
Diabetes insipidus, následovaný po 4 letech dysartrií a lehkou pravostrannou hemiparézou – první klinické příznaky Erdheimovy‑ Chesterovy nemoci. Popis a zobrazení případu s přehledem informací o této nemoci
Diabetes insipidus followed, after 4 years, with dysarthria and mild right ‑ sided hemiparesis – the first clinical signs of Erdheim - Chester disease. Description and depiction of a case with a review of information on the disease
In 2004, diabetes insipidus was the first clinical sign of Erdheim ‑ Chester disease in our patient. Following introduction of substitution therapy with adiuretin, the patient had no further health complaints for four years until 2008 when he gradually developed dysarthria and, consequently, movement disorder in the form of mild right hemiparesis. The first CNS CT scan (2004) did not reveal any pathology. The first pathological MRI of the brain in 2006 – thickening of pituitary stalk by pathological infiltration to 4 – 5 mm. During the following year, further infiltrates were detected in the CNS. The number and size of CNS infiltrates increased gradually on MRIs performed repeatedly up to 2008. Erdheim ‑ Chester disease has become suspected based on PET‑CT examination at the end of 2008. CT showed irregular structure of the skeleton with noticeable sclerotic foci in otherwise osteoporotic bone structure; changes were the most evident in the long bones of lower limbs, in the pelvic bones, skull and arms, while only one vertebra was affected from within the entire spine. Finding of thickened aortic wall (up to 8 mm) as another pathological circumstance was consistent with the Erdheim ‑ Chester disease‑associated changes described as coated aorta. CT scan revealed clear fibrotic changes in the area of retroperitoneum. Applied fluorodeoxyglucose has accumulated in the bone foci described on CT scans as well as in the thickened wall of the thoracic and abdominal aorta (SUV 3.6). Tc ‑ pyrophosphonate skeleton scintigraphy showed the same bone foci as PET‑CT. Full body MRI showed pathological signal from the bone marrow of the above mentioned locations, particularly during STIR imagining, where there was clear abnormal signal corresponding to accumulated histiocytes, the higher signal of which was well‑differentiated from the normal bone marrow. Measurement of bone mineral density with DEXA confirmed reduced density in lumbar vertebrae to the average value of – 2.7 SD (the lowest value was – 3.1SD). The disease is associated with elevated inflammatory parameters: leucocytosis, thrombocytosis, elevated CRP and fibrinogen levels. Diagnosis was verified following histological assessment of iliac bone marrow, where focal infiltrations with foamy histiocytes of typical immunophenotype (CD68+, CD1a – , S100 – ) were confirmed. Treatment was initiated with chemotherapy consisting of 2 g/ m2 of cyclophosphamide on day 1 and 200 mg/ m2 of etoposide IV infusion on days 1 – 3, and followed by administration of 5 μg/ kg of G‑CSF and collection of haematopoietic peripheral blood stem cells (PBSC). PBSC collection was followed by 5 ‑ day administration of 5 mg/ m2/ day of 2 ‑ chlorodeoxyadenosine (Litac) administered to the patient at monthly intervals.
Key words:
Erdheim ‑ Chester disease – juvenile xanthogranuloma – osteosclerosis – skeletal scinigraphy – PET‑CT – multiple myeloma – 2 ‑ chlorodeoxyadenosine – cladribin – retroperitoneal fibrosis
Authors:
Z. Adam 1; K. Balšíková 2; L. Pour 1; M. Krejčí 1; P. Svačina 2; M. Dufek 3; L. Křen 4; M. Hermanová 5; M. Moulis 4; J. Vaníček 6; J. Neubauer 7; M. Mechl 7; J. Prášek 8; J. Staníček 9; R. Koukalová 9; R. Hájek 1; J. Mayer 1
Authors‘ workplace:
Interní hematologická klinika Lékařské fakulty MU a FN Brno, pracoviště Bohunice, přednosta prof. MU Dr. Jiří Vorlíček, CSc.
1; II. interní klinika Lékařské fakulty MU a FN u sv. Anny Brno, přednosta prof. MU Dr. Miroslav Souček, CSc.
2; Neurologická klinika Lékařské fakulty MU a FN u sv. Anny Brno, přednosta prof. MU Dr. Ivan Rektor, DrSc.
3; Ústav patologie Lékařské fakulty MU a FN Brno, pracoviště Bohunice, přednosta doc. MU Dr. Josef Feit, CSc.
4; Patologicko‑anatomický ústav Lékařské fakulty MU a FN u sv. Anny Brno, přednostka doc. MU Dr. Markéta Hermanová, Ph. D.
5; Klinika zobrazovacích metod Lékařské fakulty MU a FN u sv. Anny Brno, přednosta doc. MU Dr. Petr Krupa, CSc.
6; Radiodiagnostiká klinika Lékařské fakulty MU a FN Brno, pracoviště Bohunice, přednosta prof. MU Dr. Vlastimil A. Válek, CSc.
7; Klinika nukleární medicíny Lékařské fakulty MU a FN Brno, pracoviště Bohunice, přednosta doc. MU Dr. Jiří Prášek, CSc.
8; Oddělení PET CT Masarykova onkologického ústavu Brno, přednosta prim. MU Dr. Karol Bolčák, Ph. D.
9
Published in:
Vnitř Lék 2009; 55(12): 1173-1188
Category:
Case Reports
Overview
Prvním klinickým příznakem Erdheimovy ‑ Chesterovy nemoci u našeho pacienta byl v roce 2004 diabetes insipidus. Po zavedení substituce adiuretinem byl bez dalších zdravotních potíží po další 4 roky, do roku 2008, kdy se postupně rozvinula dysartrie a následně porucha hybnosti ve formě frustní pravostranné hemiparézy. První CT zobrazení CNS (roku 2004) bylo bez patologie. V roce 2006 byl první patologický nález na MR mozku – zesílení stopky hypofýzy patologickou infiltrací na 4 – 5 mm. V následujícím roce nález dalších infiltrátů v CNS. Na opakovaných MR zobrazeních do roku 2008 narůstal počet infiltrátů CNS a zvětšovala se jejich velikost. Podezření na Erdheimovu ‑ Chesterovu chorobu vyplynulo z PET‑CT vyšetření koncem roku 2008. CT zobrazila nepravidelnou strukturu skeletu s nápadnými sklerotickými ložisky v jinak prořídlé kostní struktuře, změny byly nejvíce zřetelné v dlouhých kostech dolních končetin, dále pak v kostech pánve, kalvy, pažních kostí, zatímco z páteře byl postižen pouze jeden obratel. Dalším patologickým nálezem bylo zesílení stěny aorty (až na 8 mm), což odpovídá změnám popisovaným u Erdheimovy ‑ Chesterovy nemoci jako coated aorta. Na CT obrazu byly zřetelné fibrotické změny v oblasti retroperitonea. Aplikovaná fluorodeoxyglukóza se kumulovala v kostních ložiscích, popsaných na CT zobrazení, a dále v zesílené stěně hrudní a břišní aorty (SUV 3,6). Scintigrafie skeletu Tc ‑ pyrofosfonátem znázornila stejná kostní ložiska jako PET‑CT. Celotělové MR zobrazení popsalo patologický signál z kostní dřeně ve výše uvedených lokalizacích, zejména při STIR zobrazení, kde byl zřetelný abnormální signál, který odpovídal nakumulovaným histiocytům, jejichž vyšší signál byl dobře diferencovatelný od normální kostní dřeně. Měření kostní hustoty metodou DEXA prokázalo snížení denzity bederních obratlů na průměrnou hodnotu – 2,7 SD (nejnižší hodnota byla – 3,1 SD). Nemoc provází zvýšení parametrů zánětu: leukocytóza, trombocytóza, zvýšená hladina CRP a fibrinogenu. Diagnóza byla stanovena histologickým hodnocením kostní dřeně získané z lopaty kosti pánevní, v níž byla prokázána ložisková infiltrace pěnitými histiocyty s typickým imunofenotypem (CD68+, CD1a – , S100 – ). Léčba byla zahájena chemoterapií cyklofosfamidem 2 g/ m2 den 1 a etoposidem 200 mg/ m2 i.v. infuze den 1 – 3, s následnou aplikací G‑CSF v dávce 5 μg/ kg a sběrem kmenových krvetvorných buněk z periferní krve (PBSC). Po sběru PBSC byla zahájena aplikace 2 ‑ chlordeoxyadenosinu (Litac), v dávce 5 mg/ m2 s.c., cyklofosfamidu 150 mg/ m2 i.v. a dexametazonu 24 mg i.v., vše 1. – 5. den. Tyto cykly se opakovaly po 28 dnech, celkem 6krát. Účinnost léčby byla hodnocena kontrolním celotělovým PET‑CT, celotělovým MR zobrazením s difuzí, scintigrafií skeletu a MR zobrazením CNS. Po 6 cyklech chemoterapie zůstává patologický nález v dlouhých kostech beze změny. Pouze na MR mozku bylo zřetelné zmenšení ložiskových infiltrátů.
Klíčová slova:
Erdheimova ‑ Chesterova choroba – juvenilní xantogranulom – osteoskleróza – scintigrafie skeletu – PET‑CT – mnohočetný myelom – 2 ‑ chlordeoxyadenosin – cladribin – retroperitoneální fibróza
Úvod
Maligní histiocytární nemoci tvoří svým výskytem nepatrný zlomek ze všech krevních chorob. Nejčastější z této skupiny nemocí je histiocytóza z Langerhansových buněk (LCH), ale i ta je velmi vzácná. Pediatři v ČR ročně diagnostikují asi 26 nových případů LCH [1–3]. V dospělosti je však popisována také [4,5]. Výskyt LCH u dospělých v ČR není znám.
Erdheimova Chesterova choroba jepodstatně vzácnější než LCH. Početpopsaných případů Erdheimovy Chesterovy choroby ve světě se v roce 2004 pohyboval kolem jednoho sta, v roce 2006 jich v lékařské literatuře bylo napočítáno přes 200.
Dle WHO klasifikace maligních krevních chorob patří Erdheimova Chesterova nemoc do kategorie diseminovaného juvenilního xantogranulomu, který tvoří samostatnou jednotku ve skupině histiocytárních nemocí.
Erdheimova Chesterova nemoc máněkteré společné rysy s LCH. Prvním příznakem obou nemocí může být diabetes insipidus, neboť obě nemoci mohou postihnout hypotalamus, stopkuhypofýzy a cerebellum. Obě nemoci mohou poškodit i plicní parenchym. Jinými projevy se však tyto nemocí liší a liší se také průměrným věkem výskytu. Erdheimova Chesterova nemoc se pravidelně vyskytuje v dospělém věku.
Podstatou Erdheimovy Chesterovy nemoci je proliferace histiocytů, obsahujících tukové inkluze. Díky nim mají histiocyty pak pěnitý charakter (foamy histiocytes). Tyto patologické histiocyty pravidelně proliferují v dlouhých kostech dolních končetin, což vede k typickému obrazu zbytnění trabekul a kortikalis v tibiích a femorech. Dále mohou pěnité histiocyty infiltrovat retroperitoneum a způsobit zánětlivé a fibrotické změny, mohou infiltrovat mediastinum a struktury v něm uložené, srdce a cévy. Proliferace patologických histiocytů může také postihnout plicní parenchym, kůži i mozek. Mezi jednotlivými nemocnými jsou rozdíly v rozsahu postižení. Pro léčbu této nemoci nejsou pro její výjimečnost vypracována žádná doporučení, v publikovaných popisech byla použita léčba podobná jako u LCH [6].
V následujícím textu popisujeme pacienta s touto chorobou a v diskuzi přinášíme podrobné informace o této nemoci, zatím zřejmě nejobsáhlejší pojednání o této nemoci v české literatuře.
Popis případu
Příznaky nemoci
Centrální diabetes insipidus – iniciální příznak Erdheimovy Chesterovy nemoci
Muž narozený roku 1965 byl až do roku 2004 (do svých 39 let) zcela zdráv. Prvním příznakem nemoci byla polydipsie a polyurie. Diagnóza centrálního idiopatického diabetes insipidus byla potvrzena koncentračním testem v září roku 2004 za hospitalizace na II. interní klinice FN u sv. Anny v Brně. Tvorba ostatních hypofyzárních hormonů byla v mezích normy, pacient byl tedy bez nálezu panhypopituitarizmu. První MR mozku bylo provedeno v roce 2004. V té době nebyla popsána žádná patologie v oblasti hypofýzy. Oční vyšetření a vyšetření perimetru bylo zcela normální. Po zavedení substituce adiuretinem v dávce 2 kapky denně byl mladý muž opět bez potíží. Z kompletního laboratorního vyšetření v roce 2004 uvádíme pouze hladiny hormonů v séru: tyreotropin (ultras. TSH) 2,26 mU/l, volný tyroxin (fT4) 14,28 pmol/l, trijódtyronin (T3) 1,84 nmol/l, folikuly stimulační hormon (FSH) 3,8 U/l, luteinizační hormon (LH) 3,5 U/l, testosteron 11,57 nmol/l, kortizol (ráno) 772,6 nmol/l, adrenokortikotropin (ACTH) 28,9 pg/ml. Osmolalita moče byla 72 mmol/kg a séra 322 mmol/kg. V roce 2004 měl zcela normální krevní obraz, leukocyty 8,5 × 109/l, hemoglobin 162g/l, trombocyty 355 × 109/l s normální diferenciálním rozpočtem, CRP bylo 4,5mg/l, fibrinogen 3,78g/l a normální sedimentace. Tedy žádné zvýšení parametrů zánětu.
Při dalších kontrolách na endokrinologické ambulanci byl mladý muž až do června roku 2006 bez dalších zdravotních problémů, při výše uvedené terapii adiuretinem byl bez obtíží, měl vyrovnanou a přiměřenou bilanci tekutin.
MR mozku a diferenciální diagnostika infiltrátu stopky hypofýzy
V červnu roku 2006 bylo provedeno kontrolní MR zobrazení mozku. Poprvé se objevil patologický nález v oblasti stopky hypofýzy. Stopka byla v obou rovinách zesílená na 4–5mm, MR obraz odpovídal patologické infiltraci. Diferenciálně diagnosticky připadaly v úvahu následující choroby: sarkoidóza, histiocytóza, germinom, lymfocytární hypofyzitida.
Nález byl konzultován na Neurochirurgické klinice FN u sv. Anny v Brně a v červenci roku 2006 byla provedena endoskopická, stereotakticky navigovaná ventrikulocisternoanastomóza s parciální resekcí (elektrokoagulací) stopky hypofýzy a byl odebrán drobný vzorek k histologickému vyšetření.
Ve vyšetřeném vzorku z oblasti stopkyhypofýzy byla zastižena glioneuronální tkáň s mírnou proliferací gliové komponenty, ložisková lymfocytární celulizace tvořená smíšenou populací T a B lymfocytů, s mírnou převahou T lymfocytů a perivaskulární akcentací.
Nález byl z hlediska patomorfologie zcela nespecifický, a byl proto hodnocen jako lymfoidní zánětlivá infiltrace v oblasti hypofýzy, avšak vzhledemke klinickému obrazu nebyla vyloučenatotožnost s lymfocytární infundibulo-neurohypofizitidou.
Morfologie příznačná pro diagnózu Erdheimovy Chesterovy choroby (infiltrace pěnitými makrofágy, mnohojaderné obrovské buňky Toutonovy, fibroblastické pozadí) nebyla prokázána.
Po neurochirurgickém zákroku došlo u pacienta k rozvoji kompletního panhypopotuitarizmu v ose tyreotropní, gonadotropní, kortikotropní, s nutností substituční terapie (Hydrocortizon 10–10–5mg, Euthyrox 75 μg 1krát denně, Sustanon 1 amp i.m. ve 3týdenním intervalu, Minirin spray 2krát denně), při které byl pacient dobře kompenzován.
Uvedeme hodnoty hormonů z 18. 7. 2006, tedy z vyšetření před nasazením substituční léčby: folikuly stimulující hormon (FSH) 1,9 U/l, lutei-nizační hormon (LH) 2,1 U/l, testosteron 2,34 nmol/l, tyreotropin (ultras. TSH) 1,17 mU/l, volný tyroxin (fT4) 7,71 pmol/l, kortizol (ráno) 176,3 nmol/l, sérový adrenokortikotropin (ACTH) < 10,0 pg/ml, růstový hormon (hSTH) < 0,12 mU/l, IGF1 (Insulin like growth) 162 μg/l.
Při dalších kontrolních MR zobrazeních mozku v letech 2006, 2007 i 2008 dochází postupně k přibývání ložisek v mozkové tkáni a v lednu roku 2008 se objevilo největší ložisko v mesencefalu, jak uvádí obr. 1–6.
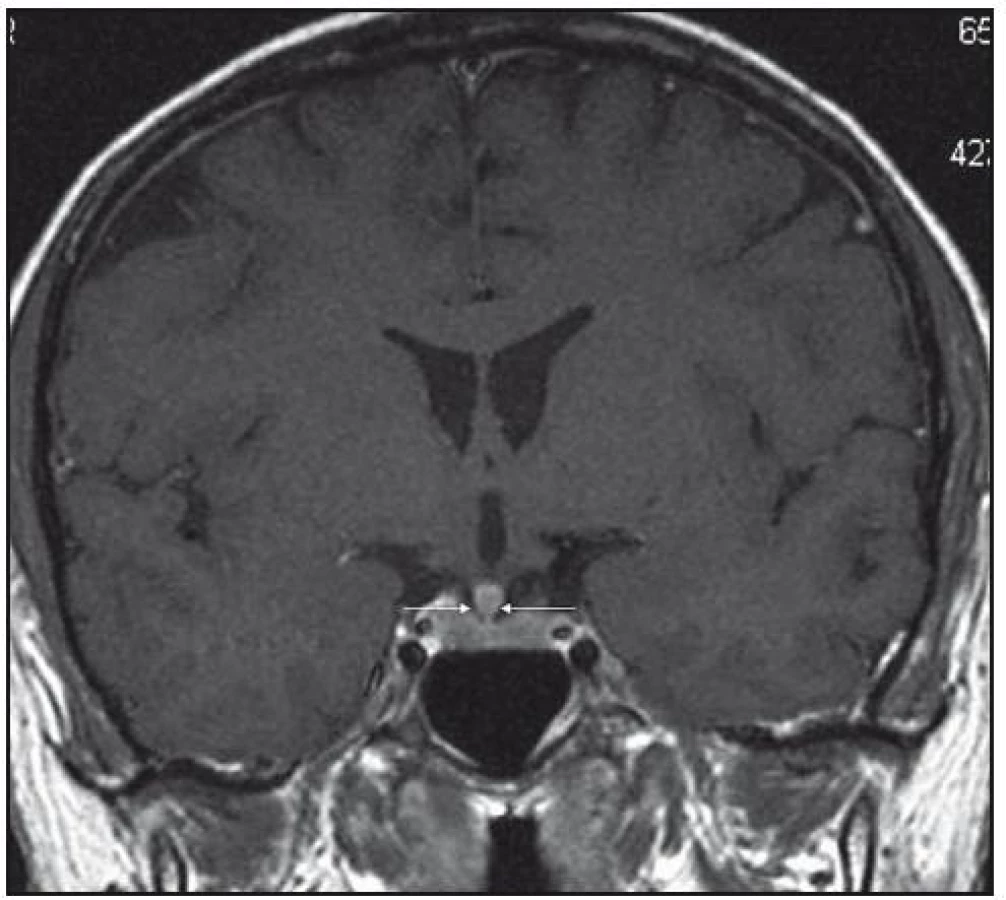
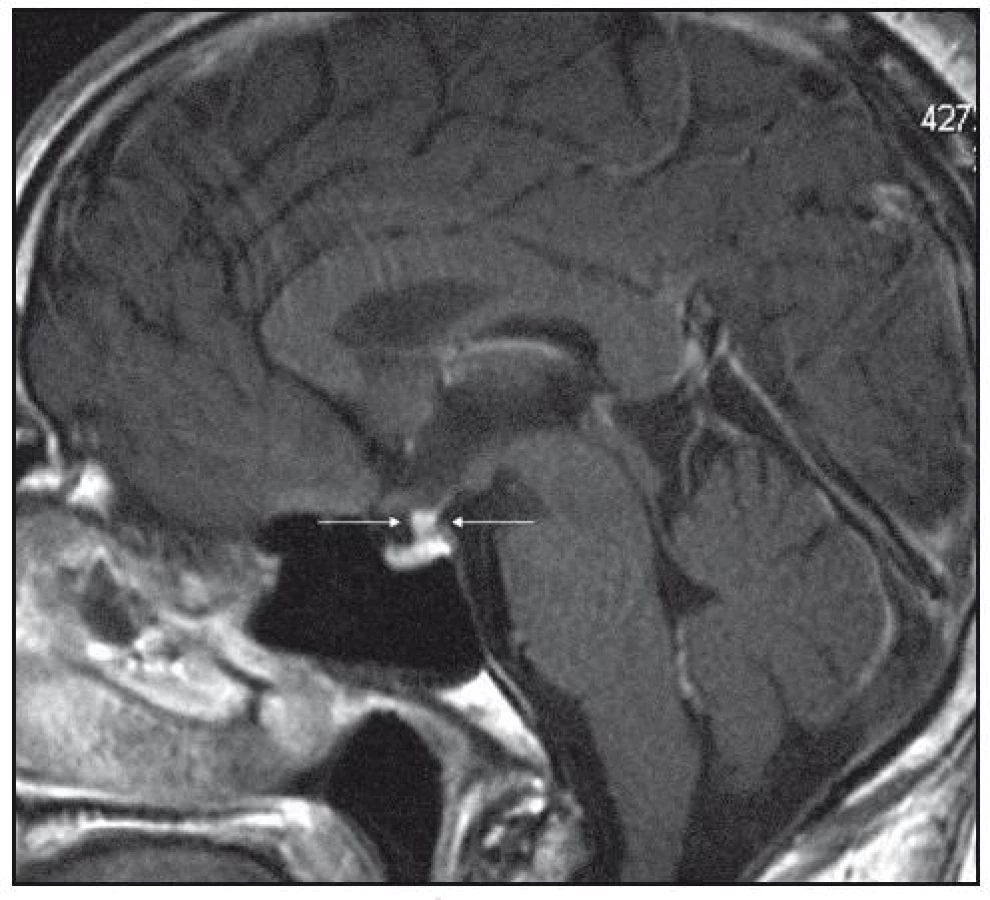
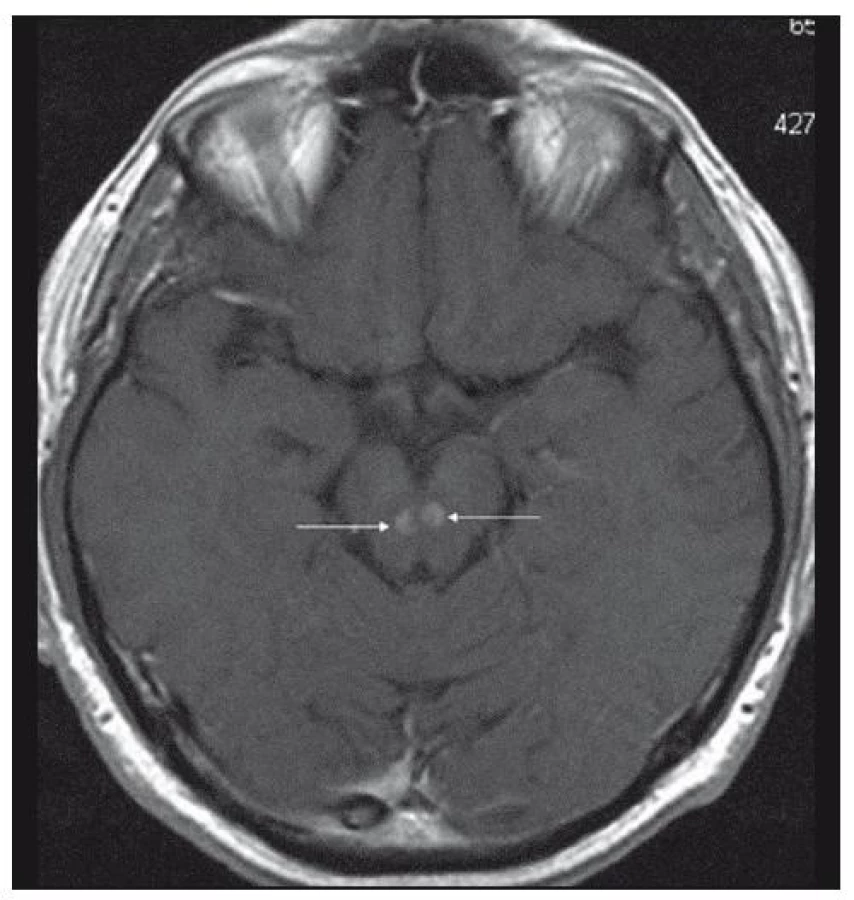
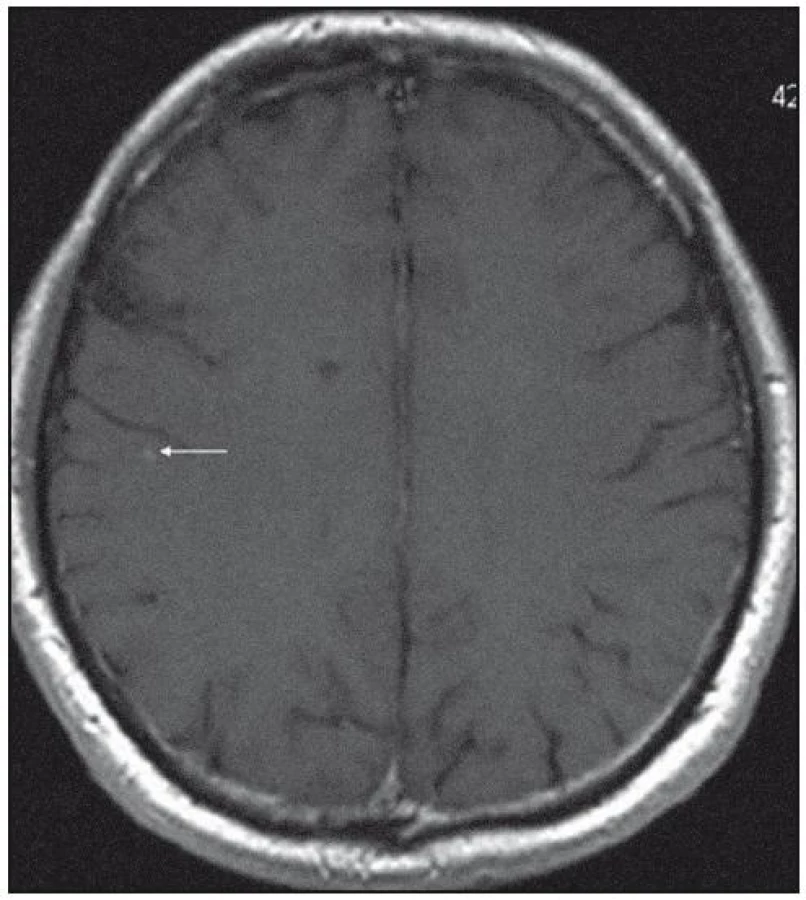
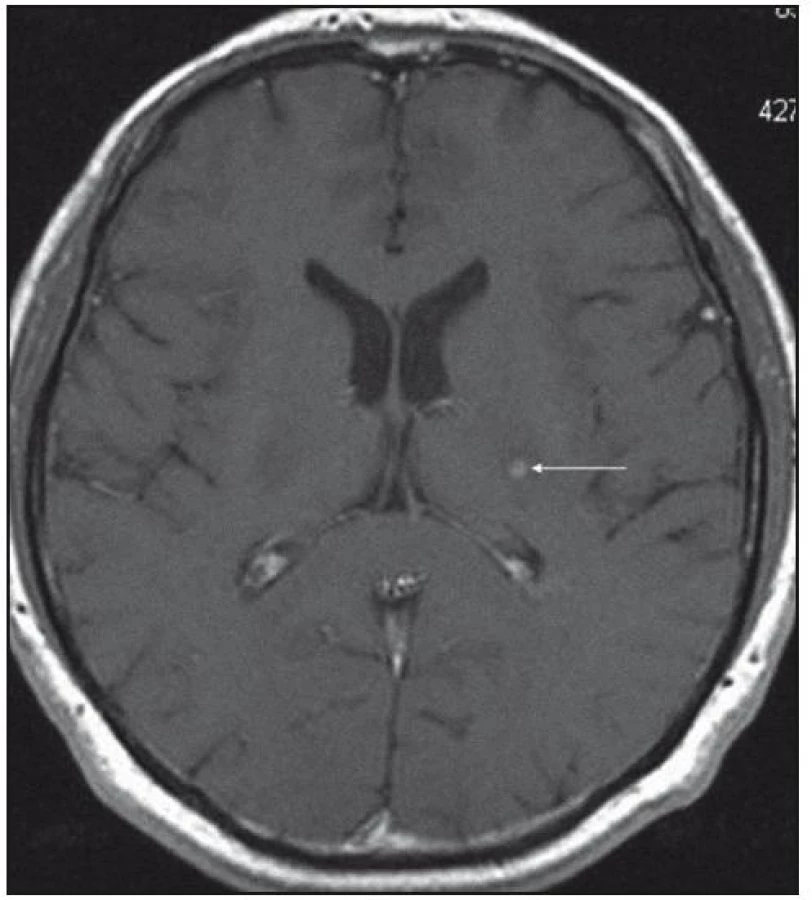
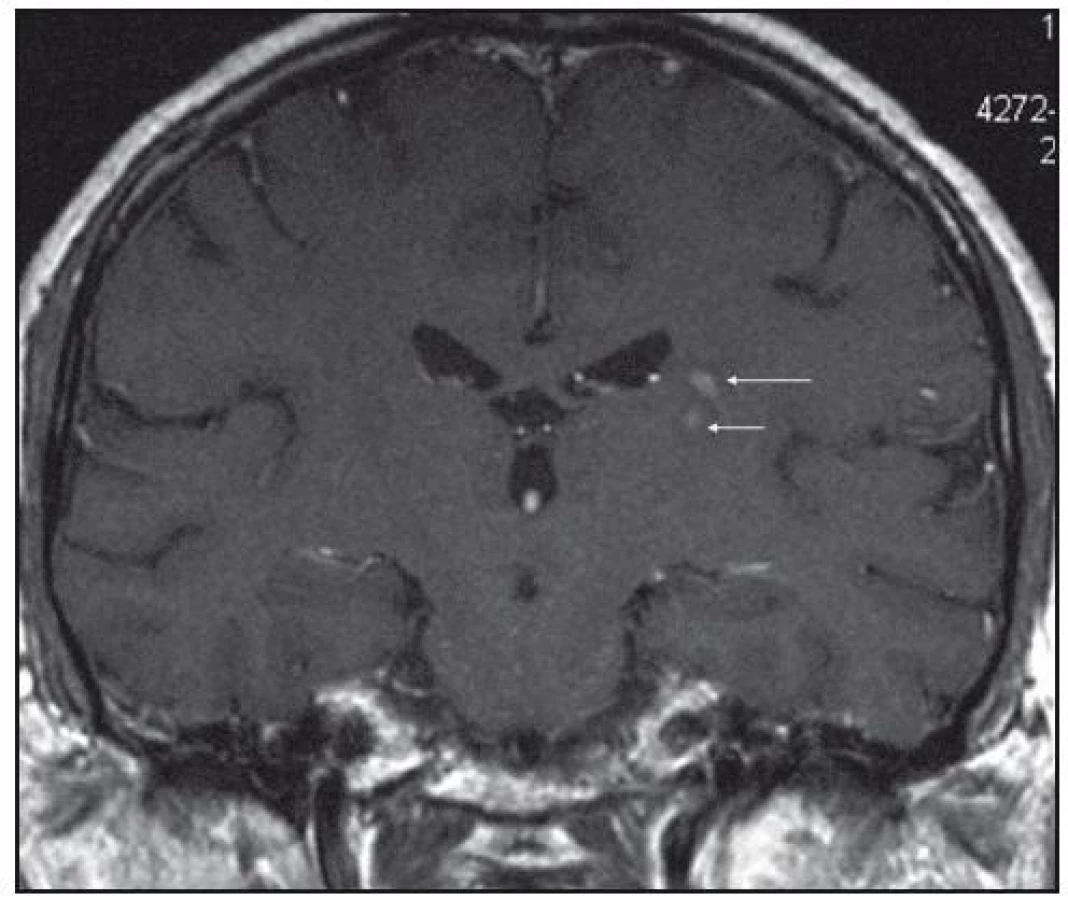
Dle hodnocení MR specialistů byla cévní etiologie těchto změn vyloučena, metastatické postižení a postižení lymfomem bylo velmi málo pravděpodobné. Proto tyto změny byly v té době neurology hodnoceny jako zánětlivá ložiska, stejného charakteru a vzhledu jako původní ložisko ve stopce hypofýzy, které bylo bioptováno a histologicky vyšetřeno.
Dysartrie a porucha hybnosti pravostranných končetin – další příznaky nemoci
Od ledna roku 2008 se u pacienta začala objevovat porucha řeči charakteru dysartrie, zprvu velmi mírná, a proto pacientem přecházená, postupně však porucha získávala na závažnosti. Pacient se tedy znovu dostavil v červenci roku 2008 k neurologickému vyšetření na Neurologickou kliniku FN u sv. Anny v Brně k vyloučení demyelinizačního onemocnění. V objektivním neurologickém nálezu byla mimo dysartrie středního stupně dále reflexologická převaha na pravé dolní končetině a porucha alternujících pohybů levé ruky. Končetiny byly bez parézy. Subjektivně pacient popisoval ještě parestezie pravostranných končetin a zvýšenou únavnost.
Na neurologické klinice byla povedena diagnostika demyelinizačního onemocnění – lumbální punkce, která neprokázala oligoklonální pásy (průkaz intrathekální syntézy Ig) a byla provedena kontrolní MR mozku s kontrastní látkou – výsledek rovněž nebyl pro demyelinizaci typický – chyběl hypointenzní korelát sledovaných ložisek v T1 vážených obrazech a navíc pro toto onemocnění nesvědčil současný enhancement všech ložisek.
Na konci srpna roku 2008 byl pacient přeléčen pulzem metylprednisolonu v dávce 5g beze změny klinického obrazu a beze změny MR nálezu, včetně postkonstrastního enhancementu, proto bylo rozhodnuto o doplnění dalšího pulzu metylprednisolonu v dávce 3g, který opět nepřinesl zlepšení klinického stavu. Z endokrinologického hlediska pacient pokračuje ve výše zmíněné substituční terapii, klinicky i laboratorně celkem uspokojivě kompenzován.
Výsledky dalších zobrazovacích a dalších vyšetření
Celotělové FDG PET zobrazení a scintigrafie skeletu
V rámci hledání příčiny těchto výše popsaných mozkových změn bylo mimo mnoha dalších, zde neuváděných vyšetření provedeno celotělové FDG PET, kde byla difuzně (nespecificky) vyšší aktivita v kostní dřeni femurů.
V listopadu roku 2008 byla provedena celotělová scintigrafie skeletu. Byla patrná patologická kumulace v dlouhých kostech. Zřetelně zvýšená kumulace techneciumpyrofosfátu byla v obou klavikulách, obou humerech, v kostech předloktí (radiu a ulně), v obou femurech (maximálně v distálním části), v obou tibiích (maximálně v proximální části), v pánvi, v os ischiadicum vlevo, os ilium a os pubis, u pravého kyčelního kloubu, v oblasti sakroiliakálního skloubení a v přilehlé části lopaty. Dále bylo zřetelné ložisko v pravé lopatce mediálně, v maxille vlevo. V kalvě byla zřetelná difuzně nehomogenně vysoká aktivita s ložisky vpravo parietálně a frontálně bilaterálně (obr. 7–9).
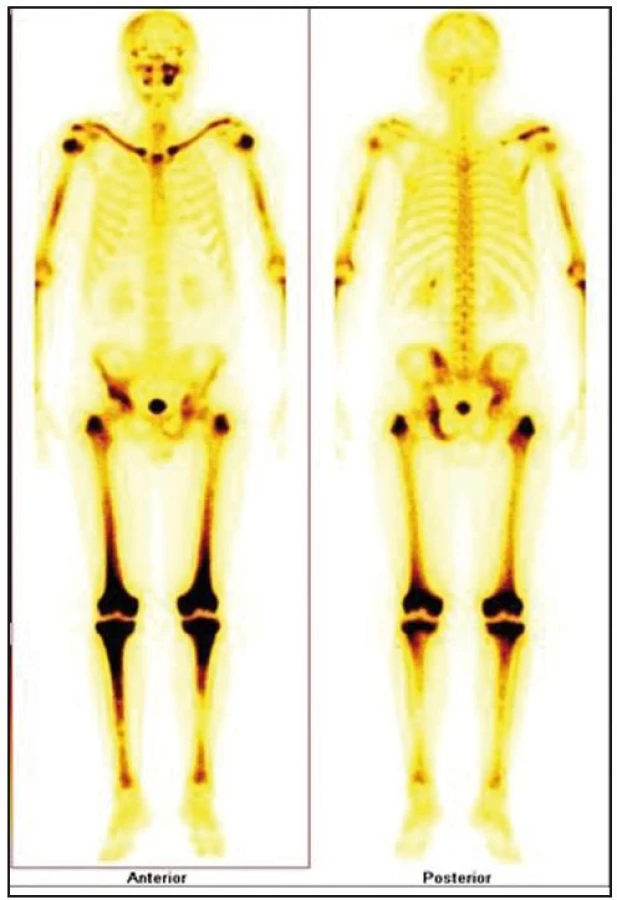
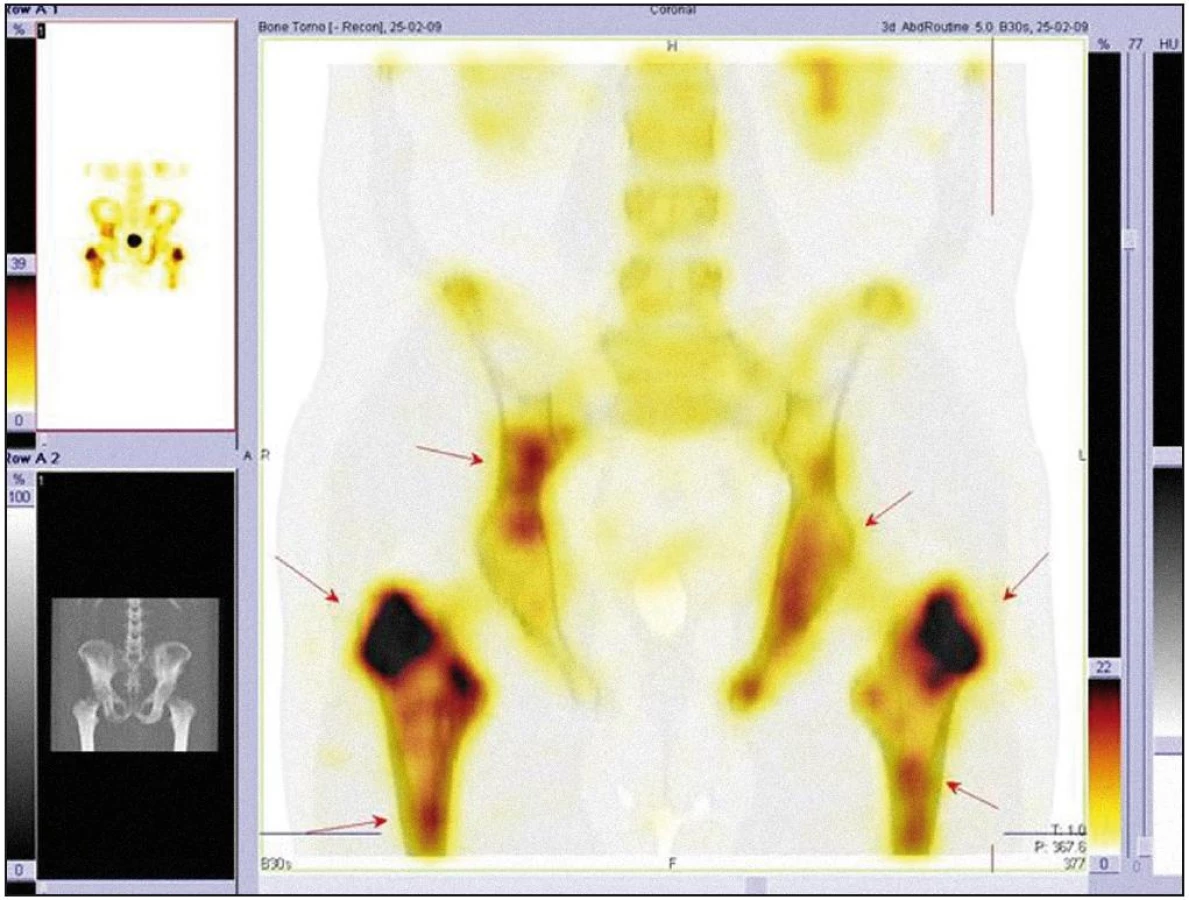
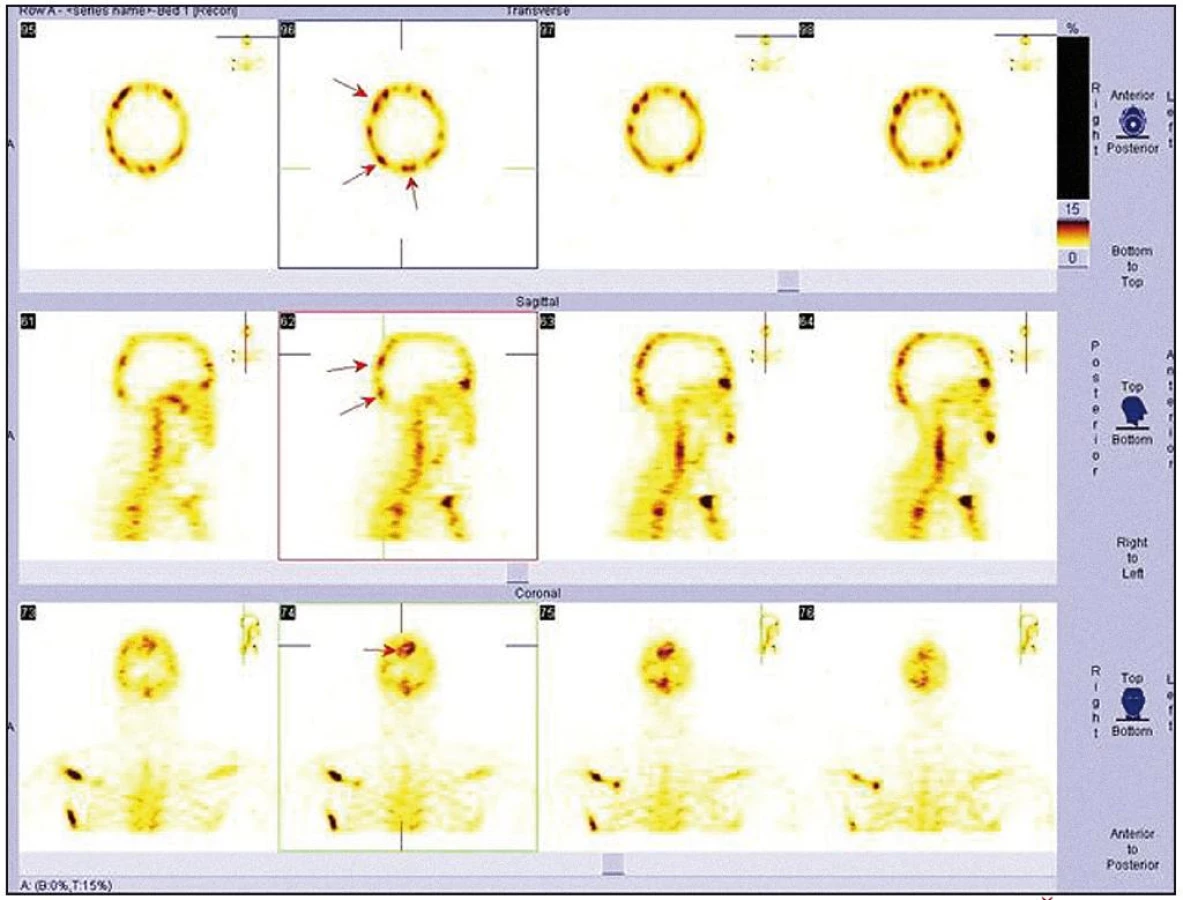
Koncem listopadu roku 2008 vysloveno podezření na hematologické onemocnění a pacient byl odeslán na Interní hematoonkologickou kliniku FN Brno Bohunice. Při prvním vyšetření na tomto pracovišti si muž stěžoval na zvýšenou únavu a slabost pravostranných končetin až typu pravostranné hemiparézy. Na cílenou otázku, zda pociťoval bolest kostí, odpověděl, že ne. Neurolog konstatoval přetrvávající pravostrannou hemiparézu lehkého stupně.
Vzhledem k tomu, že scintigrafieskeletu byla typická pro Erdheimovu -Chesterovu chorobu – difuzně zvýšená aktivita v oblasti tibií a femorů, byla provedena trepanobiopsie lopaty kosti kyčelní.
Průkaz pěnitých histiocytů typickéhoimunofenotypu (CD68+, CD1a–, S100–) v kostní dřeni
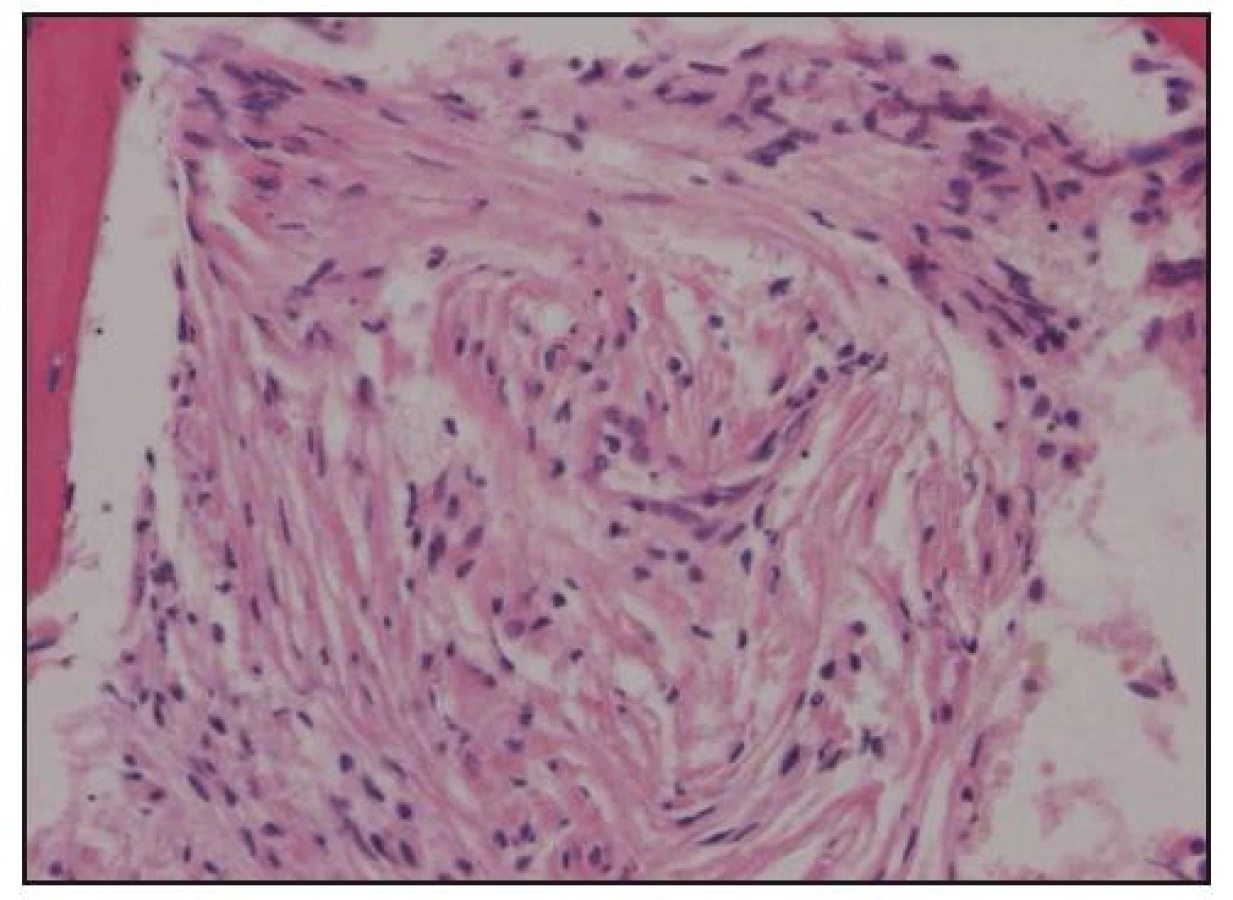
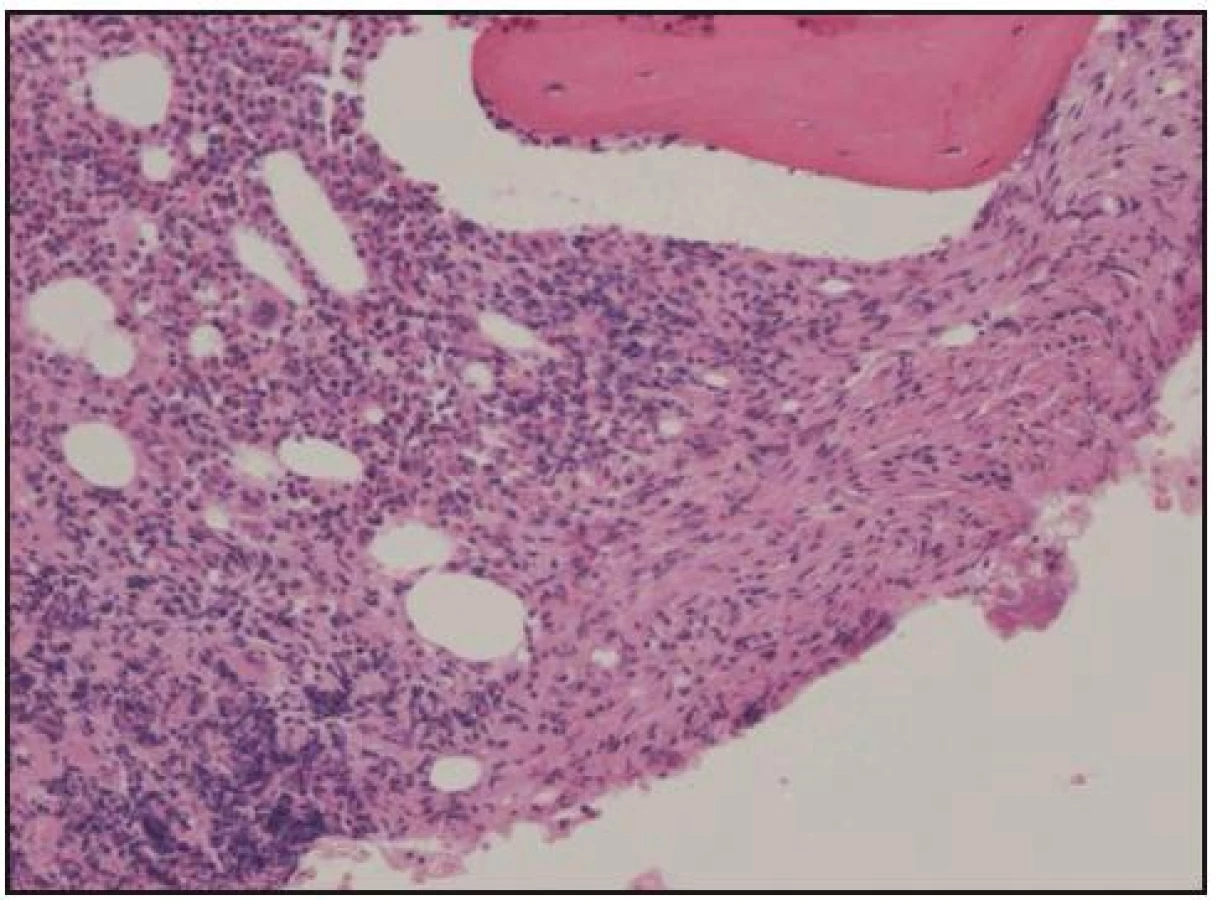
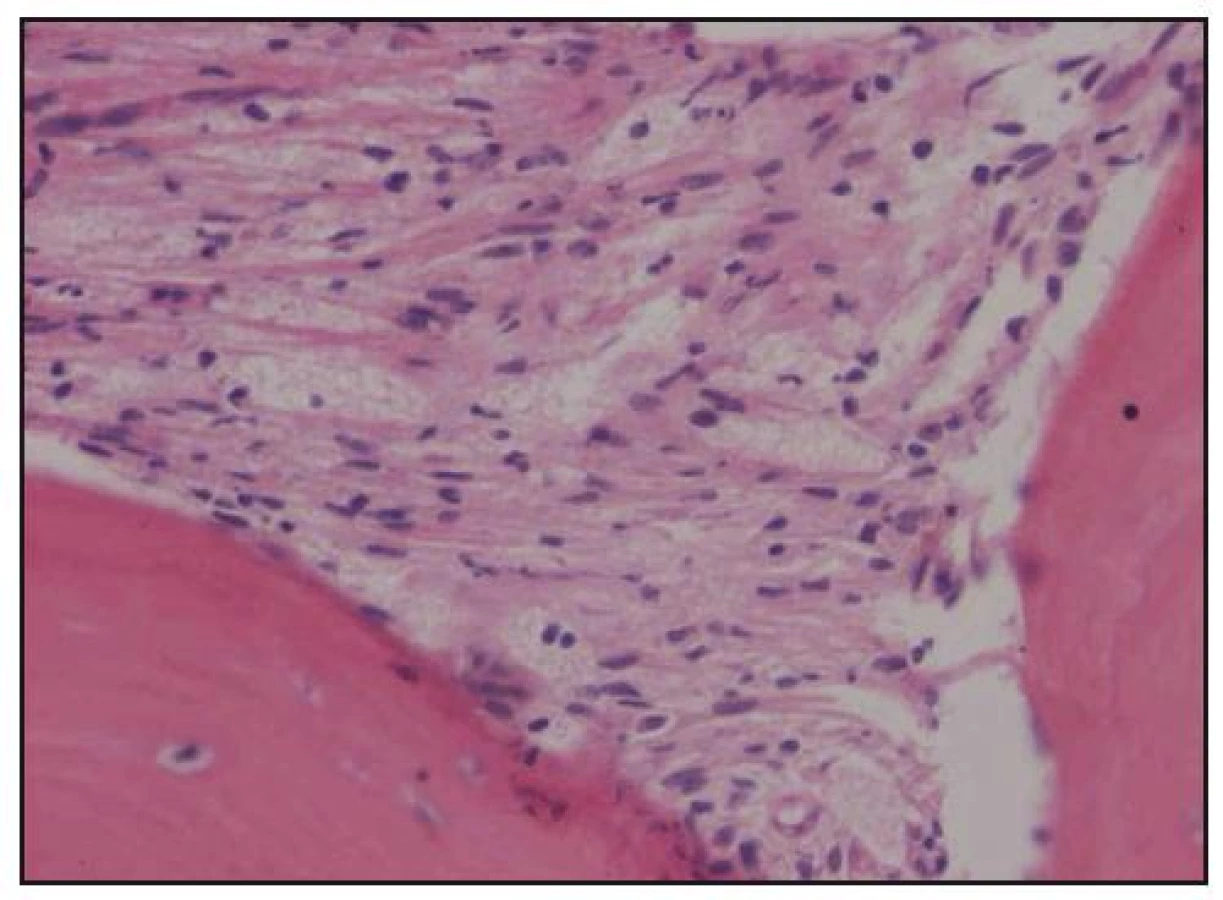
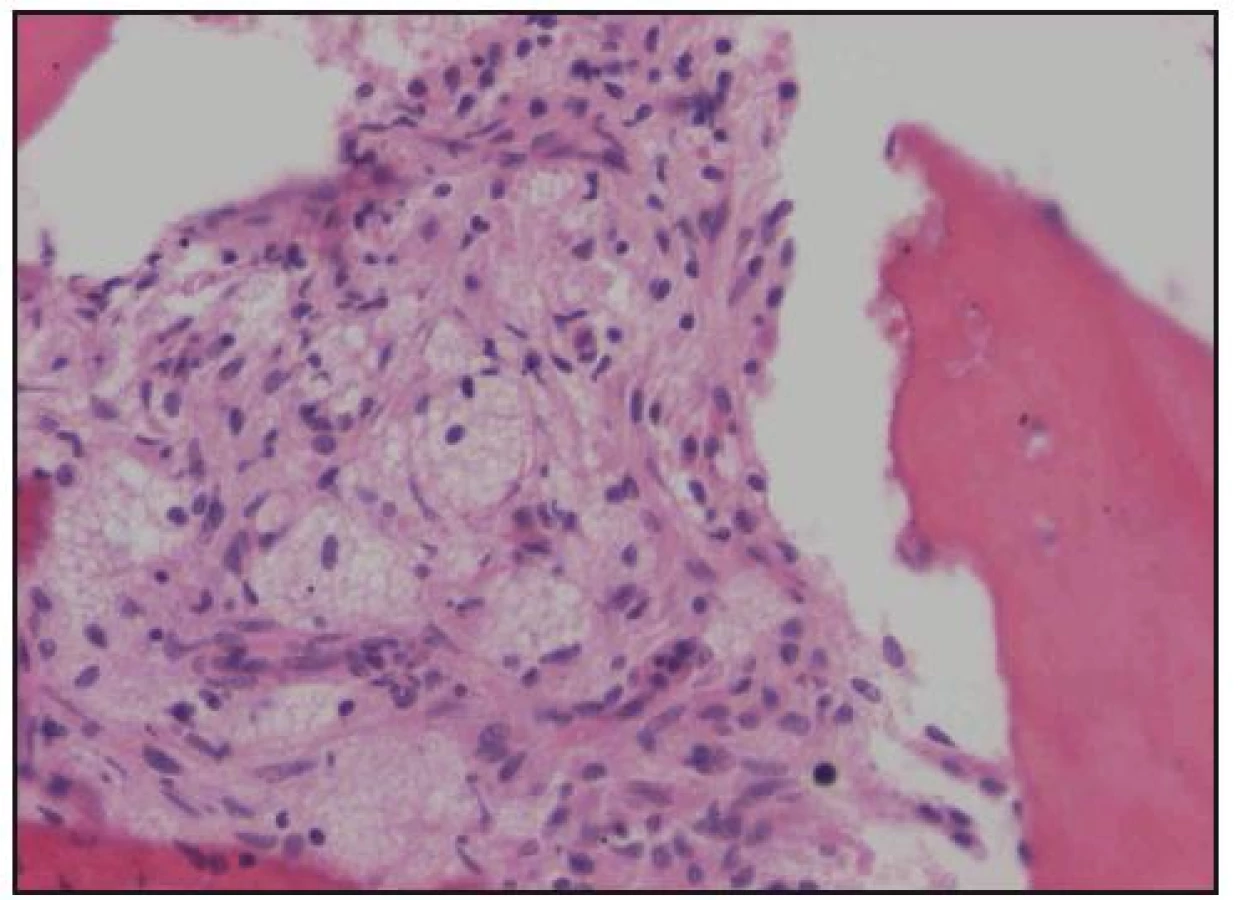
Při histologickém hodnocení válečku kostní dřeně, získaného trepanobiopsií, byla zjištěna ložiskovitá infiltrace kostní dřeně shluky histiocytů s abundantní pěnitou cytoplazmou na pozadí osteosklerózy a sekundární myelofibrózy. Ložiska zaujímala 40–60% délky trepanobioptického válečku. Imunohistochemicky byly pěnité histiocyty CD68+, CD1a–, vyšetření proteinu S 100 nebylo hodnotitelé (obr. 10–13). Při cytologickém hodnocení roztěru na sklíčko nebyly tyto patologické buňky zachyceny.
Změny typické pro tuto nemoc na RTG snímcích a sonografickém vyšetření
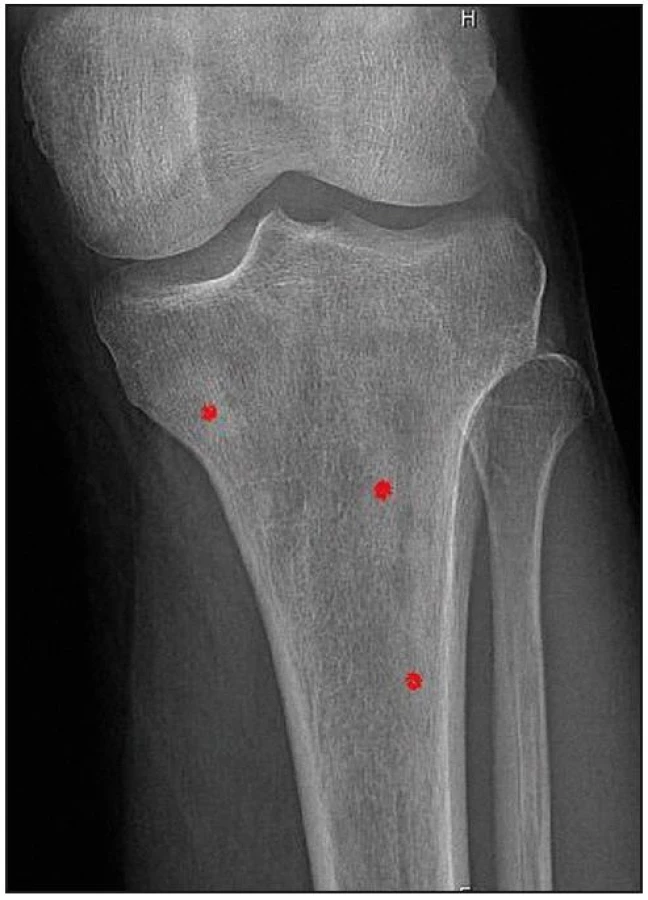
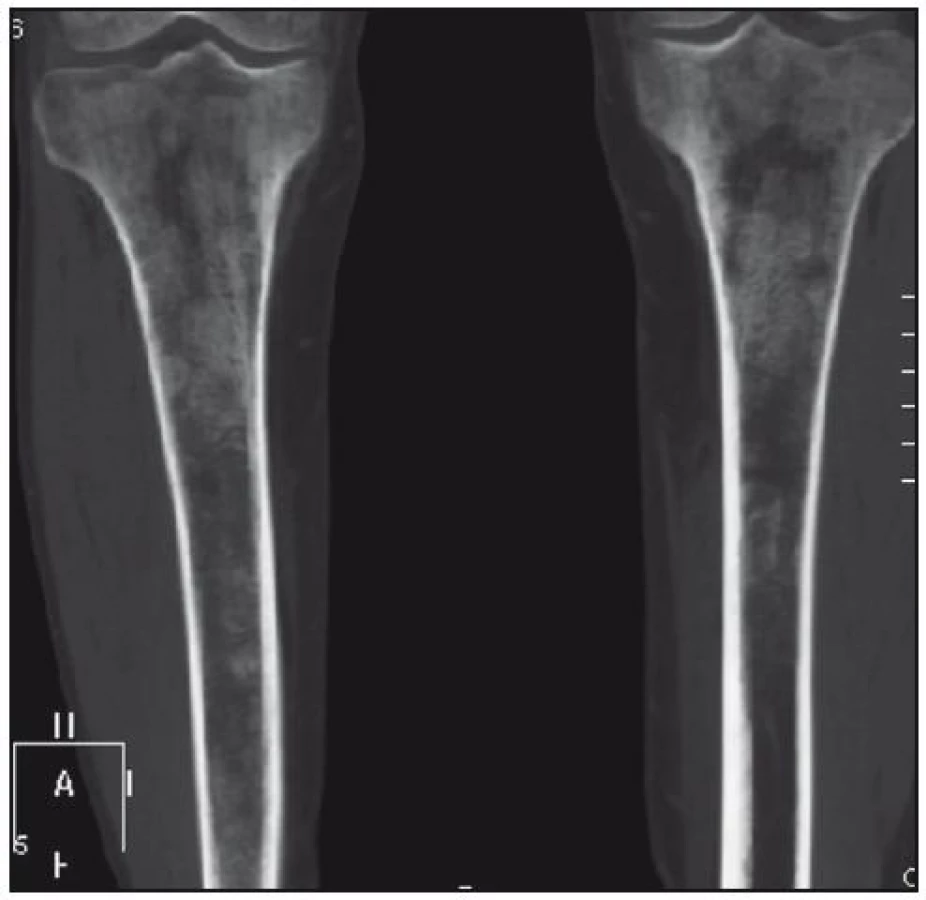
U pacienta jsme provedli RTG vyšetření celého skeletu podobně, jak se provádí u pacientů s mnohočetným myelomem. Patologické odchylky byly zřetelné pouze v oblasti pánve, kde bylo zřetelné zahuštění spongiózy. Nejvíce patologických změn bylo patrných na kostech stehenních, ale i v tibiích – oboustranně zvýšení opacity stehenních kostí, zesílení kompakty, která zužuje projasnění nitrodřenové dutiny, jak je zřetelné z klasického RTG snímku (obr. 14) a dále z CT zobrazení (obr. 15 a 16).
RTG snímek plic a sonografické vyšetření dutiny břicho bylo bez patologického nálezu.
Vyšetření kostní hustoty metodou DEXA
Současně provedené měření kostní hustoty metodou DEXA v oblasti 4 bederních obratlů prokázalo osteoporózu, T skóre mělo hodnotu –2,7 SD (standard deviation) a stejně tak Z skóre –2,7 SD, nejnižší kostní hustota byla naměřena v oblasti bederního obratle –3,1 SD. Toto vyšetření signalizuje skutečnost, že zatímco v dlouhých kostech dochází ke zvýšení hustoty, k projevům osteosklerózy, tak v osovém skeletu naopak dochází k úbytku denzity obratlů na hodnoty odpovídají osteoporóze.
CT vyšetření mediastina a břišní dutiny
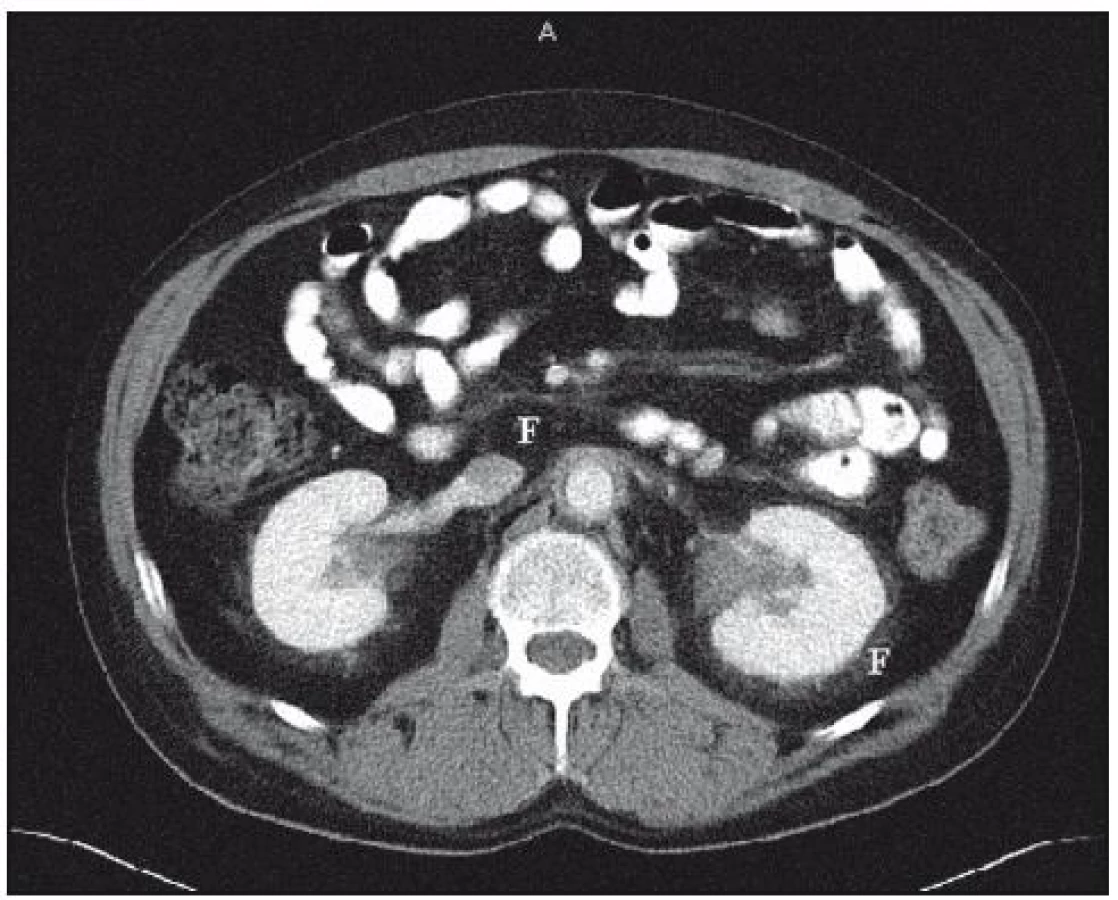
V rámci základního vyšetření bylo provedeno CT plic s aplikací kontrastní látky. V plicním parenchymu nebylo popsáno nic patologického, v mediastinu nebyly zřetelné zvětšené uzliny. Bylo ale popsáno zesílení dorzální stěny sestupné aorty až na 6mm. Nález na abdominálních orgánech, vyjma difuzního zesílení stěny sestupné aorty, byl normální. V oblasti retroperitonea však byla popsána zvýšená denzita tuku a fibrózní změny perirenálně oboustranně, čili obraz retroperitoneální fibrózy (obr. 17). V zachycených kostech bylo popsáno sklerotické ložisko v obratli Th4.
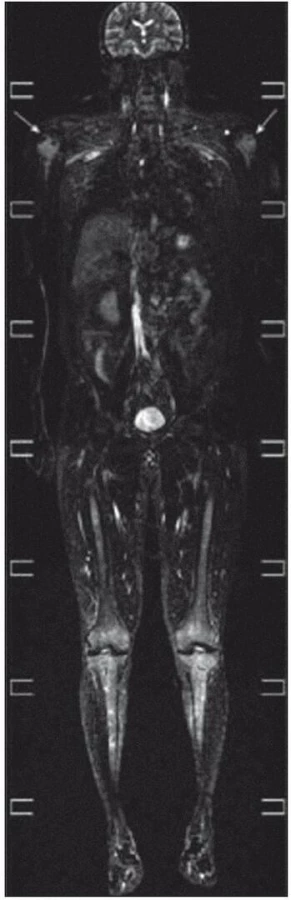
Celotělové MR zobrazení skeletu
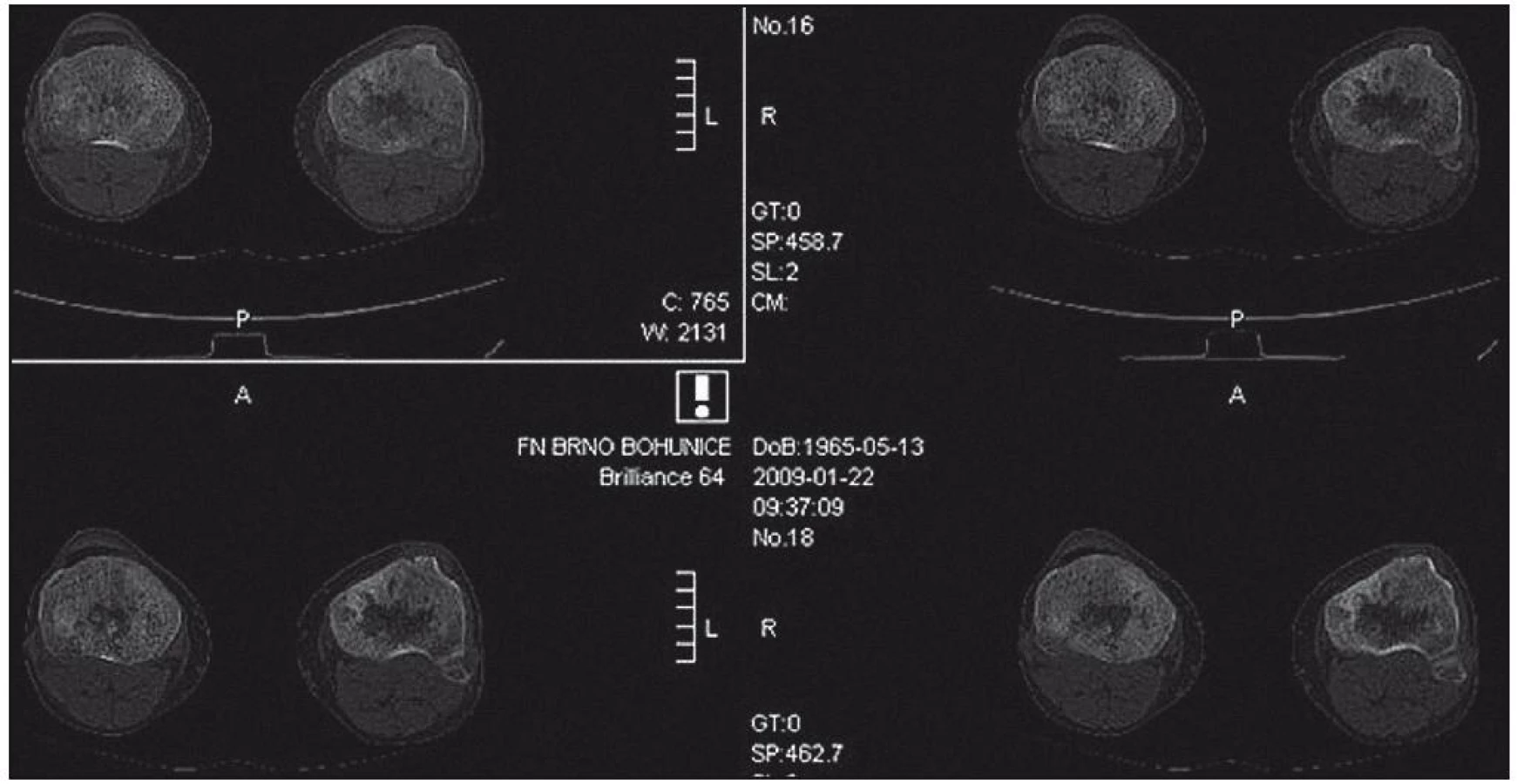
Zhotoveny koronární T1 STIR, DWIBS obrazy a sagitální T1 a STIR na oblast páteře. V kostní dřeni dlouhých kostí je výrazně abnormní nález, splývavá ložiska smíšené intenzity v T1 v obou tibiích, fibulách, femurech, humerech, ulnách i radiích. Dolní končetiny jsou postiženy více než horní, ložiska částečně šetří epifýzy rukou. Skelet pánve je postižen jen částečně, patrné jsou nehomogenity oblastí ilických kostí s převahou vlevo. Na difuzních vážených obrazech je jednoznačná restrikce difuze ve všech popsaných oblastech. V oblasti páteře je abnormní signál pouze v oblasti těla Th4 stejně jako v periferii. V těle Th11 je 20mm hemagniom. Páteřní kanál je volný. Obraz odpovídá rozsáhlému postižení kostní dřeně periferního skeletu, zatímco v páteři je patrné pouze jedno ložisko (obr. 18–20).
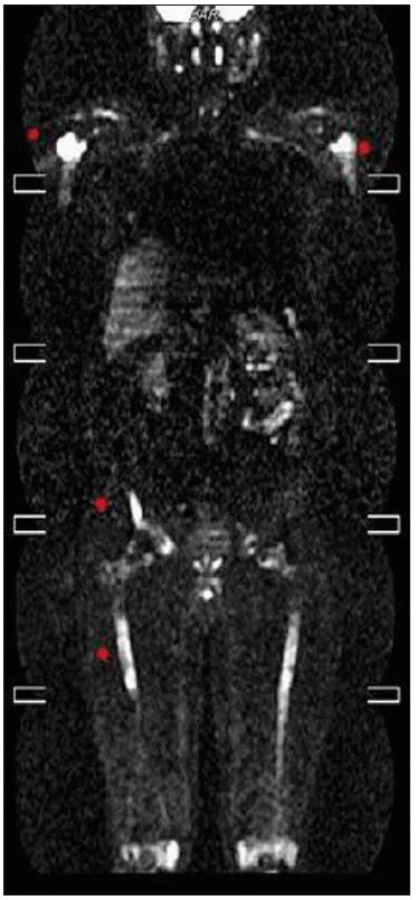
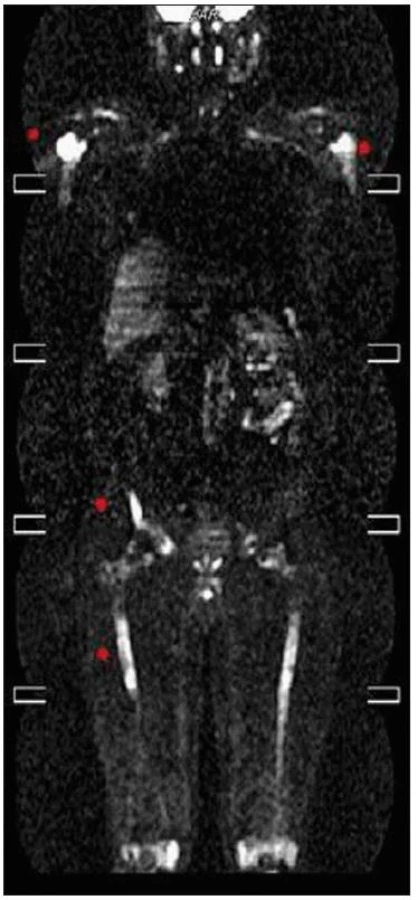
Celotělové PET CT
Několik měsíců před stanovením diagnózy bylo provedeno PET vyšetření, kombinované PET CT vyšetření bylo provedené až v průběhu léčby a prokázalo postižení celého těla.
CT zobrazení: zesílené měkkotkáňové struktury v levém maxilárním sinu, vzhledem ke sklerotickým změnám okolního skeletu suspektní infiltrace. Zřetelná sklerotizace mediální části levé jařmové kosti a suspektní přesah patologických změn na baze orbit. Plicní parenchym a mediastinum v normě, místy pleurální změny. V břiše zvýšení denzity perirenálního tuku, zneostření kontur ledvin, bez lymfadenopatie. Cirkulární zesílení stěny břišní aorty místy až na 8mm, méně výrazný je nález na hrudní aortě a na společných ilických tepnách.
Struktura skeletu je zřetelně nepravidelná, s nápadnými sklerotickými ložisky, místy v terénu prořídlé struktury (zřetelné zesílené skeletu levé maxilární dutiny a jařmové kosti vlevo, hlavic obou humerů a těla obratle Th4, obou lopat kyčelních, vlevo v blízkosti sakroiliakálního skloubení, levé kosti stydké, levé sedací kosti). Je výrazné postižení skeletu obou dolních končetin, výrazně nepravidelná, převážně sklerotická struktura diafýz dlouhých kostí dolních končetin.
FDG-PET sken prokázal difuzně zvýšenou aktivitou fluorodeoxyglukózy (FDG) v místech CT strukturálních změn skeletu, míra kumulované aktivity do výše SUV 3,6. Zvýšená kumulace FDG také v zesílených stěnách hrudní a bederní aorty.
Laboratorní a další vyšetření
Laboratorní vyšetření před zahájením léčby: leukocyty 14,9 × 109/l, erytrocyty 4,67 × 1012/l, hemoglobin 125g/l, trombocyty 524 × 109/l, v diferenciálním krevním rozpočtu převládala neutrofilie, patologické krvinky nebyly zachyceny. Dále byla zvýšena koncentrace fibrinogenu na 5,9g/l. Základní biochemické vyšetření bylo zcela normální, pouze hodnota CRP byla zvýšena na 72 mg/l, aniž by byla přítomna infekce. Vzhledem k pacientem uváděné snížení fyzické zdatnosti jsme provedli echokardiografické vyšetření, které nezachytilo nic patologického, ejekční frakce byla 65%.
Léčba
Léčba byla zahájena stimulačním cytostatickým režimem (cyklofosfamid 2g/m2 den 1 a etoposid 200mg/m2 i.v. infuze den 1–3) se sběrem kmenových hemopoetických buněk (PBSC). Stimulační režim se sběrem kmenových krvetvorných buněk jsme použili v první linii léčby proto, že po léčbě 2 chlordeoxyadenosinem (cladribinem) se často sběr PBSC nezdaří.
Měsíc od ukončení sběru PBSC byla zahájena léčba 2-chlordeoxyadenosinem (Litac) v dávce 5mg/m2 s.c. + cyklofosfamid 150mg/m2 + dexametazon 24mg i.v., vše 1.–5. den. Tyto cykly se opakovaly ve 28denních intervalech. Pro osteoporózu v oblasti páteře jsme zahájili pravidelnou aplikaci zoledronátu ve 28denních intervalech. Šestý, poslední cyklus kladribinového režimu byl aplikován koncem července roku 2009.
Přínos zvolené léčby první linie
Za tři měsíce od ukončení léčby byla provedena kontrolní zobrazovací vyšetření. Dle celotělového PET CT nedošlo k očekávané léčebné odpovědi, kumulace v dolních končetinách se v průběhu léčby zvýšila, hodnoty uvádíme v tab. 1. V oblasti retroperitonea a ledvin jsou stále fibrotické změny zřetelné při CT zobrazení.

Na klasické scintigrafii skeletu, provedené po ukončení léčby, bylo zřetelné nové ložisko zvýšené aktivity ve 3. žebru vlevo, jinak byl nález co do rozsahu a intenzity kumulace radiofarmaka shodný s nálezem před léčbou.
Změny ve skeletu byly také hodnoceny metodou celotělové MR s difuzí. Kontrolní vyšetření po ukončení léčby neprokázalo žádné zásadní změny proti stavu před zahájením léčby.
Jedině na MR mozku bylo zřetelné částečné zmenšení infiltrátů roztroušených bílé hmotě mozkové a parciální ústup patologického sycení.
Pacient hodnotí svůj stav jako mírné zlepšení – dominantně zlepšení hybnosti pravostranných končetin, toto zlepšení by mohlo korespondovat s drobným zmenšením infiltrátů dle MR mozku.
Celkově však výsledek léčby hodnotíme jako bez léčebné odpovědi.
Diskuze a přehled publikovanýchinformací o Erdheimově - Chesterově nemoci
Úvod
Z maligních histiocytárních chorob se vyskytuje nejčastěji histiocytóza z Langerhansových buněk, na našem pracovišti jsme se v průběhu 19 let registrovali 20 pacientů [5]. S Erdheimovou Chesterovou nemocí jsme se v průběhu 19 let existence našeho pracoviště setkali poprvé. Jde o velmi vzácnou nemoc, v roce 2004 bylo evidováno méně než 80 popisů případů [7], v roce 2006 se počet popsaných případů zvýšil na více než 200 [8]. V databázi české a slovenské literatury jsme našli pouze několik sdělení zabývajících se touto nemocí [9–14,17].
První popis této nemoci je z roku 1930, kdy byl zveřejněn popis dvou nemocných s „lipoidgranulomatosis“, kterážto nemoc se odlišovala od Handovy Schillerovy Christianovy nemoci. Oba popsaní pacienti měli symetrickou sklerózu diafýzy a metafýzy dolních končetin a mimokostní postižení [18]. O 42 roků později popsal Jaffe podobný případ a vytvořil jméno Erdheimova Chesterova nemoc tím, že k prvnímu autorovi připojil jméno jeho vídeňského kolegy, Jakoba Erdheima, který se na prvním popisu podílel [19].
Původně byla tato nemoc považována za jednu z forem histiocytóz z Langerhansových buněk, ale tento pohled byl postupně pozměněn a Erdheimova Chesterova nemoc byla přirazena do skupiny diseminovaného juvenilního xantogranulomu [20,21].
Definice
Erdheimova Chesterova nemoc je řazena do kategorie „non Langerhans cell histiocytosis“, představuje systémovou formu diseminovaného juvenilního xantogranulomatózního onemocnění dle WHO klasifikace (tab. 2). Nemoc způsobují proliferující pěnité histiocyty, které obsahují hojně lipidů. Klinické příznaky jsou způsobeny osteosklerózou dolních končetin a dále histiocytární infiltrací oblasti retroperitonea, mediastina, plic, srdce, sleziny a jater kůže či orbity a CNS se vznikem zánětlivých a fibrotických změn. Postižení CNS má obvykle za následek neurologické příznaky a panhypopituarizmus. Někteří nemocní mají i kožní projevy [22,138].
![Klasifikace histiocytárních chorob dle WHO klasifi kace maligních krevních chorob [27] a dále dle International Histiocyte Society [28].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/8e257825922300e317a028e96fdf9f46.jpg)
Erdheimova Chesterova nemoc má velmi variabilními příznaky, a proto je obtížně rozpoznatelná. Systémová forma juvenilního xantogranulomu s postižením viscerálních orgánů se popisuje ve věku do 10 let, v polovině případů do jednoho roku. Medián výskytu juvenilního xantogranulomu je jeden rok, průměrný věk výskytu 3,3 roky [23]. Pouze Erdhaimova Chesterova nemoc se objevuje až v dospělém věku. Kenn uvádí věkové rozpětí nemocných s Erdheimovou Chesterovou chorobou 21–77 let s věkovým průměrem 54 let [24–26].
Klinické příznaky
Klasickým a téměř vždy přítomným projevem je osteoskleróza tibií a femorů, která může působit bolesti. Mimokostní postižení je u této nemoci popisováno v 50% případů.
Více než 1/2 nemocných má další příznaky nemoci: horečku nejasného původu, úbytek hmotnosti, slabost, exoftalmus, diabetes insipidus, dysurii, bolesti břicha způsobené obstrukční nefropatií. Mozek je postižen u jedné třetiny nemocných (u 60 z 203 popsaných případů) [8]. Postižení CNS způsobuje ataxii, parézy a diabetes insipidus. Pokud je postižen mozek, tak cerebellární (41%) příznaky (ataxie) a pyramidální příznaky (45%) (parézy) jsou nejčastějšími problémy, méně často jsou přítomny bolesti hlavy, neuropsychiatrické či kognitivní poruchy, senzorické poruchy či parézy hlavových nervů [29]. Diabetes insipidus bývá přítomen u 47% nemocných, kteří mají postižení CNS [8,30 32]. V některých případech byly prvními příznaky bolesti kostí [33] nebo kožní příznaky [34].
U našeho pacienta byl prvním příznakem diabetes insipidus, který byl od roku 2004 po celé další 4 roky jediným příznakem této nemoci, teprve později se přidaly další neurologické příznaky, poruchy řeči a lehká pravostranná hemiparéza.
Projevy nemoci v jednotlivých lokalizacích
Hlava
V oblasti hlavy je nejčastěji popisována infiltrace orbity vedoucí k exoftalmu a dále xantogranulomatózní kožní projevy kolem očnice [35,36]. Byly i popsány projevy nitrolební hypertenze a přítomnost městnavé papily [37,38]. Triáda příznaků, diabetes insipidus, exoftalmus a kostní změny, připomíná Handovu Schillerovu Christianovu chorobu neboli jednu z klinických forem histiocytózy z Langerhansových buněk. U našeho pacienta však nebyla přítomna ani městnavá papila, ani jakékoliv projevy v obličejové části hlavy.
Centrální nervový systém (CNS)
Mimokostní postižení se uvádí v 50% případů, neurologické příznaky byly popsány u 60 u 203 zveřejněných případů [8]. Postižení CNS je tvořeno ložisky, které vychytávají kontrastní látky, a ty v nich setrvávají až po dobu dvou týdnů [39–42]. MR zobrazení může pomoci v diferenciální diagnostice cerebrálního postižení [43].
Neurologické postižení je možno rozdělit do několika typů. První typ je charakteristický infiltráty v oblasti kmene, v cerebellu a v mozku. Vyskytuje se u 44% pacientů. Druhým typem (37%) je meningeální postižení s tumory, které odpovídají tvarem meningeomům a s nodulárním zduřením dura mater. Třetí typ (19%) je spojením předchozích, infiltrativního postižení CNS a meningeálního postižení. Optimálním způsobem jejich znázornění je MR, ložiska dávají zvýšený signál v T2 váženém obraze a sytí se po aplikaci Gadolinia v T1 váženém obraze. Byla popsána prolongovaná retence kontrastní látky [8,44–47].
Hypofýza a hypotalamus
Diabetes insipidus bývá přítomen asi u 25% případů. Tito nemocní mívají na MR vyšetření ztrátu hyperintenzního signálu z posteriorního laloku hypofýzy a zesílení infundibula, případně infiltraci stopky hypofýzy. Tyto změny vedou k atrofii zadního laloku hypofýzy [41,48].
Diabetes insipidus bývá v některých případech prvním projevem nemoci, pak následují MR vyšetření prokazující infiltraci v oblasti stopky hypofýzy. Někdy bývá v době stavení diagnózy zjištěna i snížená hladina gonadotropinů a dalších hormonů [49–53]. Nález infiltrace v oblasti stopky hypofýzy je velký diferenciální diagnostický problém. Je sice možné provedení biopsie tohoto infiltrátu, ale ne vždy biopsie infiltrátu vede k diagnóze. Sheu popisuje punkci infiltrátů s nálezem pseudoinflamatorní infiltrace a teprve až sekčně se potvrdila souvislost s Erdheimovou Chesterovou nemocí [54]. V diferenciální diagnostice infiltrátů v oblasti infundibula a stopky hypofýzy připadá v úvahu neurosarkoidóza, Erdheimova Chesterova nemoc, histiocytóza z Langerhansových buněk a lymfocytární hypofyzitida. Sheen doporučuje vždy při diferenciální diagnostice infiltrace stopky hypofýzy provedení vyšetření dolních končetin, zda nejsou známky Erdheimovy Chesterovy nemoci a dále vyšetření lokalizací typických pro systémové projevy histiocytózy z Langerhansových buněk a vyšetření plicních hilů, zda nejsou zvětšené hilové uzliny, což je zase typické pro sarkoidózu [55–65].
Také u tohoto pacienta budila izolovaná infiltrace hypofýzy značné diagnostické rozpaky. Provedená stereotakticky navigovaná biopsie s odběrem materiálu k histologickému vyšetření přinesla pouze morfologický obraz zánětlivé infiltrace, podobně jak popsali i výše citovaní autoři. Proč cílená biopsie nepřinesla v našem případě ani v jiných, v literatuře popsaných případech histologickou diagnózu, můžeme pouze spekulovat a uvést analogii s neurodegenerativními projevy histiocytózy z Langerhansových buněk (LCH), kde byly i sekčně nalezeny pouze lymfocytární infiltráty, a bylo spekulováno, že celé neurodegenerativní poškození může mít podklad maligní chorobou indukované degenerativní změny. Je možné, že infiltrace CNS pěnitými histiocyty provází intenzivní zánětlivá reakce, a proto cílené biopsie tuto diagnózu nemusí odhalit.
Hrudník
V oblasti hrudníku může nemoc infiltrovat plíce a vést k plicní fibróze a může způsobit respirační insuficienci. Při zobrazovacích vyšetřeních jsou popisovány intersticiální infiltráty [66–71].
Infiltrace perikardu způsobuje perikardiální výpotek, který je nejčastějším kardiálním projevem této nemoci [72,73]. Dále byla popsána infiltrace myokardu, chlopní, koronárních arterií. Specifickým projevem je periaortální fibróza, tzv. coated aorta [6,74–79]. K monitorování postižení srdce byla použita scintigrafie pomocí 67Ga [80].
Selhání plic či kardiální slabost jsou nejčastějšími příčinami úmrtí těchto osob [75].
Při vyšetření hrudníku u našeho pacienta metodou CT s aplikací kontrastu, stejně jako při vyšetření břišní dutiny bylo popsáno zesílení stěny aorty, což odpovídá literárnímu popisu coated aorta.
Břicho
V oblasti břicha se nemoc nejčastěji projevuje retroperitoneálními infiltráty a fibrotizací. Tyto projevy vedou k obstrukční nefropatii. Zobrazovacími vyšetřeními je prokazatelná retroperitoneální a perirenální infiltrace a fibrotické změny. Pokud dominuje aktivita nemoci v oblasti retroperitonea, je možná záměna s Ormondovou chorobou (idiopatická retroperitoneální fibróza) [81,82]. Literatura popisuje případy, kdy byly provedeny biopsie této zdánlivě maligní infiltrace retroperitonea a histologické hodnocení odebraného materiálu vyznělo nespecificky: „pojivová fibrózní tkáň s fokálními známkami zánětu“ [83,84]. Diferenciálně diagnostické postupy při nálezu retroperitoneální fibrózy uvádí citované publikace [85–87]. V některých případech právě retroperineální infiltrace, fibrotizace byla první známkou této nemoci [88,89], která případně způsobila hydronefróza [90] či akutní renální selhání [91–93]. Zcela výjimečně je přítomna infiltrace jater a dalších parenchymatózních orgánů [94].
Popisovaný pacient má sice již na CT zřetelné známky retroperitoneální fibrózy, ale zatím není narušen odtok moče z ledvin. Odtok moče však budeme monitorovat radioizotopovým vyšetřením ledvin a sonograficky, abychom zavčas zjistili vznik obstrukční nefropatie.
Dolní a horní končetiny
Radiografické abnormality kostí končetin jsou typické, pravidelně se vyskytují symetricky na dolních, méně často horních končetinách. Denzita kosti je ložiskovitě či difuzně zvýšena, je patrné zesílení trabekul, skleróza a zesílení kortikalis. Proces postihuje hlavně diafýzy, metafýzy s minimálními změnami v epifýzách. Chester se domníval, že infiltrace kostní dřeně histiocyty naplněnými tukem byl primární jev a kostní reakce se sklerózou byla sekundární. To zůstalo pouze domněnkou a doposud není jasné, co spouští procesy vedoucí ke sklerotizaci kosti [95–98]. Při MR vyšetření je obraz patologického signálu z postižených kostí [99].
Odchylky metabolizmu skeletu od normálu je možné zobrazit jak pomocí scintigrafie skeletu, tak pomocí FDG PET vyšetření, scintigrafie skeletu však znázorňuje změny kostního metabolizmu intenzivněji než FDG PET zobrazení, to však může prokázat mimokostní patologie [100–107].
U našeho pacienta je již na RTG snímcích patrné zesílení kompakty stehenní kosti a dále v oblasti pánve, při CT vyšetření byly popsány i sklerotické změny v obratli, byť změny páteře nejsou pro tuto nemoc typické.
Scintigrafický obraz byl zcela jednoznačně patologický, s vysoce zvýšeným vychytáváním technecia pyrofosfátu a patologických kostních ložisek.
Zobrazení PET CT bylo provedeno až v průběhu léčby, ale zvýšená akumulace fluorodeoxyglukózy je patrná nejen v kostech se změněnou strukturou (SUV 3,6), ale i ve stěnách velkých tepen. Nález zcela koreluje s nálezem celotělového MR, jehož výsledek uvádíme na obr. 18–20.
Laboratorní známky
S průběhem nemoci souvisejí zvýšení zánětlivých parametrů (CRP), leukocytóza, trombocytóza, vysoká hodnota fibrinogenu a sedimentace erytrocytů [108,109].
Tyto parametry (CRP, fibrinogen, leukocytóza, trombocytóza) jsou zvýšené i u našeho pacienta.
Stanovení diagnózy
Morfologická diagnóza
Tato nemoc nejasného původu je histologicky charakterizována infiltrací histocyty obsahujícími tuk (pěnitými histiocyty) a dále Toutonovými obrovskými mnohojadernými buňkami. Tyto buňky tvoří xantogranulomatózní a infiltrativní ložiska. Imunohistochemické vyšetření prokazuje u těchto buněk typický imunofenotyp (CD68+, CD1a–, S100–). Dle literatury je zde kolísání v expresi, a tedy pozitivitě znaku S 100. U minoritního počtu pacientů s Erdheimovou Chesterovou nemocí byl pozitivní, zatímco u většiny byl negativní [24,26,110,111]. Naproti tomu v expresi CD1a antigenu, jehož průkaz je typický pro LCH, byla shodně popsána negativita ve všech případech Erdheimovy Chesterovy nemoci [24,112]. Zcela výjimečný je popis průkazu nekrózy kostní dřeně [113], anebo hemofagocytóza [114,115].
V námi popisovaném případě trepanobiopsie s histologickým hodnocením válečku jasně popsalo patologickou infiltraci kostí dřeně, značně masivní, dosahující 40–60% s odpovídajícími imunofenotypickými znaky.
Diferenciální diagnóza
V rámci diferenciální diagnózy je třeba odlišit Rosaiovu Dorfmanovu nemoc (sinusovou histiocytózu s masivní lymfadenopatií), která způsobuje oboustranné zvětšení krčních uzlin. Histiocyty jsou u této nemoci podobné jako při popisovaném nemoci, ale na rozdíl od ní je často přítomný kontakt histiocytů s lymfocyty (emperipolesis). Buňky sinusové histiocytózy jsou silně pozitivní pro S 100 [116–118]. Podobně může vypadat střádací nemoc způsobená polyvinylpyrolidinem [24].
Od histiocytózy z Langerhansových buněk (LCH) se Erdheimova Chesterova choroba odlišuje v několika aspektech. Jednoznačným rozdílem je symetrická distribuce sklerotických změn skeletu u Erdheimovy Chesterovy nemoci, zatímco u LCH se vyskytují nesymetrická osteolytická ložiska. LCH se vyskytuje u dětí a mladých dospělých, zatímco Erdheimova Chesterova nemoc u osob mezi 21 a 77 lety věku. Průměrný věk postižených byl 54 let [24,26].
Léčba
Optimální léčba není známá a prognóza je nejistá. Chemoterapie, radioterapie a steroidy nemají ve všech případech léčebný efekt a ke spontánní remisi na rozdíl od eozinofilního granulomu nedochází [26].
Nemoc se léčí podobně jako histiocytóza z Langerhansových buněk. Někteří autoři popisují příznivý účinek vinka alkaloidů a kortikoidů [106]. Další testovali s různými výsledky léčbu pomocí cyklofosfamidu případně metotrexátu a etoposidu [107–120]. V jedné publikaci je popsán přínos kombinace prednisonu, vinblastinu a mycofenolat mofetilu [7].
Také interferon α byl u těchto pacientů používán v dávce 3–9 milionů jednotek 3krát týdně. Dočasná částečná regrese nemoci byla popsána v nadpolovičním počtu nemocných [121,122]. V jednom případně navodil interferon α roky trvající kompletní remisi choroby [119]. Pokud se podávaly kortikoidy, byly účinné ve vysokých pulzních dávkách. Nicméně uvádí se, že léčebný efekt kortikosteroidů i chemoterapie je podstatně menší než u LCH [121].
Cladribin, synonymem 2-chlordeoxyadenosin, je s úspěchem používán u histiocytózy z Langerhansových buněk a v několika případech bylo popsáno zásadní zlepšení u nemocných s Erdheimovou Chesterovou nemocí [123,124] i u postižení CNS juvenilní xantogranulomatózou [125]. Používá se v dávce 0,14mg/kg/den pět dní po sobě. Přehled léčebných postupů uvádí Heroche v roce 2004 [75] a informace o léčbě shrnuje tab. 3.
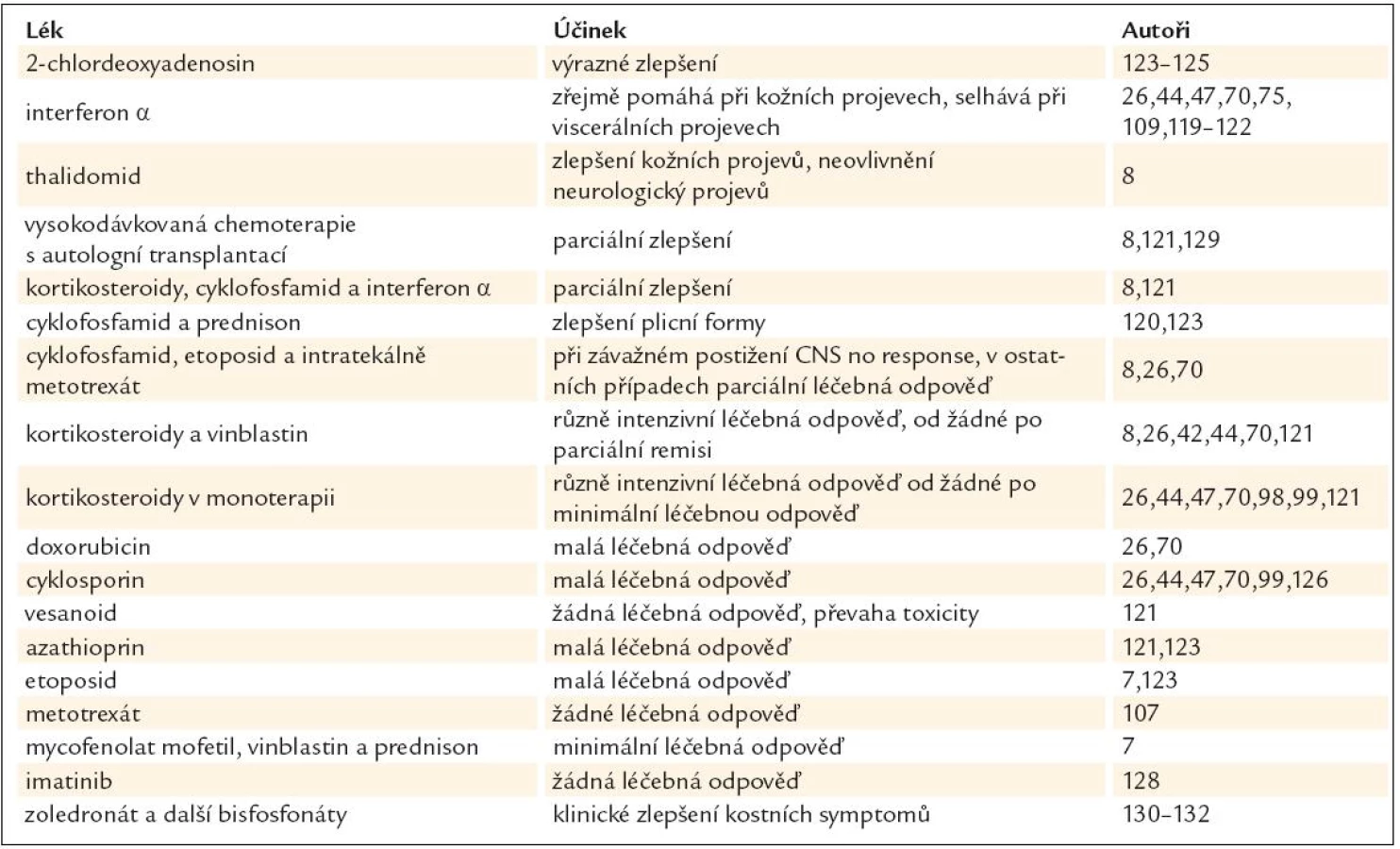
Testována byla i léčebná aplikace stroncia pro bolesti v dolních končetinách [119]. Radioterapie má prokázaný paliativní a analgetický efekt při ozáření bolestivých končetin v dávce 24 Gy, zatímco dávka 10 Gy neměla žádoucí účinek. Zkušenosti s radioterapií CNS infiltrátů jsou malé, literatura popisuje 7 pacientů léčených cílenou radioterapií na CNS infiltráty, z toho u 3 byla zaznamenána progrese a 3 měli stabilní nález po dalších 6 měsíců. Radioterapii lze využít k léčbě retrobulbárních infiltrátů, ale i tam je úspěch malý, Hoffmann uvádí, že záření v dávce 20 Gy nepřineslo efekt [98]. Z uvedené publikace vyplývá, že naděje na dosažení dlouhodobé regrese infiltrátů v CNS radioterapií není velká [126,127], lze ji použít na bolestivé změny končetin [108]. Z biologické léčby byl testován imatinib. Z 37 vyšetřených histologických preparátů byla u 32 prokázána v histiocytech přítomnost genu PDGFRβ (86,5%). Celkem bylo léčeno imatibiniben 6 pacientů s Erdheimovou Chesterovou nemocí. V průběhu sledování se ukázalo, že léčebná odpověď na imitinib závisí od lokalizace procesu. V oblasti CNS došlo u 75% léčených k progresi, zatímco v postižení kardiovaskulárního aparátu zůstalo v průběhu léčby stabilní. Autoři uzavírají, že v uvedených případech nebyl prokázán zásadní přínos imatinibu pro tyto nemocné [128]. V několika případech byla s malých efektem použita i vysokodávkovaná chemoterapie s autologní transplantací kostní dřeně [129].
Do komplexní léčby patří také bisfosfonáty, výrazné zlepšení bylo popsáno po zoledronátu zřejmě díky jeho brzdivému vlivu na produkci zánětlivých cytokinů makrofágy a jejich migraci [130–132].
Z uvedeného přehledu léčebných zkušeností vyplývá, že rozhodování o léčbě je individuální a obvykle se po zbytek života pacientů testují různé léčebné modality, většinou s malým, středním či žádným léčebným efektem. Optimální léčba není u této nemoci zatím definována.
Vzhledem k tomu, že jde o mladého pacienta, rozhodli jsme se pro léčbu 2 chlordeoxyadenosin napřed v monoterapii, při neúspěchu případně v kombinaci. A protože 2 chlordeoxyadenosin by zablokoval pozdější provedení sběru kmenových buněk krvetvorby z periferní krve, provedli jsme iniciálně sběr kmenových krvetvorných buněk po stimulačním režimu složeném z cyklofosfamidu 2g/m2 1. den a etoposid 200mg/m2 1.–3. den. Výsledek léčby budeme hodnotit pomocí zobrazovacích vyšetření a vyšetření kostní dřeně, jak popisuje literatura [133–138]. První kontrolní vyšetření jsme provedli ihned po sběru PBSC. Zvýšené vychytávání techneciapyrofosfátu v dlouhých kostech zůstala beze změny. Kontrolní PET CT vyšetření, provedené po 2. cyklu a po ukončení cladribinového režimu, prokázalo patologické vychytávání glukózy v patologických ložiscích dlouhých končetin beze změny, stejně tak se neměnil patologických signál z těchto oblastí při MR zobrazení.
Z uvedeného popisu je zřetelné, že nemoc progreduje pomalu, jinak by nebylo možné, aby diagnóza byla stanovena až po 4 letech od prvních příznaků, ale je poměrně rezistentní na léčbu.
Prognóza
Průběh nemoci je velmi individuální a odpovídá stupni poškození organizmu, nezřídka byl popisován fatální průběh. Údajů o prognóze je málo, největší soubor zveřejnil Veyssier Belot, který uvádí, že ze 37 nemocných 22 zemřelo v průběhu 2,7letého sledování [26]. Plicní fibróza s dušností a srdeční selhání jsou nejčastější příčiny úmrtí. Neurologické postižení může způsobovat ataxii či parézy, poruchu kognitivních funkcí a vést k postupnému zhoršování funkce CNS [8].
Závěr
Jak Erdheimova Chesterova nemoc, tak i LCH mají afinitu k stopce hypofýzy a hypotalamu. Při postižení těchto struktur je nutno vždy diferenciálně diagnosticky myslet na histiocytární onemocnění. A protože biopsie stopky hypofýzy či hypotalamu může způsobit trvalé poškození pacienta, je nutné vždy pátrat, zda nejsou přítomny extrakraniální příznaky některé z nemocí, které připadají v úvahu. Cílem tohoto textu bylo seznámit čtenáře s klinickými, zobrazovacími a histologickými nálezy, které jsou typické pro Erdheimovu Chesterovu nemoc. S odstupem času se k případu vrátíme a vyhodnotíme použité léčebné postupy.
Tato práce vznikla a byla podporována v rámci projektu MŠMT: LC 06027 a VZ 0021622434.
prof. MUDr. Zdeněk Adam, CSc.
www.fnbrno.cz
e mail: z.adam@fnbrno.cz
Sources
1. Mottl H, Koutecký J, Ganevová M. Strategie léčby histiocytózy z Langerhansových buněk u dětí. Čes Slov Pediat 1994; 49 : 81.
2. Mottl H, Mracek J Kabelka Z et al. Histiocytóza z Langerhansových buněk u dětí. Čs Pediat 1992; 47 : 530 – 533.
3. Mottl H, Starý J. Histiocytóza z Langerhansových buněk u dětí – klinická diagnostika a současná léčba. Čes Pediatrie 2007; 62 : 220 – 225.
4. Ščudla V, Roček V, Dušek B et al. Multifokální eozinofilní granulom v dospělosti. Vnitř Lék 1987; 33 : 1078 – 1086.
5. Adam Z, Pour L, Krejčí M et al. Histiocytóza z Langerhansových buněk u osob dospělého věku – nemoc s mnoha tvářemi. Zkušenosti jednoho pracoviště a přehled projevů nemoci. Vnitř Lék 2008; 54 : 1063 – 1081.
6. Dickson BC, Pethe V, Chung CT et al. Systemic Erdheim - Chester disease. Virchows Arch 2008; 452 : 221 – 227.
7. Jendro MC, Zeidler H, Rosenthal H et al. Improvement of Erdheim ‑ Chester disease in two patients by sequential treatment with vinblastine and mycophenolate mofetil. Clin Rheumatol 2004; 23 : 52 – 56.
8. Lachenal F, Cotton F, Desmurs - Clavel Het al. Neurological manifestations and neuroradiological presentation of Erdheim‑Chester disease: report of 6 cases and systematic review of the literature. J Neurol 2006; 253 : 1267 – 1277.
9. Kinkor Z, Koudela K, Koudela K et al. Warfarinem vyvolaná hemorhagická pseudocysta malé pánve u ženy s vrozeným genetickým defektem koagulace komplikovaná usuračním pseudoxanthomem pánevní kosti napodobující Echeimovu Chesterovu nemoc. Acta Chir Ortop Traum Čechooslov 2007; 74 : 114 – 117.
10. Kinkor Z. Severe pulmonary involvement in Erdheim‑Chester disease (case report). Cesk Patol 2001; 37 : 114 – 117.
11. Kinkror Z. Závažné plicní postižení u Erheim - Chesterovy nemoci. Čes Slov Patol 2001; 37 : 114 – 117.
12. Kolár J, Kucera V, Povýsil C et al. Erdheim‑Chester disease. Rofo 1984; 141 : 698 – 701.
13. Mergancová J, Kubes L, Elleder M. Xanthogranulomatous processes in the area of the large vessels. Cesk Patol 1986; 22 : 145 – 150.
14. Mergancová J, Kubes L Elleder M. A xantogranulomatous process encircling large blood vessels (Erheim - Chester disease?). Czech Med 1988; 11 : 57 – 64.
15. Kučera V, Čáp V, Kužel J et al. Vzácná příčina osteosklerózy: Erheimův ‑ Chesterův syndrome. ČS Radiol 1984; 38 : 393 – 402.
16. Janková H, Říhová E. Juvenilní xantogranulom. Oftalmologie v kasuistikách 2007; 3 : 214 – 218.
17. Vašáková M. Co je to Erheimova nemoc? Kazuist Alergol Pneumol ORL 2006; 3 : 22 – 25.
18. Chester W. Über lipoigranulomatóse. Virchows Arch 1930; 279 : 561 – 602.
19. Jaffe HL. Metabolic, degenerative and inflammatory disease of bones and joints. Philadelphia: PA Lea and Febiger 1972 : 531 – 541.
20. Miller RL, Sheeler LR, Bauer TW et al. EC diseae. Am J Med 1986; 80 : 1230 – 1236.
21. Alper MG, Zimmermann LE, Piana FG. Orbital manifestatitons of Erdheim‑Chester disease. Trans Am Ophthalmol Soc 1983; 81 : 64 – 85.
22. Garg T, Chander R, Gupta T et al. Erdheim‑Chester disease with cutaneous features in an Indian patient. Skinmed 2008; 7 : 103 – 106.
23. Dehner LP. Juvenile xanthogranulomas in the first two decades of life: a clinicopathologic study of 174 cases with cutaneous and extracutaneous manifestations. Am J Surg Pathol 2003; 27 : 579 – 593.
24. Kenn W, Eck M, Allolio B et al. Erheim - Chester disease: evidence for disease entity different from Langerhans cell histiocytosis? Three cases with detailed radiological and immonohistochemical analysis. Hum Pathol 2000; 31 : 734 – 739.
25. Kenn W, Stäbler A, Zachoval R et al. Erdheim Chester disease: a case report and literature overview. Eur Radiol 1999; 9 : 153 – 158.
26. Veyssier - Belot C, Cacoub P, Caparros - Lefebvre B et al. Erdheim Chester disease: Clinical and radiological characteristics of 59 cases. Medicine (Baltimore) 1996; 75 : 157 – 169.
27. Swerdlow SH, Campo E, Harris NL et al. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon: IARC Press 2008.
28. Favara EB. Contemporary classification of histiocytis disorders. Med Pediatr Oncol 1997; 29 : 157 – 166.
29. Perras B, Petersen D, Lorch H et al. Psychoneuroendocrine disturbances in a patient with a rare granulomatous disease. Exp Clin Endocrinol Diabetes 2002; 110 : 248 – 252.
30. Brodkin CL, Wszolek ZK. Neurologic presentation of Erdheim‑Chester disease. Neurol Neurochir Pol 2006; 40 : 397 – 403.
31. De Abreu MR, Chung CB, Biswal S et al. Erdheim‑Chester disease: MR imaging, anatomic, and histopathologic correlation of orbital involvement. AJNR Am J Neuroradiol 2004; 25 : 627 – 630.
32. Salsano E, Savoiardo M, Nappini S et al. Late - onset sporadic ataxia, pontine lesion, and retroperitoneal fibrosis: a case of Erdheim‑Chester disease. Neurol Sci 2008; 29 : 263 – 267.
33. Sistermann R, Katthagen BD. Erdheim Chester disease: a rare cause of knee and leg pain. Arch Orthop Trauma Surg 2000; 120 : 112 – 113.
34. Yanagi T, Kato N, Yamane N et al. Verruca plana‑like papules as a new manifestation of Erdheim‑Chester disease. Arch Dermatol 2007; 143 : 952 – 953.
35. Sivak ‑ Callcott JA, Rootman J, Rasmussen SL et al. Adult xanthogranulomatous disease of the orbit and ocular adnexa: new immunohistochemical findings and clinical review. Br J Ophthalmol 2006; 90 : 602 – 608.
36. Watermann DF, Kiesewetter F, Frosch PJ. Skin manifestations of Erdheim‑Chester disease. Case report and review of the literature. Hautarzt 2001; 52 : 510 – 517.
37. Hammond MD, Niemi EW, Ward TP et al. Adult orbital xanthogranuloma with associated adult ‑ onset asthma. Ophthal Plast Reconstr Surg 2004; 20 : 329 – 332.
38. Karcioglu ZA, Sharara N, Boles TL et al. Orbital xanthogranuloma: clinical and morphologic features in eight patients. Ophthal Plast Reconstr Surg 2003; 19 : 372 – 381.
39. Kujat C, Martin J, Püschel W. Erdheim‑Chester disease. Radiologe 1991; 31 : 297 – 306.
40. Fink MG, Levinson DJ, Brown NL et al. Erdheim‑Chester disease. Case report with autopsy finding. Arch Pathol Lab Med 1991; 21 : 1714 – 1716.
41. Tien RD, Brasch RC, Jackson DE et al. Cerebral Erdheim‑Chester disease: persistent enhancement with Gd ‑ DTPA on MR images. Radiology 1989; 172 : 791 – 792.
42. Martinez R. Erdheim‑Chester disease: MR of intraaxial and extraaxial brain stem lesion. AJNR Am J Neuroradiol 1995; 16 : 1787 – 1790.
43. Ernemann U, Skalej M, Hermison M et al. Primary cerebral non Langerhans cell histiocytosis MR and diferential diagnosis. Neuroradiology 2002; 44 : 759 – 763.
44. Babu RP, Lansen TA, Chadburn A et al. Erdheim‑Chester disease of the central nervous system. Report of two cases. J Neurosurg 1997; 86 : 888 – 892.
45. Bohlega S, Alwatban J, Tulbah A et al. Cerebral manifestation of Erdheim‑Chester disease. Clinical and radiological findings. Neurology 1997; 49 : 1702 – 1705.
46. Weidauer S, von Stuckrad‑Barre S, Dettmann E et al. Cerebral Erdheim‑Chester disease: case report and review of the literature. Neuroradiology 2003; 45 : 241 – 245.
47. Wright RA, Hermann RC, Parisi JE. Neurological manifestation of Erdheim‑Chester disease. J Neurol Neurosurg Psychiatry 1999; 66 : 72 – 75.
48. Shimada S, Ono K, Hashizume Y et al. Intracranial lesion of Erdheim‑Chester disease. Hum Pathol 2007; 38 : 950 – 951.
49. Grothe C, Urbach H, Bös M et al. Cerebellar syndrome, exophthalmos and secondary hypogonadism in Erdheim‑Chester disease. Nervenarzt 2001; 72 : 449 – 452.
50. Khamseh ME, Mollanai S, Hashemi F et al. Erdheim‑Chester syndrome, presenting as hypogonadotropic hypogonadism and diabetes insipidus. J Endocrinol Invest 2002; 25 : 727 – 729.
51. Kovacs K, Bilbao JM, Fornasier VL et al. Pituitary pathology in Erdheim‑Chester disease. Endocr Pathol 2004; 15 : 159 – 166.
52. Oweity T, Scheithauer BW, Ching HS et al. Multiple system Erdheim‑Chester disease with massive hypothalamic ‑ sellar involvement and hypopituitarism. J Neurosurg 2002; 96 : 344 – 351.
53. Tritos NA, Weinrib S, Kaye TB. Endocrine manifestations of Erdheim‑Chester disease (a distinct form of histiocytosis). J Intern Med 1998; 244 : 529 – 535.
54. Sheu SY, Wenzel RR, Kersting R et al. Erdheim‑Chester disease: A case report with multisystemic manifestation including testes, thyroid, and lymph nodes and review of literature. J Clin Pathol 2004; 57 : 1225 – 1228.
55. Bullmann C, Faust M, Hoffmann E et al. Five cases with central diabetes insipidus and hypogonadism as first presentation of neurosarcoidosis. Eur J Endocrinol 2000; 142 : 365 – 372.
56. Mahnel R, Tan KH, Fahlbusch R et al. Problems in differential diagnosis of non Langerhans cell histiocytosis with pituitary involvement: case report and review of literature. Endocr Pathol 2002; 13 : 361 – 368.
57. Sheen KC, Chang CC, Chant TC et al. Thickened pituitary stalk with central diabetes insipidus. Report of 3 cases. J Formos Med Assoc 2001; 100 : 198 – 204.
58. Takao T, Asaba K, Tanaka H et al. A case of lymphocytic infundibuloneurohypophysitis hypophisitits schowing diabetes insipidus followed by anterior hypopituitarism associated with trombnasthenia. Endocr J 2000; 47 : 285 – 291.
59. Tashiro T, Sano T, Xu B et al. Spectrum of different types of hypophysitis. A clinicopathologic study hypophysitis in 31 cases. Endocr Pathol 2002; 13 : 183 – 185.
60. Mohn A, Fahlbusch R, Dörr HG. Panhypopituaitarism associated with diabetes insipidus in a girl with suprasellar arachnoid cyst. Horm Res 1999; 52 : 35 – 38.
61. Folkerth RD, Price DL jr, Schwartz M et al. Xanthomatous hypophysitis. Am J Surg Pathol 1998; 22 : 736 – 741.
62. Athanasou NA, Barbatis C. Erdheim‑Chester disease with epiphyseal and systemic disease. J Clin Pathol 1993; 46 : 481 – 482.
63. Augoustides JG, Szeto WY. Unmasked diabetes insipidus after pericardial drainage and biopsy for pericardial effusion in association with Erdheim‑Chester disease. J Thorac Cardiovasc Surg 2008; 136 : 217 – 218.
64. Rushing EJ, Kaplan KJ, Mena H et al. Erdheim‑Chester disease of the brain: cytological features and differential diagnosis of a challenging case. Diagn Cytopathol 2004; 31 : 420 – 422.
65. Reithmeier T, Trost HA, Wolf S. Xanthogranuloma of the Erdheim‑Chester type within the sellar region. A case report. Clin Neuropathol 2002; 21 : 24 – 28.
66. Allen TC, Chevez‑Barrios P, Shetlar DJ et al. Pulmonary and ophthalmic involvement with Erdheim‑Chester disease: a case report and review of the literature. Arch Pathol Lab Med 2004; 128 : 1428 – 1431.
67. Kambouchner M, Colby T, Domenge C et al. Erdheim‑Chester disease with prominent pulmonary involvement associated with eosinophillic granuloma of mandibular bone. Histopathology 1997; 30 : 353 – 358.
68. Krüger S, Krop C, Wibmer T et al. Erdheim‑chester disease: a rare cause of interstitial lung disease. Med Klin (Munich) 2006; 101 : 573 – 576.
69. Protopapadakis C, Antoniou KM, Nicholson AG et al. Erdheim‑Chester disease: pulmonary presentation in a case with advanced systemic involvement. Respiration 2009; 77 : 337 – 340.
70. Shamburek RD, Brewer HB jr, Gochuico BR et al. Erdheim‑Chester disease. A rare multisystem histiocytic disorder associated with interstitial lung disease. Am J Med Sci 2001; 321 : 66 – 75.
71. Rush WL, Andriko JA, Galateau - Salle F et al. Pulmonary pathology of Erdheim‑Chester disease. Mod Pathol 2000; 13 : 747 – 754.
72. Vasáková M, Fiala P, Kinkor Z. Erdheim‑Chester disease. A case report. Monaldi Arch Chest Dis 2001; 56 : 115 – 117.
73. Vaglio A, Corradi D, Maestri R et al. Pericarditis heralding Erdheim‑Chester disease. Circulation 2008; 118: e511 – e512.
74. Serratrice J, Granel B, De Roux C et al. “Coated aorta” a new sign of Erdheim‑Chester disease. J Rheumatol 2000; 27 : 1550 – 1553.
75. Haroche J, Amoura Z, Dion E et al. Cardiovascular involvement, an overlooked feature of Erdheim‑Chester disease: report of 6 new cases and a literature review. Medicine (Baltimore) 2004; 83 : 371 – 392.
76. Bassou D, El Kharras A, Amezyane TT et al. Cardiac Erdheim‑Chester. Intern Med 2009; 48 : 83 – 84.
77. Dion E, Graef C, Haroche J et al. Imaging of thoracoabdominal involvement in Erdheim‑Chester disease. AJR Am J Roentgenol 2004; 183 : 1253 – 1260.
78. Granier M, Micheau A, Serre I. A rare cause of cardiac tumour: an Erdheim‑Chester disease with cardiac involvement co - existing with an intracerebral Langerhans cell histiocytosis. Eur Heart J 2008; 29 : 1929 – 1935.
79. Loeffler AG, Memoli VA. Myocardial involvement in Erdheim‑Chester disease. Arch Pathol Lab Med 2004; 128 : 682 – 685.
80. Kudo Y, Iguchi N, Sumiyoshi T et al. Dramatic change of Ga ‑ 67 citrate uptake before and after corticosteroid therapy in a case of cardiac histiocytosis (Erdheim‑Chester disease). J Nucl Cardiol 2006; 13 : 867 – 869.
81. Loddenkemper K, Hoeyr B, Loddenkemper C et al. A case of Erdheim‑Chester disease initially mistaken for Ormond’s disease. Nat Clin Pract Rhematol 2008; 4 : 50 – 55.
82. Bangard C, Lotz J, Rosenthal H et al. Erdheim‑Chester disease versus multifocal fibrosis and Ormond‘s disease: a diagnostic dilemma. Clin Radiol 2004; 59 : 1136 – 1141.
83. Murray M, Marshall M, England E et al. Erdheim‑Chester disease. Clin Radiol 2001; 56 : 481 – 484.
84. Haroche J, Amoura Z, Touraine P et al. Bilateral adrenal infiltration in Erdheim‑Chester disease. Report of seven cases and literature review. J Clin Endocrinol Metab 2007; 92 : 2007 – 2012.
85. Pickhardt PJ, Bhalla S. Unusual nonneoplastic peritoneal and subperitoneal conditions: CT findings. Radiographics 2005; 25 : 719 – 730.
86. Scheer M, Hon M, Fruauff AA et al. Perinephric xanthogranulomatosis: CT diagnosis and confirmation by CT ‑ guided percutaneous biopsy. Clin Imaging 2000; 24 : 64 – 67.
87. Surabhi VR, Menias C, Prasad SR et al. Neoplastic and non‑neoplastic proliferative disorders of the perirenal space: cross ‑ sectional imaging findings. Radiographics 2008; 28 : 1005 – 1017.
88. Colin P, Ballereau C, Lambert M et al. Retroperitoneal infiltration as the first sign of Erdheim‑Chester disease. Int J Urol 2008; 15 : 455 – 456.
89. Moore FO, Berne JD, Fox AD. Mesenteric panniculitis and Erdheim‑Chester disease: xanthogranulomatous diseases confused with malignancy. J Am Coll Surg 2007; 204 : 326 – 327.
90. Droupy S, Attias, D, Eschwege P. Bilateral hydronephrosis in a patient with Erdheim Chester disease. J Urol 1999; 162 : 2084 – 2085.
91. O‘Rourke R, Wong DC, Fleming S et al. Erdheim‑Chester disease: a rare cause of acute renal failure. Australas Radiol 2007; 51: B48 – B51.
92. Verdalles U, Goicoechea M, García de Vinuesa H et al. Erdheim‑Chester disease: a rare cause of renal failure. Nephrol Dial Transplant 2007; 22 : 1776 – 1777.
93. Wimpissinger TF, Schernthaner G, Feichtinger H et al. Compression of kidneys in Erdheim‑Chester disease of retroperitoneum: Open surgical approach. Urology 2005; 65 : 798.
94. Gupta A, Aman K, Al ‑ Babtain M et al. Multisystem Erdheim‑Chester disease; a unique presentation with liver and axial skeletal involvement. Br J Haematol 2007; 138 : 280.
95. Canbaz F, Dabak N, Baris S et al. Erdheim‑Chester disease: 99mTc ‑ MDP bone scan provides the diagnosis. Eur J Nucl Med Mol Imaging 2005; 32 : 998.
96. Dion E, Graef C, Miquel A et al. Bone involvement in Erdheim‑Chester disease: imaging findings including periostitis and partial epiphyseal involvement. Radiology 2006; 238 : 632 – 639.
97. Girszyn N, Arnaud L, Villain D et al. Usefulness of combined positron emission tomography and computed tomography imaging in Erdheim‑Chester disease. Rev Med Interne 2007; 28 : 770 – 774.
98. Hoffmann EM, Müller – Forel W, Pitz S et al. Erdheim‑Chester disease: a case report. Graefes Arch Clin Exp Ophthalmol 2004; 242 : 803 – 807.
99. Gottlieb R, Chen A. MR findings of Erdheim‑Chester disease. J Comput Assist Tomogr 2002; 26 : 257 – 261.
100. Nakahara T, Suzuki T, Uno K et al. 18F - FDG positron emission tomographic imaging in Erdheim‑Chester disease with skeletal and extra - skeletal involvement. Leuk Lymphoma 2006; 47 : 935 – 937.
101. Namwongprom S, Núñez R, Kim EE et al. Tc - 99m MDP bone scintigraphy and positron emission tomography/ computed tomography (PET/ CT) imaging in Erdheim‑Chester disease. Clin Nucl Med 2007; 32 : 35 – 38.
102. Blum R, Seymour JF, Hicks RJ. Role of 18FDG positron emission tomography scanning in the management of histiocytosis. Leuk Lymphoma 2002; 43 : 2155 – 2157.
103. Palotás A, Bogáts G, Lázár M et al. Radiopharmaceutical diagnosis of Erdheim‑Chester‘s disease. Nucl Med Commun 2007; 28 : 63 – 65.
104. Spyridonidis TJ, Giannakenas C, Barla Pet al. Erdheim‑Chester disease: a rare syndrome with a characteristic bone scintigraphy pattern. Ann Nucl Med 2008; 22 : 323 – 326.
105. Zanglis A, Valsamaki P, Fountos G. Erdheim‑Chester disease: Symmetric uptake in the 99mTc ‑ MDP bone scan. Hell J Nucl Med 2008; 11 : 164 – 167.
106. Kim EE, Romero JA. Erdheim Chester disease demonstrated with bone radiographs and scans. Clin Imaging 1997; 21 : 328 – 331.
107. Koziolek MJ, Kunze E, Müller A et al. Erdheim‑Chester disease. Dtsch Med Wonchenschr 2005; 132 : 25 – 28.
108. Matsui K, Nagata Y, Hiraoka M. Radiotherapy for Erdheim‑Chester disease. Int J Clin Oncol 2007; 12 : 238 – 241.
109. Perlat A, Decaux O, Sébillot M et al. Erdheim‑Chester disease with predominant mesenteric localization: Lack of efficacy of interferon alpha. Joint Bone Spine 2009; 76 : 315 – 317.
110. Vanichaniramol N, Kingpetch K, Buranasupkajorn P et al. Erdheim‑Chester disease. Intern Med 2008; 47 : 1633 – 1634.
111. Ono K, Oshiro M, Uemura K et al. Erdheim‑Chester disease: case report with imunohistochemical and biochemical examination. Hum Pathol 1996; 27 : 91 – 95.
112. Egan AJ, Boardman LA, Swensen SJ et al. Erdheim Chester disease: clinical, radiologic, and histopathologic findings in five patients with interstitial lung disease. Am J Surg Pathol 1999; 23 : 17 – 26.
113. Kim NR, Ko YH, Choe EY et al. Erdheim Chester disease Extesive marrow necrosis: a case report and review of the literature. Intern J Surg Pathol 2001; 9 : 73 – 79.
114. Busemann C, Kallinich B, Schwesinger G et al. Erdheim‑Chester disease with hemophagocytosis. Ann Hematol 2007; 86 : 847 – 849.
115. Rao RN, Chang CC, Uysal N et al. Fulminant multisystem non‑langerhans cell histiocytic proliferation with hemophagocytosis: a variant form of Erdheim‑Chester disease. Arch Pathol Lab Med 2005; 129: e39 – e43.
116. Lopez P, Estes ML. Immunohistochemical characterization of the histiocytes in sinus histiocytosis with massive lymphadenopathy. Analysis of an extranodal case. Hum Pathol 1989; 20 : 711 – 715.
117. Bonnetti F, Chilosi M, Menestrina Fet al. Immunohistological analysis of Rosai ‑ Dorfman histiocytosis. A disease of S ‑ 100 + CD1 – histiocytes. Virchows Arch Apathol Anat Histopathol 1987; 411 : 129 – 135.
118. Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai ‑ Dorfman disease). Review of the entity. Semin Diagn Pathol 1990; 7 : 19 – 73.
119. Braiteh F, Boxrud C, Esmaeli B et al. Successful treatment of Erdheim‑Chester disease, a non‑Langerhans ‑ cell histiocytosis, with interferon‑alpha. Blood 2005; 106 : 2992 – 2994.
120. Bourke SC, Nicholson AG, Gibson GJ. Erdheim‑Chester disease: pulmonary infiltration responding to cyclophosphamide and prednisolone. Thorax 2003; 58 : 1004 – 1005.
121. Haroche J, Amoura Z, Trad SG. Variability in the efficacy of interferon alpha in Erdheim Chester disease by patient and site of involvement results in eight patients. Arthritis Rheum 2006; 54 : 3330 – 3336.
122. Esmaeli B, Ahmadi A, Tang R et al. Interferon therapy for orbital infiltration secondary to Erdheim‑Chester disease. Am J Ophthalmol 2001; 132 : 945 – 947.
123. Myra C, Sloper L, Tighe PJ et al. Treatment of Erdheim‑Chester disease with cladribine: a rational approach. Br J Ophthalmol 2004; 88 : 844 – 847.
124. Sheidow TG, Nicolle DA, Heathcote JG. Erdheim-Chester disease: two cases of orbital involvement. Eye (Lond). 2000; 14 : 606–612.
125. Rajendra B, Duncan A, Parslew R et al. Successful treatment of central nervous system juvenile xanthogranulomatosis with cladribine. Pediatr Blood Cancer 2009; 52 : 423 – 415.
126. Mascalchi M, Nencini P, Nistri M et al. Failure of radiation therapy for brain involvement in Erdheim Chester disease. J Neurooncol 2002; 59 : 169 – 172.
127. Miller RC, Villà S, Kamer S et al. Palliative treatment of Erdheim‑Chester disease with radiotherapy: a Rare Cancer Network study. Radiother Oncol 2006; 80 : 323 – 326.
128. Haroche J, Amoura Z, Charlotte F et al. Imatinib mesylate for platelet ‑ derived growth factor receptor‑beta‑positive Erdheim‑Chester histiocytosis. Blood 2008; 111 : 5413 – 5415.
129. Boissel N, Wechsler B, Leblond V. Treatment of refractory Erdheim‑Chester disease with double autologous hematopoietic stem ‑ cell transplantation. Ann Intern Med 2001; 135 : 844 – 845.
130. Srikulmontree T, Massey HD, Roberts WN et al. Treatment of skeletal Erdheim‑Chester disease with zoledronic acid: case report and proposed mechanisms of action. Rheumatol Int 2007; 27 : 303 – 307.
131. Eyigör S, Kirazli Y, Memis A et al. Erdheim‑Chester disease: the effect of bisphosphonate treatment – a case report. Arch Phys Med Rehabil 2005; 86 : 1053 – 1057.
132. Mossetti G, Rendina D, Numis FG et al. Biochemical markers of bone turnover, serum levels of interleukin‑6/ interleukin‑6 soluble receptor and bisphosphonate treatment in Erdheim‑Chester disease. Clin Exp Rheumatol 2003; 21 : 232 – 236.
133. Yahng SA, Kang HH, Kim SK et al. Erdheim‑Chester disease with lung involvement mimicking pulmonary lymphangitic carcinomatosis. Am J Med Sci 2009; 337 : 302 – 304.
134. de Abreu MR, Castro MO, Chung C et al. Erdheim‑Chester disease: case report with unique postmortem magnetic resonance imaging, high‑resolution radiography, and pathologic correlation. Clin Imaging 2009; 33 : 150 – 153.
135. Salsano E, Savoiardo M, Nappini S et al. Late ‑ onset sporadic ataxia, pontine lesion, and retroperitoneal fibrosis: a case of Erdheim‑Chester disease. Neurol Sci 2008; 29 : 263 – 367.
136. Monfredi O, Jones C, Warnock N. No way in & no way out: a case of renal failure due to both pre ‑ and post‑renal obstruction. Nephrol Dial Transplant 2008; 23 : 2406 – 2408.
137. Lin E. FDG PET/ CT for biopsy guidance in Erdheim‑Chester disease. Clin Nucl Med 2007; 32 : 860 – 862.
138. Yanagi T, Kato N, Yamane N et al. Verruca plana‑like papules as a new manifestation of Erdheim‑Chester disease. Arch Dermatol 2007; 143 : 952 – 953.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2009 Issue 12
Most read in this issue
- Preeklampsie z hlediska mezioborové spolupráce
- Program řízené ambulantní rehabilitace u pacientů po operaci chlopenních srdečních vad
- Komplexita interakcií nádorového procesu
- Obojstranná flebotrombóza dolných končatín zapríčinená vrodenou malformáciou dolnej dutej žily