také účinné [21,67,68].
Plazmaferéza [21,26] a nitrožilní imunoglobuliny [59] také nepomáhaly. Stejně tak chemoterapeutické léčebné režimy nepřinášely dlouhodobý prospěch [12,70] a ani v našem případně léčba 2‑chlordeoxyadenosinem nebyla přínosem, stejně jako jednorázová aplikace cyklofosfamidu nepřinesla zlepšení.
Kortikosteroidy snižují intenzitu kožních projevů, ale pro dosažení léčebného efektu je zapotřebí přiměřeně vysokých dávek. U našeho nemocného byly potíže dlouhodobě mírněny prednisonem v průměrné dávce 20 (10–30) mg. Dávku si pacient sám upravoval dle intenzity potíží.
V předchozích letech bylo popsáno několik alespoň středně účinných alternativ. Léčba interferonem α snižovala v několika případech dlouhodobě intenzitu svědění a rozsah kopřivkového výsevu [71–73]. Nicméně tato léčba je účinná jen po dobu aplikace a po přerušení se navracejí původní projevy nemoci. Stejně to bylo u našeho nemocného, který si aplikoval interferon α po dobu téměř 2 let. Zpočátku přínos interferonu α hodnotil jednoznačně pozitivně, po 2 letech však začaly nabývat na intenzitě nežádoucí účinky této léčby, až převážily nad jejím přínosem, takže léčba byla ukončena.
Thalidomid navodil parciální remisi ve 3 popsaných případech [74–76], ale jeho dlouhodobé použití bylo limitováno jeho neuroxicitou. Obdobně tomu bylo i v našem případě, kdy po delší době užívání převážily nežádoucí účinky nad žádoucími a léčba byla ukončena.
Fototerapie metodou PUVA v několika případech zmírnila kožní projevy této nemoci [12,78,79]. Lze říci, že i u našeho nemocného měla léčba metodou PUVA tlumící vliv na kožní projevy, ale jen po dobu jejího provádění.
Byly testovány i další léky, jejich přehled je uveden v tab. 4: pefloxacin [80], extrakorporální imunoadsorbce [80], cyklosporin [81,82], infuze monoklonální anti‑TNF protilátky [15,83], rituximab [84,85].
Zásadní změnu přinesl až zcela nový preparát anakinra, což je antagonista receptoru pro interleukin‑1. V literatuře jsme našli informace o 8 pacientech léčených tímto lékem. Ve všech popsaných případech navodila 1. injekce ihned kompletní remisi nemoci. Anakinra odstranila zcela svědění kůže a kožní projevy [73,74,86–92]. Nejdelší zkušenost s podáváním preparátu anakinra, popsaná v literatuře, je 3letá. Po celou dobu podávání tohoto léku zůstal nemocný v kompletní remisi nemoci [74]. Preparát anakinra se osvědčil také u IL-1 dependentních periodických horeček (Muckle-Well syndrom a jiné), k nimž má Schnitzlerův syndrom blízko a které rovněž odpovídají na anti‑IL-1 léčbu.
Také náš pacient měl ihned od počátku této léčby okamžitou úlevu od potíží. Nyní je již 12 měsíců na udržovací léčbě, při dávkách 100 mg denně je zcela bez kožních projevů a laboratorních příznaků (normální hodnoty hemoglobinu a CRP). Při prodloužení intervalu aplikací na 48 hod potíže ihned mírně recidivovaly, asi na 30–50 % původní intenzity.
Díky vymizení urtiky při aplikaci anakinry mohla být ukončena symptomatická léčba prednisonem, která jako jediná z dříve použitých léků dlouhodobě mírnila kožní projevy. Upravil se steroidní diabetes mellitus, celková denní dávka inzulinu poklesla na 1/4 dávky nutné při používání prednisonu.
Přehled publikovaných zkušeností s léčbou Schnitzlerova syndromu uvádí tab. 4.
![Přehled zkušeností s léčbou Schnitzlerova syndromu publikovaný skupinu Schnitzler Syndrome Study Group [14], www.schnitzlersyndrome.com.](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/c99c2f756e1bef373565775ec5884a8a.png)
Průběhy a prognóza nemoci
Nemoc má chronický průběh, spontánní remise nebyly popsány. Z hlediska mortality je průběh příznivý, 91 % nemocných žije déle než 15 let. Zajímavé je, že ač jde o chronické zánětlivé onemocnění, byla jen výjimečně popsána v jeho průběhu AA-amyloidóza [93,94]. Prognóza nemocných se Schnitzlerovým syndromem je odvislá od toho, zda se vyvine, či nevyvine maligní lymfoproliferativní onemocnění (maligní nehodgkinský lymfom, Waldenströmova makroglobulinemie). U 11 z 94 popsaných případů byl zjištěn přechod do lymfoproliferativní choroby [3,14,93,95–100]. Nicméně je možné, že počet nemocných se Schnitzlerovým syndromem, jejichž nemoc přešla v maligní lymfoproliferaci, bude vyšší, protože většina publikovaných případů byla zveřejněna časně po zjištění diagnózy, takže sledování bylo velmi krátké pro vyjádření se o vývoji nemoci. Maligní lymfomy nebo Waldneströmova nemoc se objevují nejdříve po 10–20 letech od prvních příznaků nemoci. Pacient, poprvé popsaný Schnitzlerem, zemřel pod obrazem lymfoplazmocytárního lymfomu s přítomnou infiltrací kostní dřeně a jater 23 let od stanovení diagnózy [100]. Pro předpověď přechodu v maligní lymfoproliferaci nebyly popsány žádné prognostické znaky.
Proto základní vyšetření u této nemoci má vždy obsahovat vyšetření kostní dřeně a dále elektroforézu a imunofixaci moči a krve k zastižení přítomnosti monoklonálního Ig. Případné zvětšené uzliny by měly být vždy odebrány k histologickému vyšetření. Monoklonální imunoglobulin by měl být pravidelně sledován a při výrazném vzestupu je vhodné přešetření s cílem diagnostikovat případný přechod v maligní lymfoproliferace.
Závěr
V průběhu 14 let jsme u našeho pacienta docílili vždy parciálního zlepšení při použití kontinuálních nízkých až středních dávek prednisonu, posléze léčbou interferonem α, světloléčbou PUVA a thalidomidem. Uvedené léčebné postupy sice přinesly parciální zlepšení, ale délka jejich aplikace byla vždy limitována postupně se zvýrazňujícími nežádoucími účinky.
Zásadní zlepšení, úplné odstranění kožních příznaků, přinesla až léčba preparátem anakinra, který z pohledu roku 2008 je vhodným lékem pro Schnitzlerův syndrom. Je třeba jej však používat trvale do té doby, než bude nalezena kauzální léčba této nemoci.
Poděkování
Velmi vzácné nemoci nemohou být předmětem zkoumání klinických studií. Registrace léku pro tyto nemoci nemohou být tedy obsaženy v SPC dokumentaci a přitom je nutné tyto choroby také léčit. Proto ani Schnitzlerův syndrom nebude v registrační dokumentaci léčiv. Se žádostí o schválení této zcela výjimečné léčby jsme se obrátili na ředitele VZP, který ji schválil. Chceme proto poděkovat MUDr. Pavlu Horákovi, CSc., MBA, řediteli VZP, za velmi vstřícný přístup vedení Všeobecné zdravotní pojišťovny při zajišťování úhrady léčby preparátem anakinra – Kineret – pro dlouhodobě trpícího člověka.
Tato práce vznikla a byla podporována v rámci projektu MŠMT: LC 06027 a VZ 0021622434.
prof. MUDr. Zdeněk Adam, CSc.
www.fnbrno.cz
e‑mail: z.adam@fnbrno.cz
Doručeno do redakce: 12. 5. 2008
Přijato po recenzi: 22. 7. 2008
Sources
1. Schnitzler L, Hurez D, Verret JL. Urticaire chronique osteoconcensation-macroglo-bulinemie. Cas Princeps Etude sur 20 ans Ann. Dermatol Venereol 1989; 116 : 547–550.
2. Schnitzler L, Schubert B, Boasson M et al. Urticaire chronique lesions osseuses macroglobulinemie IgM: Maladie de Waldenström? Bull Soc Fr Dermatol Syph, 1974; 81 : 363–366.
3. Altmeyer P, Welke S. Macroglobulinemiae Waldenström associirt mit einem chronisch rezidivierenden urtikariellen Exanthem. Akt Dermatol 1977; 3 : 71–76.
4. Baty V, Hoen B, Hudziak H et al. Schnitzler’s syndrome. Two case reports and review of literature. Mayo Clin Proc 1995; 70 : 570–572.
5. de Kleijn EM, Telgt D, Laan R. Schnitzler’s syndrome presenting as fever of unknown origin (FUO). The role of cytokines in its systemic features. Neth J Med 1997; 51 : 140–142.
6. Puddu P, Cianchini G, Girardelli CR et al. Schnitzler’s syndrome: report of a new case and a review of the literature. Clin Exp Rheumatol 1997; 15 : 91–95.
7. Kropp JD, Czarnetzki BM. Utica, vasculitis, und Schnitzler’s syndrome. Allergologie 1994; 17: S17–S20.
8. Shibolet O, Schata O, Krieger M et al. Schnitzler syndrome: chronic urticaria, fever and immunoglobulin M monoclonal gammopathy. Isr Med Assoc J 2002; 4 : 466–467.
9. Verella TC, Nishimura MY, Machado MC et al. Schnitzler’s syndrome without monoclonal gammopathy. Acta Derm Venereol 2005; 85 : 272–273.
10. Harati A, Brockmeyer NH, Altmeyer P et al. Skin disorders in association with monoclonal gammopathies. Eur J Med Res 2005; 10 : 93–104.
11. Zuberbier T, Bindeslev-Jensen C, Canonica W et al. EAACI/GA2LEN/EDF Guidelines: definition, classification and diagnosis of urticaria. Allergy 2006; 61 : 316–320.
12. Lipsker D, Veran Y, Grunenberger F et al. The Schnitzler’s syndrome. Four new cases and review of the literature. Medicine (Baltimore) 2001; 80 : 37–44.
13. Lipsker D. The Schnitzler syndrome – a treatment at last? Dermatology 2002; 205 : 1–2.
14. de Koning HD, Bodar EJ, van der Meer JW et al. Schnitzler’s Syndrome Study Group. Schnitzler’s syndrome: beyond the case reports: review and follow‑up of 94 patients with an emphasis on prognosis and treatment. Semin Arthritis Rheum 2007; 37 : 137–148.
15. de Koning HD, van der Meer JW, Simon A. Comment on: Schnitzler’s syndrome – exacerbation after anti‑TNF treatment. Rheumatology (Oxford) 2007; 46 : 1741.
16. Lipsker D, Boeckler P. Cutaneous manifestations of paraproteinemia and their mechanisms. Presse Med 2007; 36 : 1135–1140.
17. Lipsker D, Cribier B, Maloisel F et al. Chronic urticaria and IgA myeloma. Acta Derm Venereol (Stockholm) 1998; 78 : 395.
18. Lipsker D, Imrie K, Simon A et al. Hot and hobbling with hives: Schnitzler’s syndrome. Clin Immunol 2006; 119 : 131–134.
19. Lipsker D, Spehner D, Drillien R et al. Schnitzler’s syndrome: heterogeneous immunopathological findings involving IgM-skin interactions. Br J Dermatol 2000; 142 : 954–959.
20. de Castro FR, Masouyé I, Winkelmann RK et al. Urticarial pathology in Schnitzler’s syndrome. Dermatology 1996; 193 : 94–99.
21. Berdy SS, Bloch KJ. Schnitzler’s syndrome: A broader clinical spectrum. J Allergy Clin Immunol 1991; 87 : 849–854.
22. Borradori L, Rybojad M, Puissant A et al. Urticarial vasculitis associated with monoclonal IgM gammopathy, Schnitzler’s syndrome. Brit J Dermatol 1990; 123 : 113–118.
23. Morita A, Sakakibara S, Yokota M et al. A case of urticarial vasculitis associated with macroglobulinemia (Schnitzler’s syndrome). J Dermatol 1995; 22 : 32–35.
24. Lautenschlager S, Itin PH. Das Schnitzler Syndrom. Hautarzt 1993; 44 : 781–784.
25. Janier M, Bonvalet D, Blanc MF et al. Chronic urtica and macroglobulinemia. Schnitzler’s syndrome. Report of two cases. J Am Acad Dermatol 1989; 20 : 206–211.
26. Olsen E, Førre O, Lea T et al. Unique antigenic determinants (idiotypes) used as markers in a patient with macroglobulinemia urticaria. Similar idiotypes demonstrated in the skin and on peripheral blood lymphocytes. Acta Med Scand 1980; 207 : 379–384.
27. Gallo R, Sabroe RA, Black AK et al. Schnitzler’s syndrome: no evidence for autoimmune basis in two patients. Clin Exp Dermatol 2000; 25 : 281–284.
28. Eiling E, Schröder JO, Gross WL et al. The Schnitzler’s syndrome: Chronic urticaria and monoclonal gammopathy – an autoinflammatory syndrome? J Dtsch Dermatol Ges 2008; 6 : 626–631 [Epub ahead of print].
29. Tomková H, Shirafuji Y, Arata J. Schnitzler’s syndrome versus adult onset Still’s disease. Eur J Dermatol 1998; 8 : 118–121.
30. Sanmartín O, Febrer I, Botella R et al. Urticarial lesions and monoclonal IgM gammopathy. Schnitzler’s syndrome. Arch Dermatol 1994; 130 : 1193–1198.
31. Almerigogna F, Giudizi MG, Cappelli F et al. Schnitzler’s syndrome: what’s new? J Eur Acad Dermatol Venereol 2002; 16 : 214–219.
32. Clauvel JP, Brouet JC, Danon F et al. Chronic urticaria with monoclonal IgM Reprot of five cases. Clin Immunol Immunopathol 1982; 25 : 348–353.
33. Sánchez G, Añó M, García-Avilés C et al. Schnitzler syndrome: a case study. J Investig Allergol Clin Immunol 2000; 10 : 41–43.
34. deKleijn EM, Telg D, Laan R. Schnitzler’s syndrome presenting as a fever of unknown origin. The role of Cytokines in its systemic features. Neth J Med 1997; 51 : 140–142.
35. Winckelmann G, Nagel HG, Maier R et al. The Schnitzler’s syndrome as a cause of recurrent fever of unknown origin. Dtsch Med Wochenschr 1996; 121 : 860–864.
36. Nashan D, Sunderkötter C, Bonsmann G et al. Chronic urticaria, arthralgia, raised erythrocyte sedimentation rate and IgG paraproteinemia? A variant of Schnitzler’s syndrome? Brit J Dermatol 1995; 133 : 132–135.
37. Akimoto R, Yoshida M, Matsuda R et al. Schnitzler’s syndrome with IgG κ gammopathy. J Dermatol 2002; 29 : 735–738.
38. Husak R, Nestoris S, Goerdt S et al. Severe course of chronic urticaria, arthralgia, fever and elevation of erythrocyte sedimentation rate: Schnitzler’s syndrome without monoclonal gammopathy? Br J Dermatol 2000; 142 : 581–582.
39. Rybojad M, Moraillon I, Cordoliani F et al. Schnitzler syndrome with genetic C4 deficiency. 2 cases. Ann Dermatol Venereol 1993; 120 : 783–785.
40. Ščudla V, Adam Z, Ščudlová M. Současné možnosti diagnostiky a léčby anémie chronických chorob. Vnitř Lék 2001; 47 : 400–406.
41. Lecompte M, Blais G, Bisson G et al. Schnitzler’s syndrome. Skeletal Radiol 1998; 27 : 294–296.
42. De Saint-Pierre V, Ehrhart A, Baron D et al. Systemic urticaria, sclerosing osteopathy, monoclonal gammopathy (Schnitzler’s syndrome). Apropos of a case. Rev Rheum Mal Osteoartic 1992; 59 : 288–292.
43. De Waele S, Lecouvet FE, Malghem J et al. Schnitzler’s syndrome: an unusual cause of bone pain with suggestive imaging features. AJR Am J Roentgenol 2000; 175 : 1325–1327.
44. Flórez AF, Gallardo Agromayor E, García‑Barredo R et al. Radiological aid to clinical diagnosis of Schnitzler’s syndrome: multimodality imaging approach. Clin Rheumatol 2008; 27 : 107–110.
45. Ferrando FJ, Pujol J, Hortells JL et al. Schnitzler’s syndrom: Report of a case with bone osteolysis. J Invest Allergol Clin Immunol 1994; 4 : 203–205.
46. Bertrand A, Feydy A, Belmatoug N et al. Schnitzler’s syndrome: 3-year radiological follow‑up. Skeletal Radiol 2007; 36 : 153–156.
47. Dupuy O, Pinede L, Coppere B et al. Schnitzler’s syndrome with stable course over a 18-year period. Report of a case. Presse Med 1995; 24 : 1402.
48. Germain P, Fach J, Bui N et al. Schnitz-ler’s syndrome: a rare cause of systemic urticaria. Rev Med Interne 2000; 21 : 285–289.
49. Singh B, Ezziddin S, Rabe E et al. Bone scan appearance supportive of Schnitzler’s syndrome: report of two new cases. Clin Nucl Med 2006; 31 : 151–153.
50. Soubrier M. Schnitzler syndrome. Joint Bone Spine 2008; 75 : 263–236 [Epub ahead of print].
51. Doutre MS, Beylot C, Bioulac P et al. Monoclonal IgM and chronic urticaria: two cases. Ann Allergy 1987; 58 : 413–414.
52. Welsh B, Tate B. Schnitzler’s syndrome: report of a case with progression to Waldenström’s macroglobulinaemia. Australas J Dermatol 1999; 40 : 201–203.
53. Barriere M, Schnitzler L, Moulin G et al. Lesions uricariennes chroniques at macroglobulinemie. Sem Hop Paris 1976; 52 : 221–227.
54. Goupille P, Pizzuti P, Diot E et al. Schnitzler’s s syndrome (urticaria macroglobulinemia) dramatically improved with corticosteroids. Clin Exp Rheumatol 1995; 13 : 95–98.
55. Machet L, Vaillant L, Machet MC et al. Schnitzler’s syndrome and associated with pseudoxanthoma elasticum. Acta Derm Venereol (Stockholm) 1992; 72 : 22–24.
56. Machet L, Wattier H, Vaillant L. Urticaria and systemic diseases. Ann Dermatol Venereol 2001; 128 : 1156–1160.
57. Adam Z, Šmardová J, Ščudla V. Waldenströmova makroglobulinémie: klinické projevy a diferenciální diagnostika a prognóza nemoci. Vnitř Lék 2007; 53 : 1325–1337.
58. Wilhelmi ML, Wilhelmi J, Sierra J. Schnitzler syndrome and pseudoxanthoma elasticum 3rd Congress EADV 26–30 Sept. 1993 Kopenhagen, Abstract FC 20 159.
59. Lebbe C, Rybojad M, Klein F et al. Schnitzler’s syndrome with sensomotor neuropathy. J Amer Acad Dermatol 1994; 30 : 316–318.
60. Blaise S, Vallat JM, Tabaraud F et al. Sensitive chronic inflammatory demyelinating polyradiculoneuropathy in Schnitzler’s syndrome. Ann Dermatol Venereol 2003; 130 : 348–351.
61. Gossrau G, Pfeiffer C, Meurer M et al. Schnitzler’s syndrome with neurological findings. J Neurol 2003; 250 : 1248–1250.
62. Nagy L, Hannema A, Swaak A. Acquired C1 inhibitor deficiency associated with systemic lupus erythematosus, secondary antiphospholipid syndrome and IgM monoclonal paraproteinaemia. Clin Rheumatol 1999; 18 : 56–58.
63. Famularo G, Barracchini A, Minisola G. Severe thrombophilia with antiphospholipid syndrome and hyperhomocysteinemia in a patient with Schnitzler’s syndrome. Clin Exp Rheumatol 2003; 21 : 366–368.
64. Lauwers A, Chouvy V, Mosnier JF et al. A case of Schnitzler’s syndrome with nodular regenerative hyperplasia of the liver. Rev Rhum Engl Ed 1999; 66 : 281–283.
65. Doutre MS, Beylot C. Chronic urticaria and monoclonal IgM: Treatment with ibuprofen. J Am Acad Dermatol 1990; 22 : 143–144.
66. Bonnetblanc JM, Drouet M, Laplaud P et al. Urticaria with macroglobulinaemia. Disease activity associated alterations in immunoglobulins profile and bone marrow hypodiploidy. Dermatologica 1990; 181 : 41–43.
67. Lewicki M, Lewicka K, Kotyla PJ et al. Schnitzler’s syndrome. Pol Arch Med Wewn 2004; 111 : 735–741.
68. Lozano GF, Aquirre PA, Rivera Civico JM et al. The Schniztler’s syndrome. A case report. Med Clin (Barc) 1999; 112 : 158–159.
69. Obořilová A, Adam Z. Schnitzler’s syndrome. Vnitř Lék 1998; 44 : 423–427.
70. Peterlana D, Puccetti A, Tinazzi E et al. Schnitzler’s syndrome treated successfully with intravenous pulse cyclophosphamide. Scand J Rheumatol 2005; 34 : 328–330.
71. Schartz NE, Buder S, Sperl H et al. Report of a case of Schnitzler’s syndrome treated successfully with interferon α 2b. Dermatology 2002; 205 : 54–56.
72. Kuenzli S, Buchet S, Saurat JH. Successful treatment of Schnitzler’s syndrome with interferon α-2b. Dermatology 2002; 205 : 74.
73. Martinez-Taboada VM, Fontalba A, Blanco R et al. Successful treatment of refractory Schnitzler’s syndrome with anakinra. Arthritis Rheum 2005; 52 : 2226–2227.
74. de Koning HD, Bodar EJ, Simon A et al. Beneficial response to anakinra and thalidomide in Schnitzler’s syndrome. Ann Rheum Dis 2006; 65 : 542–544.
75. Worm M, Kolde G. Schnitzler’s syndrome: successful treatment of two patients using thalidomide. Br J Dermatol 2003; 148 : 601–602.
76. Dalle S, Balme B, Sebban C et al. Schnitzler’s syndrome associated with systemic marginal zone B-cell lymphoma. Br J Dermatol 2006; 155 : 827–829.
77. Modiano P, Bauraud-Laveine E et al. Efficacite de la puva therapei das um syndrome de Schnitzler. Nouv Dermatol 1995; 14 : 362–363.
78. Cianchini G, Colonna L, Bergamo F et al. Efficacy of Psoralen-UV-A therapy in 3 cases of Schnitzler’s syndrome. Arch Dermatol 2001; 137 : 1536–1537.
79. Asli B, Bienvenu B, Cordoliani F et al. Chronic urticaria and monoclonal IgM gammopathy (Schnitzler syndrome): report of 11 cases treated with pefloxacin. Arch Dermatol 2007; 143 : 1046–1050.
80. Blasum J, Wozel G, Patzak A et al. Schnitzler’s syndrome successful treatment with extracorporal immunoadsorption. Ann Dermatovenereol 1997; 124 (Suppl): 117.
81. Carbone J, Paravisini A, Sarmiento E et al. Partial response to cyclosporine in a patient with Schnitzler’s syndrome. Allergol Immunopathol (Madr) 2007; 35 : 71–73.
82. Pascual-López M, Hernández-Núñez A, Sánchez-Pérez J et al. Schnitzler’s syndrome with monoclonal IgG κ gammopathy: good response to cyclosporin. J Eur Acad Dermatol Venereol 2002; 16 : 267–270.
83. Thonhofer R, Uitz E, Graninger W. Schnitzler’s syndrome-exacerbation after anti‑TNF treatment. Rheumatology (Oxford) 2007; 46 : 1041–1042.
84. Eiling E, Möller M, Kreiselmaier I et al. Schnitzler syndrome: treatment failure to rituximab but response to anakinra. J Am Acad Dermatol 2007; 57 : 361–364.
85. Ramadan KM, Eswedi HA, El-Agnaf MR. Schnitzler syndrome: a case report of successful treatment using the anti‑CD20 monoclonal antibody rituximab. Br J Dermatol 2007; 156 : 1072–1074.
86. Gilson M, Abad S, Larroche C et al. Treatment of Schnitzler’s syndrome with anakinra. Clin Exp Rheumatol 2007; 25 : 931.
87. Klemmer N, Lenain P, Balguerie X et al. Effectiveness of anti‑IL1 in Schnitzler’s syndrome. Joint Bone Spine 2007; 74 : 509–510.
88. Kozel MM, Sabroe RA. Chronic urticaria: aetiology, management and current and future options. Drugs 2004; 64 : 2515–2536.
89. Ryan JG, de Koning HD, Beck LA et al. IL‑1 blockade in Schnitzler’s syndrome: ex vivo findings correlate with clinical remission. J Allergy Clin Immunol 2008; 121 : 260–262.
90. Schneider SW, Gaubitz M, Luger TA et al. Prompt response of refractory Schnitzler’s syndrome to treatment with anakinra. J Am Acad Dermatol 2007; 56 (Suppl 5): S120–S122.
91. Treudler R, Kauer F, Simon JC. Striking effect of the IL‑1 receptor antagonist anakinra in chronic urticarial rash with polyclonal increase in IgA and IgG. Acta Derm Venereol 2007; 87 : 280–281.
92. Wastiaux H, Barbarot S, Gagey-Caron V et al. Schnitzler syndrome: a dramatic improvement with anakinra. J Eur Acad Dermatol Venereol 2008 [Epub ahead of print].
93. Carlioz R, Haas C, Jauber F et al. Maladie de Waldenstöm avec lymphome diffuse et urticarie chronique. Ann Med Interne 1989; 140 : 51–78.
94. Claes K, Bammens B, Delforge M et al. Another devastating complication of the Schnitzler’s syndrome: AA amyloidosis. Br J Dermatol 2008; 158 : 182–184.
95. Govindaraju S, Brochot P, Ringot AC et al. Chronic urticaria-macroglobulinemia (Schnitzler syndrome): developing to IgM myeloma. Apropos of a case. Rev Med Interne. 1993; 14 : 780–783.
96. Machet L, Vaillant L, Machet MC. Schnitzler’s syndrome: Evolution to Waldenström’s disease is non uncommon. Acta Derm Venereol (Stockholm) 1996; 76 : 413.
97. Karakelides M, Monson KL, Volcheck GW et al. Monoclonal gammopathies and malignancies in patients with chronic urticaria. Int J Dermatol 2006; 45 : 1032–1038.
98. Lim W, Shumak KH, Reis M et al. Malignant evolution of Schnitzler’s syndrome – chronic urticaria and IgM monoclonal gammopathy: report of a new case and review of the literature. Leuk Lymphoma 2002; 43 : 181–186.
99. Pujol RM, Barnadas MA, Brunet S. Urticarial dermatosis associated with Waldenström’s macroglobulinemia. J Am Acad Dermatol 1989; 20 : 588–857.
100. Verret JL, Leclech C, Rousselet MC et al. Schnitzler’s syndrome and Waldenström disease. Fatal outcome of the original case. Ann Dermatol Venereol 1993; 120 : 459–460.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2008 Issue 12
Most read in this issue
- Nová evidence‑based kritéria pro posouzení vhodnosti lékového režimu u seniorů. Kritéria STOPP (Screening Tool of Older Person’s Prescriptions) a START (Screening Tool to Alert doctors to Right Treatment)
- Schnitzlerův syndrom – popis čtrnáctiletého průběhu nemoci a přehled informací o této nemoci
- Obrovskobuněčná arteriitida manifestující se oboustrannou arteriitickou přední ischemickou neuropatií zrakového nervu (AION)
- Léčba invazivní kandidózy – doporučení odborných společností