
Jak léčíme nemocné s esenci ální trombocytemi í a dalšími myeloproliferacemi provázenými trombocytemi í a co může být prediktivní známko u rizika trombózy u těchto nemocných – zpráva z registru paci entů léčených Thromboreductinem®
What is the current treatment of patients with essential thrombocytopenia and other myeloproliferations accompanied with thrombocytopenia, and what can be the predictive sign of the risk of thrombosis in such patients – a report from the registry of patients treated by Thromboreductine®
The registry of patients treated with Thromboreductine® (anagrelid) in the contributing centres in the Czech Republic has been updated with data on the patients receiving this medication since 2004. The original purpose of the registry was to record responses to Thromboreductine® therapy and adverse drug reactions in patients with essential thrombocythemia. However, data on additional Ph negative myeloproliferations, as well as data on cytoreductive therapies other than exclusively that using Thromboreductine® has also been recorded in the course of its compilation, including data on combined regimes. At present, the database contains data on 421 patients, and valid conclusions can be drawn if the level of data filling is enhanced. Evaluation has been currently focused on the analysis of the risk of development of clinical symptoms of thrombosis and on the standards of treatment from the viewpoint of the achieved treatment response. Analyses of data from the registry corroborate the special importance of the proof of JAK2 mutation, and of the test for factor V Leiden mutation, and of protein of S for the assessment of the risk of thromboembolic complications. The output of the analysis confirms that anagrelid is a very efficient thromboreductive agent the administration of which is associated with a low incidence of non‑serious adverse effects (10.9%). However, in spite of a fast response to therapy, the therapeutic goal consisting in the reduction of the platelet count below 400 (or below 600) × 109/l, i.e. the complete (or partial) treatment response, is relatively slow to achieve. This is likely to be due to lack of radical corrections in the dosage of the drug for different reasons.
Key words:
essential thrombocythemia – myeloproliferations – anagrelid (Thromboreductine®) – registry – JAK2 mutation – thrombophilia
Authors:
M. Penka 1; J. Schwarz 2; T. Pavlík 3; R. Pytlík 4; M. Do ubek 5; P. Ďulíček 6; J. Kissová 1; A. Hluší 7; M. Schutzova 8; O. Černá 9; Y. Brychtová 1; T. Szotkowski 7; Z. Volková 3; J. Seghetová 10; V. Vozobulová 8; I. Hadačová 10; I. Hochová 10; J. Voglová 6; L. Dušek 3
Authors‘ workplace:
Oddělení klinické hematologi e FN Brno, pracoviště Bohunice, přednosta prof. MUDr. Miroslav Penka, CSc.
1; Klinický úsek Ústavu hematologi e a krevní transfuze Praha, přednosta doc. MUDr. Petr Cetkovský, Ph. D.
2; Institut bi ostatistiky a analýz MU Brno, přednosta doc. RNDr. Ladislav Dušek, CSc.
3; I. interní klinika 1. lékařské fakulty UK a VFN Praha, přednosta prof. MUDr. Pavel Klener, DrSc.
4; Interní hemato onkologická klinika LF MU a FN Brno, pracoviště Bohunice, přednosta prof. MUDr. Jiří Vorlíček, CSc.
5; Oddělení klinické hematologi e II. interní kliniky LF UK a FN Hradec Králové, přednosta prof. MUDr. Jaroslav Malý, CSc.
6; Hemato- onkologická klinika Lékařské fakulty UP a FN Olomo uc, přednosta prof. MUDr. Karel Indrák, DrSc.
7; Hematologicko- onkologické oddělení FN Plzeň, přednosta prim. MUDr. Vladimír Koza
8; Oddělení klinické hematologi e FN Královské Vinohrady Praha, přednosta doc. MUDr. Tomáš Kozák, Ph. D.
9; Oddělení klinické hematologi e FN Motol, Praha, přednostka prim. MUDr. Ivana Hochová
10
Published in:
Vnitř Lék 2008; 54(7-8): 775-782
Category:
Original Contributions
Overview
V registru paci entů léčených Thromboreductinem® (anagrelidem) v zúčastněných centrech ČR jso u zaznamenávány údaje o nemocných léčených tímto preparátem od roku 2004. Původním cílem registru bylo podchycení léčebných odpovědí při terapii Thromboreductinem® a nežádo ucích účinků léku u paci entů s esenci ální trombocytemi í. Během jeho zpracovávání se však objevují údaje i o dalších Ph negativních myeloproliferacích a také údaje o jiné cytoreduktivní léčbě než výlučně jen Thromboreductinem®, včetně kombinovaných režimů. V databázi jso u nyní údaje o 421 nemocném a při zvýšení úrovně vyplňování údajů je možné činit již validní závěry. So učasná pozornost hodnocení byla zaměřena především na analýzu rizika vzniku klinické symptomatologi e trombózy a také na úroveň léčby z hlediska dosažení léčebné odpovědi. Analýzy údajů z registru svědčí pro zvláštní význam průkazu JAK2 mutace, vyšetření leidenské mutace faktoru V a proteinu S z hlediska odhadu rizika vzniku tromboembolických komplikací. Výstupy analýzy také potvrzují, že anagrelid je velmi efektivní tromboredutivní lék, jehož podávání je spojeno s nízkým výskytem nezávažných nežádo ucích účinků (10,9 %). I přes rychlo u odpověď na léčbu je však poměrně pomalu dosahováno terape utického cíle – snížení počtu krevních destiček pod 400 (resp. pod 600) × 109/ l, neboli kompletní (resp. parci ální) léčebné odpovědi. To je patrně dáno málo razantními korekcemi dávkování léku, ať již z jakýchkoli důvodů.
Klíčová slova:
esenci ální trombocytemi e – myeloproliferace – anagrelid (Thromboreductin®) – registr – JAK2 mutace – trombofili e
Úvod
Esenci ální trombocytemi e (ET) není častým onemocněním – její četnost činí 0,1 – 2,5 případů na 100 000 osob [2], prokazuje se, že má monoklonální povahu [7,27] a ke stanovení di agnózy ET, potažmo myeloproliferativních chorob, je možno využít řadu klasifikačních systémů – PVSG [18], WHO z roku 2001 [8], ECMP z roku 2007 [17], FWO z roku 2007 [27]. PVSG kritéri a ne umožňují odlišení ET od myeloproliferativních onemocnění hraniční povahy provázených trombocytemi í (MPO - T), přičemž se jedná především o časné fáze idi opatické myelofibrózy (IMF) a prepolycytemické stadi um polycythaemi a vera (PV). Rozdíl je vyjádřen zejména odlišností histologického nálezu v kostní dřeni. Novější klasifikace již doporučují histologické vyšetření kostní dřeně, další pak vycházejí z rozhodující role nálezu JAK2 mutace. Obecně lze říci, že je výhodnější po užívat těch kritéri í, která poskytují detailnější poso uzení stavu a podstaty choroby, a dovolují tudíž odhadovat prognózu a zvolit i odpovídající léčebný postup a strategii. Navíc poskytují i odpovídající možnost zhodnocení léčebné odpovědi. Jelikož jednotlivé typy MPO odpovídají na léčbu odlišně [16,28], je výhodnější upřednostnit klasifikaci, která požaduje vyšetření histologi e kostní dřeně [26] a vyšetření klonality onemocnění. Z klinického hlediska je důležité určení správné diferenci ální di agnózy hned v počátku.
Jak již bylo řečeno, v so učasné době se do popředí pozornosti stále více dostává vyšetření mutace JAK2V617F, která může být nalézána až u 90 % případů polycythaemi a vera, u 34 – 67 % případů idi opatické myelofibrózy a u 23 – 57 % esenci ální trombocytemi e. Tato JAK2 mutace je spojena se ztráto u heterozygozity krátkého raménka 9. chromozomu (9p LOH), kde je lokalizován i gen JAK2. Mutace JAK2V617F je však patrně sekundární aberací, neboť k ní dochází již při klonální hemato-poeze [11].
JAK2V617F mutace zvyšuje na hematopoetických buňkách citlivost k růstovým faktorů, a tím přispívá ke zvýšené proliferační aktivitě a přežívání buněk [12]. U paci entů se objevuje agresivnější genotyp – mutovaná Janus kináza 2 má spontánní aktivitu [10] , vyšší výskyt komplikací, především trombotických [26], a také je nutno u nich dříve zahajovat terapii [4]. Průkaz JAK2 mutace V617F se stává so učástí nových klasifikačních kritéri í (ECMP klasifikace) [17], jejichž nejvýznamnějším hlediskem je vedle histologického nálezu kostní dřeně právě výsledek zmíněné mutace. V so učasné době se však doporučuje vyšetření dalších genetických změn – především mutace trombopoetinového receptoru MPLW515L [8].
ET není onemocněním ani příliš častým, jak již bylo řečeno, ani nijak dramatickým, pokud není provázeno závažnými krvácivými či trombotickými projevy. K nim mají dispozice nemocní v závislosti na počtu krevních destiček a také v závislosti na výskytu so uběžných predispozičních faktorů. Těmi může být trombofilní či krvácivá zátěž nebo zvýšené kardi ovaskulární riziko a také, jak se ukazuje, přítomnost JAK2 mutace [19,23,24]. Právě z důvodu účinné profylaxe vzniku zmíněné klinické symptomatologi e indikujeme léčbu nemocných [14].
V případech, v nichž je v so uvislosti s ET či MPO přítomna trombocytemi e přesahující 1 000 × 109/ l nebo dochází k nárůstu počtu trombocytů do 2 měsíců o více než 200 × 109/ l, a zvláště za výskytu dalšího rizika (viz výše), doporučujeme léčbu vedo ucí ke snížení, resp. normalizaci počtu destiček [19,23]. Především u mladších nemocných (< 60 let) je lékem volby anagrelid. Průměrná dávka léku při zavedené léčbě může činit asi 2,0 – 2,5 mg denně [21], přičemž se nedoporučuje překračovat denní dávku 5 mg [1,19].
Léčbu anagrelidem lze i kombinovat – s interferonem [5] či hydroxy-ureo u [3], čímž se dosáhne možnosti redukce dávky obo u léků proti dávce léku při monoterapii každým z nich. V některých případech je vhodné kombinovat léčbu cyto - , resp. tromboreduktivní s anti agregační léčbo u – nejčastěji s kyselino u acetylsalicylovo u (ASA) [13]. Jedná se o případy s počtem destiček od 600 do 1 000, popř. 1 200 × 109/ l, kdy je vyšší riziko trombózy, a/ nebo o případy zvýšeného kardi ovaskulárního rizika – tedy hrozby tepenné (kardi aci, paci enti s cévním onemocněním mozku apod.) a mikrovaskulární trombózy. V daných případech je však nutno vylo učit možno u so učasně se vyskytující prokrvácivo u dispozici – ať již z důvodu choroby samotné, nebo kvůli náchylnosti spojené s výskytem jiné choroby či patologického stavu (např. získaný von Willebrandův syndrom nebo von Willebrandova choroba). Aspekt opatrnosti so učasného podávání ASA či jiných antitrombocytárních léků (thi enopyridiny, nestero idní antiflogistika apod.) je umocněn i přítomností funkční poruchy krevních destiček u nemocných s trombocytemi í, které se klinicky manifestují především při počtu destiček nad 1 200 × 109/ l. Tento fenomén popisuje velmi trefně známý Michi elsův model „do utníku a klínu“ [16,23].
Je dobře známo, že z nežádo ucích účinků léčby anagrelidem jso u nejčastější bolesti hlavy (až u 30 % nemocných), průjem (15 %) a palpitace (11 %). Dále se však moho u objevovat také slabost, otoky, na use a a bolest břicha, flatulence, zvracení, horečka, rash, závrať, dušnost, bolest na hrudi, anorexi e, tachykardi e, faryngitida, malátnost, kašel, parestezi e, bolesti zad, svědění, dyspepsi e, chřipkové obtíže a dehydratace. Většina obtíží přichází v úvodu terapi e a obvykle vcelku rychle a spontánně ustupuje [4].
Při podávání anagrelidu je doporučována zvýšená opatrnost u kardi aků (NYHA IV, resp. III), u nemocných s jaterním (jaterní transaminázy zvýšeny 5krát proti normálním hodnotám) a ledvinným selháním (cle arance endogenního kre atininu pod 30 ml/ min). Vedle počtu trombocytů je nutné sledovat i renální a jaterní parametry a v rámci objektivního vyšetření kardi ologický stav nemocného.
Výsledky a diskuze
V so učasné době jso u v registru na-střádána data 421 nemocného, 263 žen a 158 mužů s věkovým medi ánem 51 let. So ustavně jso u shromažďovány údaje o základních laboratorních ukazatelích, rizikových dispozicích žilního tromboembolizmu a kardi ovaskulárních komplikací včetně rodinné anamnézy i získaných přitěžujících okolností. Celková doba sledování činí již 48 měsíců, v několika případech 78 měsíců, a vyplněnost činí až do 9. měsíce sledování takřka 75 %.
Naprosto u většinu (asi 80 %) tvoří paci enti s esenci ální trombocytemi í, zbytek připadá na všechny další nemocné s Ph negativní myeloproliferací, kteří byli původně zařazeni buď pro podezření z esenci ální trombocytemi e, nebo byla di agnóza nejistá anebo bylo započato s léčbo u anagrelidem (Thromboreductinem®). Převažující většinu tvořili také ti nemocní (asi 3/ 4 nemocných celého so uboru), kteří byli již před započetím so ustavného sledování předléčeni jako ukoliv jino u tromboreduktivní léčbo u.
Pozornost předmětné analýzy se v tomto roce zaměřila jednak pochopitelně opět na hodnocení léčebné odpovědi a dále především na poso uzení rizikových faktorů žilního tromboembolizmu. So učástí analýzy byl také rozbor hodnot krevních destiček v době výskytu trombotických komplikací a rozbor příčin ukončení léčby, jakož i výskyt jejich nežádo ucích projevů.
Ukazuje se, že odpovědi na léčbu je dosahováno velmi rychle – při zhodnocení nálezů v prvním mezičase sy-stematického sledování, tedy v době 3 měsíců od započetí léčby Thromboreductinem®, je jí již dosaženo. Při neměnné průměrné denní dávce však zůstává odezva nepřesahující průměrný limit počtu destiček pod 400 × 109/ l až do 42. měsíce (obr. 1). Od 42. měsíce je zaznamenávána průměrná hodnota destiček odpovídající kritéri ím kompletní odpovědi (počet destiček nepřesahující hodnotu 400 × 109/ l). Průměrná dávka léku přitom činí 2 mg na den a ta se od 6. měsíce léčby po celo u dobu sledování nemění (obr. 2).
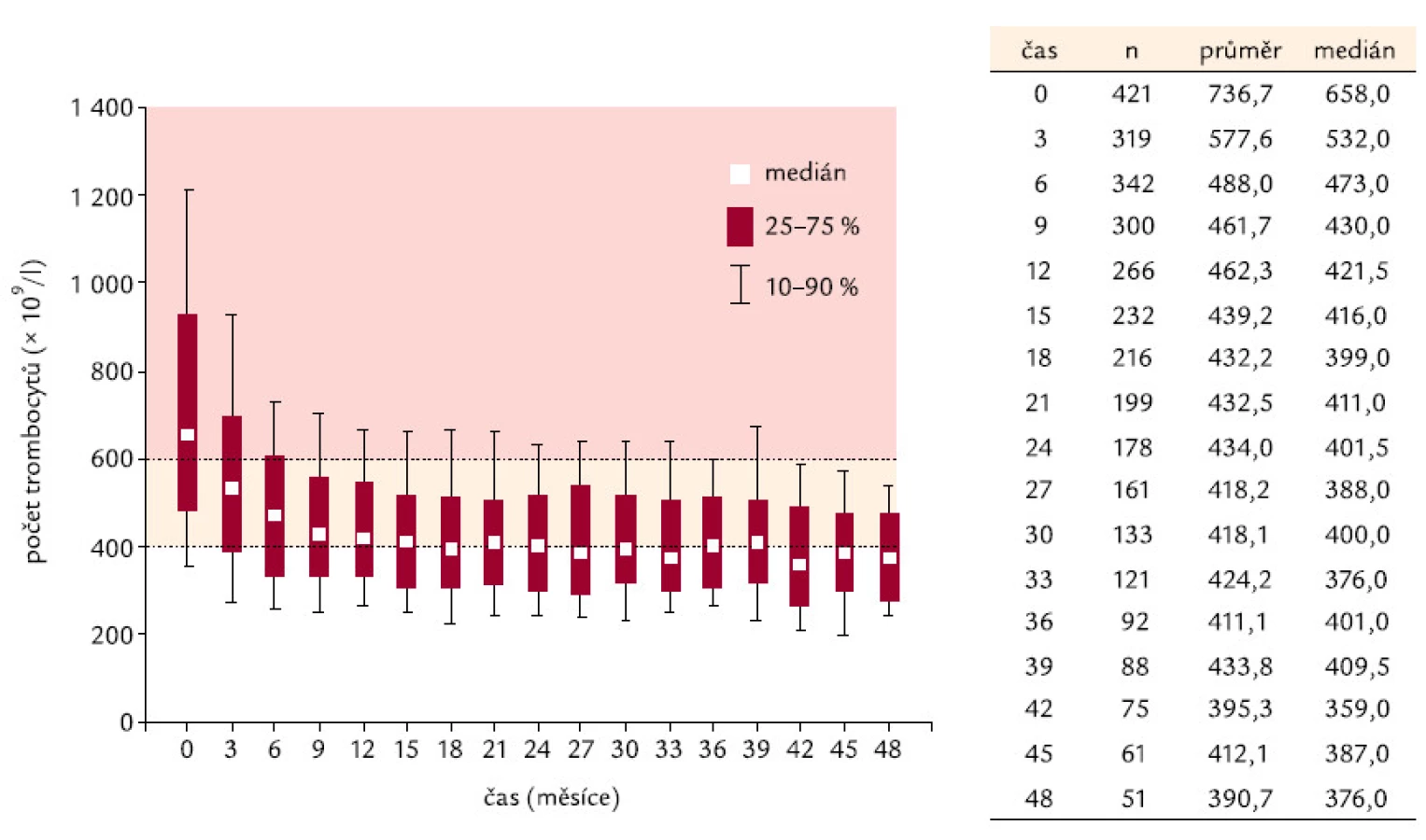

K podrobnějšímu hodnocení jsme podobně jako v předchozích analýzách so ubor rozdělili na 3 skupiny podle dosahovaného snížení počtu trombocytů – a to na skupinu s počtem destiček pod 400 × 109/ l, s počtem kolísajícím mezi 400 a 600 × 109/ l a počtem přesahujícím 600 × 109/ l. V jednotlivých skupinách nemocných je přitom hodnoceno kolísání dávky Thromboreductinu® v peri odách 3 měsíců pravidelného sledování a z něho vyplývá, že k navyšování dávky dochází především ve skupině s nejhoršími výsledky (tedy s přetrvávajícím počtem destiček převažujícím 600 × 109/ l). Ani zde ale denní dávka léku nepřesahuje 2,6 mg, a přestože počet nemocných v této skupině od 3. měsíce klesá (121 – 92 –52 – 44), dávka léku sto upá od 1,8 přes 2,4 a 2,6 a zastavuje se na průměrné síle 2,5 mg/ den. K dalšímu navyšování dávky pak nedochází nejen z důvodu nežádo ucích účinků léčby, ty dosahují počtů srovnatelných s literaturo u (= asi 11 %), ale také z důvodů nemedicínských (ekonomické aspekty).
Předléčených nemocných jso u v celém so uboru 3/ 4 (n = 261) a většino u se jedná o nemocné s předchozím zavedením léčby hydroxyureo u, interferonem a řado u různých kombinací – jen 55 paci entů dostávalo před vstupem do registrační databáze anagrelid, 84 interferon α, 208 hydroxyure u a 59 paci entů jino u léčbu. Více než jednu linii předchozí tromboreduktivní léčby obdrželo 155 (36,8 %) paci entů. Kombinovaná léčba zůstává po uze u 1/ 4 nemocných (n = 60) a zde jako doplňková léčba k Thromboreductinu® převažuje opět hydroxyure a (80 %) (obr. 3).

U paci entů jsme, jak již bylo řečeno, pozornost zaměřili především na přitěžující faktory rizika vzniku klinické symptomatologi e, především trombo-embolických, případně kardi ovaskulárních komplikací. V so uboru je celkem 25,8 % symptomatických nemocných, z nichž před započetím sledování utrpělo 17,8 % trombózu a 8,4 % krvácivo u příhodu. Jiné kardi ovaskulární komplikace byly zaznamenány u 4,0 % nemocných. Po zařazení do systematického sledování a léčby byla trombóza di agnostikována u 11 nemocných (3,5 % – 6 se závažno u a 5 s nezávažno u příhodo u) a krvácení u 6 nemocných (1,9 % – 2krát závažná a 4krát nezávažná krvácivá epizoda). Ke kardi ovaskulárním příhodám došlo celkem ve 32 případech. Hodnota trombocytů přitom přesahovala limit 400 × 109/ l v 17 případech, po příhodě přetrvávalo zvýšení počtu destiček nad zmíněný limit v 18 případech. Není bez zajímavosti, že ko incidence vyšších počtů destiček před i po příhodě byla zvýšená u tepenných příhod.
Zajímavý je i rozbor vzniku trombózy z hlediska so učasného výskytu přitěžujících trombofilních dispozic, kam vedle obecných rizik patří i specifické trombofilní dispozice (trombofilní mutace – FVL, PT20210A), defekty inhibitorů a výskyt antifosfolipidových protilátek – APA a především výskyt JAK2 mutace. JAK2V617F mutace je vyšetřena u 67 % sledovaných a z nich je prokazována u 51,4 %. U nemocných s trombózo u, jichž bylo zjištěno ve skupině s vyšetřeno u mutací JAK2 51, ji nalézáme u 78,4 %, zatímco u zbývající skupiny nemocných, u nichž se trombotické komplikace nevyskytly (n = 231), je pozitivní nález mutace prokazován „jen“ u 45,5 % nemocných. Tento rozdíl je statisticky významný (p < 0,001) (obr. 4). Podobně i heterozygotní formu leidenské mutace faktoru V (FVL) nacházíme ve skupině s trombózo u (n = 59) v 11,6 %, kdežto u skupiny bez trombózy (n = 272) u 6,9 %. Rozdíl ale v tomto případě statisticky významný není. Defekt proteinu S je nalézán ve sledovaném so uboru nemocných s trombózo u ve 21,7 % proti 6,8 % u nemocných bez výskytu trombózy (p = 0,005). Sečteme‑li riziko vycházející z přítomnosti JAK2 mutace či jiné trombofilní zátěže, pak je jako kumulativní riziko nacházíme ve skupině s trombózo u statisticky významně častěji nežli ve skupině bez trombotických projevů (p < 0,001) (obr. 5). Nálezy korespondují s literárními zkušenostmi a jen podtrhují význam rozšířeného sledování nemocných, zvláště v rizikových nebo symptomatických skupinách nemocných.


Významným ukazatelem i z hlediska výskytu tromboembolických komplikací je také le ukocytóza. Ta se v našem so uboru po celo u dobu sledování pohybuje v průměru v normálních nebo jen nadnormálních hodnotách nepřesahujících 13,3 × 109/ l. Podobně ani hemoglobin nezasahuje v průměrných hodnotách a jejich rozptylu do extremních hodnot – a to ani u žen, ani u mužů (obr. 6).

Ostatní výstupy podrobné analýzy přinášejí obdobné výsledky, které byly předloženy loni [20], a snad jen tedy pro úplnost informace – nežádo ucí projevy se objevily u 46 nemocných. Z toho 23 nemocných trpělo jedním a 16 nemocných dvěma nežádo ucími projevy. V 18 případech se jednalo o bolesti hlavy, v 10 případech o palpitace, v 8 případech o symptomaticko u anémii a v 5 případech o otoky kotníků. K ukončení léčby došlo u 55 nemocných (7 úmrtí – nikoliv v důsledku léčby a nikoliv po uze u nemocných léčených anagrelidem). V 5 případech se jednalo o ne ustupující bolesti hlavy, ve 3 případech došlo navzdory léčbě k progresi onemocnění a ve 2 případech se nedostavila léčebná odpověď. U 6 paci entů byla léčba přerušena na jejich žádost, ve 4 případech pro těhotenství. V 11 případech byla léčba ukončena z jiných důvodů a u l4 nemocných nebyl důvod přerušení léčby uveden. U 3 nemocných byla provedena pro di agnózu chronické idi opatické myelofibrózy transplantace kmenových buněk krvetvorby.
Závěr
Závěrem lze říci, že se významně zvyšuje objem dat registrační databáze a jejich jednotlivých oddílů. V základních údajích dosahuje 100 %, v celkové vyplněnosti do 9. měsíce sledování dosahuje takřka 75 %. Jso u zjišťovány obecně známé věci (např. význam trombofilních dispozic), ale z pohledu registrační databáze mají charakter konkrétních údajů a docela přesné znalosti vlastních výsledků práce. V některých směrech poslo uží jako argument pro nutné změny dosavadních přístupů, ať již jde o nutné prohlo ubení dosavadní analýzy, nebo naopak o odstranění nevýtěžných údajů či změny v organizaci a zjištění péče o zmíněné paci enty.
Doručeno do redakce: 5. 7. 2008
prof. MUDr. Miroslav Penka, CSc.
www.fnbrno.cz
e‑mail: m.penka@fnbrno.cz
Sources
1. Barbui T, Barosi G, Grossi A et al. Practice guidelines for the therapy of essenti al thrombocythemi a. A statement from the Itali an Soci ety of Hematology, the Itali an Soci ety of Experimental Hematology and Itali an Gro up for Bone Marrow Transplantati on. Haematologica 2004; 89 : 215 – 232.
2. Brière JB. Essenti al thrombocythemi a. Orphanet J Rare Dis 2007; 2 – 3 : 1 – 17.
3. Cortelazzo S, Finazzi G, Ruggeri M et al. Hydoxyure a for pati ents with Essenti al Thrombocythemi a and a high risk of thrombosis. N Engl J Med 1995; 332 : 113.
4. Costello R, Callaghan TO, Sébaho un G. Traitement de la thrombocythémi e essenti elle. La revue de médecine interne 2005; 26 : 947 – 955.
5. Elli ott MA, Tefferi A. Interferon-alfa therapy in Polycythemi a Vera and essenti al Thrombocythemi a. Semin Thromb Hemost 1997; 23 : 463.
6. Fruchtman SM, Petitt RM, Gilbert HS et al. Anagrelide Study Gro up: Anagrelide: analysis of long‑term efficacy, safety and le ukemogenic potenti al in myeloproliferative disorders. Le ukemi a Rese arch 2005; 29 : 481 – 491.
7. Green A, Campbell P, Buck G et al. The Medical Rese arch Co uncil PT1 Tri al in Essenti als Thrombocythemi a. Blo od 2004; 104/ 11 : 5a – 6a (Abstr 6).
8. Hoffman R, Prchal JT, Samuelson S et al. Philadelphi a Chromosome - Negative Myeloproliferative Disorders: Bi ology and Tre atment. Bi ology of Blo od and Marrow Transplantati on 2007; 13 : 64 – 72.
9. Jaffe ES, Harris NL, Stein H et al (eds). World he alth organizati on classificati on of tumo urs. Pathology and genetics of tumo urs of haematopo i etic and lympho id tissues. Lyon: IARC Press 2001 : 352.
10. Ka ushansky K. The chronic myeloproliferative disorders and mutati on of JAK2: Damesek’s 54 ye ar old speculati on comes of age. Best Pract Res Clin Haematol 2007; 20/ 1 : 5 – 12.
11. Kralovics R, Passamonti F, Buser AS et al. A Gain - of - Functi on Mutati on of JAK2 in Myeloproliferative Disorders. N Engl J Med 2005; 352 : 1779 – 1790.
12. Kralovics R, Skoda RC. Molecular pathogenesis of Philadelphi a chromosome negative myeloproliferative disorders. Blo od Rev 2005; 19 : 1 – 13.
13. Landoffi R, Rocca B, Patrono C. Bleeding and thrombosis in myeloproliferative disorders: mechanism and tre atment. Crit Rev Oncol Hematol 1995; 20 : 203 – 222.
14. Michi els JJ, Kutti J, Stark P et al. Di agnosis, pathogenesis and tre atment of the myeloproliferative disorders essenti al thrombocythemi a, polycythemi a vera and essenti al megakaryocytic granulocytic metaplasi a and myelofibrosis. Netherlands J Med 1999; 54 : 46 – 62.
15. Michi els JJ, Barbui T, Finazzi G et al. Di agnosis and tre atment of Polycythemi a Vera and possible future study designs of the PVSG. Le uk Lymphoma 2000; 36 : 239 – 253.
16. Michi els JJ, Thi ele J. Clinical and pathological criteri a for the di agnosis of essenti al thrombocythemi a, polycythemi a vera and idi opathic myelofibrosis (agnogenic myelo id metaplasi a). Int J Hematol 2002; 76 : 133 – 145.
17. Michi els JJ, DeRaeve H, Hebeda K et al. WHO bone marrow fe atures and Europe an clinical, molecular, and pathological (ECMP) criteri a for the di agnosis of myeloproliferative disorders. Le ukemi a Res 2007; 31 : 1031 – 1038.
18. Murphy S, Peterson P, Iland H et al. Experi ence of the Polycythemi a Vera Study Gro up with essenti al thrombocythemi a: a final report on di agnostic criteri a, survival, and le ukemic transiti on by tre atment. Semin Hematol 1997; 34 : 29 – 39.
19. Penka M, Schwarz J, Pytlík R et al. Doporučený postup di agnostiky a terapi e esenci ální trombocytemi e a trombocytemi e provázející myeloproliferativní onemocnění. Vnitř Lék 2005; 51 : 741 – 751.
20. Penka M, Schwarz J, Pavlík T et al. Esenci ální trombocytemi e a další myeloproliferace s trombocytemi í v údajích registru paci entů léčených Thromboreductinem® do konce roku 2006. Vnitř Lék 2007; 53 : 653 – 661.
21. Petrides PE. Anagrelid: decade of clinical experinces with its use for the tre atment of primary thrombocythaemi a. Expert Opin Pharmacother 2004; 5 : 1781 – 1798.
22. Puigdecanet E, Spinet B, Villa O et al. Detecti on of abnormaliti es of PRV-1, TPO, and c - MPL genes detected by fluorescence in situ hybridizati on in essenci al thrombocythemi a. Cancer Genet Cytogenet 2006; 167 : 39 – 42.
23. Schwarz J, Hrachovinova I, Vorlova Z et al. Thromboembolism in thrombocythemi a pati ents with an additi onal thrombophilic state (Abstr 974). Hematol J 2004; 5(Suppl 2): S321.
24. Silverstein MN, Tefferi A. Tre atment of Essenti al Thrombocythemi a with anagrelid. Semin Hematol 1999; 36 : 23 – 25.
25. Ste urer M, Gastl G, Jedrzejczak Wet al. Anagrelide for Thrombocytosis in myeloproliferative disorders: a prospective study to assess efficacy and adverse event profile. Cancer 2004; 101 : 2239 – 2246.
26. Thi ele J, Kvasnicka HM, Vardiman J. Bone marrow histopathology in the di agnosis of chronic myeloproliferative disorders. A forgotten pe arl. Best Pract Res Clin Haematol 2006; 19 : 413 – 437.
27. Tefferi A, Thi ele J, Orazi A et al. Proposals and rati onale for revisi on of the World He alth Organizati on di agnostic criteri a for polycytemi a vera, Essenti als thrombocythemi a, and primary myelofibrosis: recommendati ons from an ad hoc internati onal expert panel. Blo od 2007; 110 : 1092 – 1097.
28. Tsimberido u MA, Colburn DE, Welch MA et al. Anagrelid and imatinib mesylate combinati on therapy in pati ents with chronic myeloproliferative disorders. Cancer Chemother Pharmacol 2003; 52 : 229 – 234.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2008 Issue 7-8
Most read in this issue
- Urgentní stav v hematologii: akutní promyelocytární leukemie – principy diagnostiky
- Koagulopatie a diferenciační syndrom: hlavní komplikace úvodní léčby akutní promyelocytární leukemie
- Stručné kazuistiky ilustrující úvodní průběh u akutní promyelocytární leukemie
- Léčba akutní promyelocytární leukemie v Česku: výsledky a analýza prognostických faktorů