
Chronický mírný zánět spojuje obezitu, metabolický syndrom, aterosklerózu a diabetes
Chronic mild inflammation links obesity, metabolic syndrome, atherosclorosis and diabetes
Chronic low grade inflammation is relatively new concept in metabolic medicine. This concept describes the relations between the inflammation and adipose tissue, insulin resistence, atherosclerosis and type 2 diabetes mellitus. Macrophages and lymphocytes deposed in adipose tissue produce proinflammatory cytokines which directly or through the CRP liver secretion are targeting endothelial cells, hepatocytes and β cells of Langerhans islets of pancreas. The dysfunction of these cells follows often further disturbances and in case of β cells – the cell death. The connection between the adipose tissue insulin resistence, atherosclerosis and type 2 diabetes was earlier described with endocrine and metabolic descriptors. The concept of chronic low grade inflammation creates also another description of multilateral connections in metabolic syndome. The salicylates and the drugs related to them seem to have some glucose lowering properties. The recent development in the field of chronic low grade inflammation represents also certain therapeutic hope for antiinflammatory intervention in type 2 diabetes.
Key words:
chronic low grade inflammation – metabolic syndrome – type 2 diabetes mellitus – cytokines
Authors:
M. Anděl 1,2,4; J. Polák 1,3,4; P. Kraml 1,4; P. Dlouhý 1,2; V. Štich 1,3
Authors‘ workplace:
Centrum pro výzkum diabetu, metabolismu a výživy 3. lékařské fakulty UK a FN Královské Vinohrady Praha, vedoucí prof. MUDr. Michal Anděl, CSc.
1; Ústav výživy 3. lékařské fakulty UK a FN Královské Vinohrady, Praha, přednosta prof. MUDr. Michal Anděl, CSc.
2; Ústav tělovýchovného lékařství 3. lékařské fakulty UK a FN Královské Vinohrady, Praha, přednosta doc. MUDr. Vladimír Štich, Ph. D.
3; II. interní klinika 3. lékařské fakulty UK a FN Královské Vinohrady, Praha, přednosta prof. MUDr. Michal Anděl, CSc.
4
Published in:
Vnitř Lék 2009; 55(7-8): 659-665
Category:
134th Internal Medicine Day - 23rd Vanysek's Day Brno 2009 - Vanysek's Lecture
Overview
Chronický mírný zánět představuje v metabolické medicíně relativně nový způsob popisu vztahů mezi tukovou tkání, inzulinovou rezistencí, aterosklerózou a diabetem 2. typu. Tuková tkáň produkuje zejména díky makrofágům a lymfocytům v ní deponovaným řadu prozánětlivých cytokinů, které působí jednak přímo, jednak cestou přes CRP na endotelie, na hepatocyty i na β buňky pankreatu. Poškození v těchto cílových buňkách vede k jejich dysfunkci, postupujícím mírným zánětlivým změnám a v případě β buněk pankreatu i k jejich zániku. Vedle existující metabolické teorie propojující jednotlivé jednotky metabolického syndromu se tak vynořuje další pojítko. Patogenetická spojitost dává naději na cílenější léčbu. Již nyní je snaha použít salicyláty k příznivému ovlivnění hladin glykemie u nemocných s diabetem 2. typu. Klasické používání acetylsalicylové kyseliny v prevenci tepenného postižení dostává s nárůstem poznatků o chronickém mírném zánětu nový rozměr.
Klíčová slova:
chronický mírný zánět – metabolický syndrom – diabetes mellitus 2. typu – cytokiny
Úvod
Empirická pozorování spojovala výskyt aterosklerózy a diabetu s obezitou přinejmenším již od 19. století [52]. Ještě starší jsou pozorování o vztahu mezi obezitou a diabetem, jsou však známa již od začátku renesance, od doby, kdy Thomas Willis popsal u diabetiků přítomnost glukózy v moči. Tehdy bylo dobře známo, že diabetes (dnes bychom řekli, že to byl diabetes 2. typu) postihuje především zámožné. Na zánětlivou etiologii aterosklerózy pomýšlel již Virchow a „označil totéž jako endarteriitis chronica, jakkoliv byl pamětliv rozdílů sklerotických od změn zánětlivých a na rozdíly ty zvlášť poukázal: k úsudku, že běží o zánět, vedly jej hlavně hyperplazie a malobuněčná infiltrace“ (citován doslovně Maixner [33]). Většina moderního období 20. století hledala pojítko mezi obezitou, diabetem a aterosklerózou zejména v rovině metabolických vztahů. Klasickou oporou metabolických úvah se stala práce Rendla a Halese o vzájemné metabolické kompetici glukózy a mastných kyselin [43]. Postulování pojmu inzulinová rezistence, její studium pomocí clampových technik vedlo nakonec k postulování tzv. metabolického syndromu [44], ve kterém syndrom inzulinové rezistence vzájemně propojil obezitu, poruchu glukózové tolerance, diabetes 2. typu, arteriální hypertenzi a hypertriacylglycerolemii. Významný posun přinesl začátek 90. let 20. století, kdy byla objevena hormonální produkce tukových buněk. Nejprve se jednalo o leptin, později o řadu dalších hormonů: rezistin, adiponektin či visfatin. Tyto hormony ovlivňují jak energetický metabolizmus, tak inzulinovou rezistenci, a jejich účinek tak dalším způsobem propojuje obezitu s diabetem či aterosklerózou. Volné kyslíkové radikály studované již několik desetiletí modifikují především lipidové struktury k více aterogenním formám, které přímo působí na endotelie, další struktury cévní stěny a snad i na β buňky Langerhansových ostrůvků pankreatu.
Od počátku 90. let minulého století se začaly objevovat práce spojující aterosklerózu nejprve s konkrétními infekcemi, posléze spíše s jakýmkoliv zánětem. Nález produkce prozánětlivých cytokinů v tukové tkáni a posléze popis makrofágů tukové tkáně, zejména u obézních osob, vedl k postulování koncepce spojující chronický mírný zánět, aterosklerózu a diabetes 2. typu. Vedle výše uvedených metabolických a hormonálních vztahů mezi obezitou, aterosklerózou a diabetem se objevuje nově další spojnice, která může pomoci vysvětlit složité vztahy mezi imunitním a metabolickým systémem a díky tomuto vysvětlení snad i přispět k terapii.
Úvodem je však nezbytné upřesnit použití slova „zánět“ či „zánětlivá reakce“ v následujícím textu i v literatuře vážící se k tomuto tématu. Klasická klinicko‑patologická definice zánětu uvádí, že jde o „...stereotypní, složitou, vývojem získanou schopnost reakce živých organizmů na různá poškození, sestávající z jevů charakteru alterativního, exsudativního, proliferativního a imunitního, které obvykle probíhají sukcesivně, jde‑li o zánět reparativní, anebo simultánně, jde‑li o zánět defenzivní...“ [7]. Klinická manifestace takového zánětu je pak Celsem popsaný soubor příznaků: „rubor, tumor, calor, dolor“, dále rozšířený Virchowem o „functio laesa“. V etiopatogenezi chronických metabolických onemocnění jako diabetes mellitus 2. typu či obezita se na buněčné úrovni uplatňují podobné mechanizmy a signální molekuly jako u klasického zánětu, nicméně v klinickém obraze výše uvedené projevy obvykle nenacházíme. Pro tyto chronické stavy bylo proto navrženo užívat označení „zánět o nízké intenzitě“ (low-grade inflammation) nebo „chronický zánět“. Zajímavý je i návrh na zcela nové označení „metaflammation“ pocházející z anglického „metabolically triggered inflammation“ (metabolicky indukovaný zánět). V následujícím textu tedy pod pojmem zánět rozumí autoři tento metabolicky indukovaný chronický mírný zánět.
Těsná vazba mezi metabolickými a imunitními funkcemi představuje nezbytnou podmínku zajišťující dostupnost volných energetických substrátů a jejich redistribuce v organizmu během probíhající imunitní či zánětlivé reakce. Společné rysy v řízení obou systémů lze demonstrovat i na morfologické úrovni. Jaterní i tuková tkáň jsou v úzkém vztahu s imunokompetentními buňkami monocyto-makrofágového systému. Hepatocyty jsou v těsném kontaktu s Kupferovými buňkami, zatímco tuková tkáň je osídlena makrofágy (v různém stadiu aktivace). Klíčovým hormonem regulujícím metabolizmus živin je anabolicky působící inzulin. Není tedy překvapující, že mnohé procesy zahrnuté v imunitní a zánětlivé odpovědi jsou zároveň spjaty s ovlivněním inzulinové signalizace v cílových orgánech.
Tuková tkáň, chronický mírný zánět a inzulinová rezistence
Asociace mezi nadměrnou akumulací tukové tkáně a vznikem inzulinové rezistence, případně diabetes mellitus 2. typu, je známa již dlouhou dobu, nicméně poznání patogenetických mechanizmů, které jsou za tuto vazbu zodpovědné, není dosud kompletní a představuje důležitou součást současného metabolického výzkumu. Zajímavé informace poskytlo studium klinických případů lipodystrofie, tedy stavu, kdy dochází (vrozeně nebo získaně) k významné poruše funkce tukové tkáně, případně k jejímu úplnému chybění (lipoatrofie). Jedinci takto postižení se manifestují těžkou inzulinovou rezistencí se všemi typickými složkami včetně diabetes mellitus 2. typu [19]. Jedná se tedy o identické poruchy, které běžně nalézáme u stavu spojeného s chronickým nadbytkem živin – obezity. Tento zdánlivý paradox lze vysvětlit ve sjednocující teorii, která je založena na schopnosti tukové tkáně zpracovávat adekvátně tok nutrientů („nutrient flux“), tedy akumulovat lipidy za situace jejich zvýšené (ovšem nikoli trvalé) nabídky a uvolňovat je v procesu lipolýzy v období postprandiálním. Za situace absolutního chybění tukové tkáně (lipoatrofie) nebo omezení její funkce a množství (lipodystrofie) je zřejmé, že organizmus tuto pufrovací schopnost tukové tkáně pozbyl a lipidy jsou ukládány v jiných tkáních, kde významně interagují s intermediárním metabolizmem a podílejí se na vzniku inzulinové rezistence [46,49,60]. U obézních jedinců je situace jiná: chronický nadbytek energie vede postupně ke zvětšování objemu adipocytů díky depozici lipidových inkluzí (hypertrofie) a teprve při dosažení určité kritické hladiny velikosti adipocytu dochází k aktivaci a proliferaci preadipocytů [54] a vzniku nových tukových buněk (hyperplazie) [39,47]. O tom, do jaké míry se u daného jedince uplatní hypertrofie a do jaké míry hyperplazie tukové tkáně, pravděpodobně rozhoduje velmi komplexní regulace genové exprese, jejíž mechanizmy jsou předmětem intenzivního výzkumu [15]. Je ovšem nepochybné, že metabolické komplikace a inzulinová rezistence jsou úzce asociovány s velikostí adipocytu, tedy s hyperplastickou složkou expanze tukové tkáně, zatímco menší adipocyty, vzniklé např. nově z preadipocytů, jsou v tomto kontextu neutrální, nebo spíše prospěšné [21]. Negativní účinky hypertrofovaných adipocytů na celotělový metabolizmus lipidů a sacharidů jsou dány jednak vyšší mírou lipolýzy, která v takových adipocytech probíhá a uvolňuje do cirkulace volné mastné kyseliny, jednak změněnou produkcí hormonů a dalších proteinů produkovaných v tukové tkáni (souhrnně nazývané adipokiny) s následným navozením chronického zánětlivého stavu o nízké intenzitě.
Chronický mírný zánětlivý stav je charakterizován zvýšenou produkcí řady cytokinů z tukové tkáně, zejména TNF‑α, IL‑6, IL‑1, IL‑8, zatímco produkce protizánětlivých cytokinů adiponectinu a leptinu je snížena [23]. Za produkci těchto cytokinů jsou z převážné většiny zodpovědné makrofágy [57], které jsou fyziologicky v tukové tkáni přítomny a které jsou ve zvýšené míře do tukové tkáně atrahovány během rozvoje obezity. Podle současných názorů dojde při nadměrném zvětšení objemu adipocytu k produkci MCP (macrophage chemoatractant protein – makrofágový chemoatrakční protein) přímo v tukových buňkách [9,58]. Takto rekrutované makrofágy jsou nadále v tukové tkáni udržovány působením dalších faktorů produkovaných adipocyty, jako jsou MIF (macrophage migration inhibitory factor – faktor inhibující migraci makrofágů) nebo M‑CSF-1 (M-colony stimulating factor-1) [32,51]. Makrofágy se v tukové tkáni shlukují převážně kolem velkých, hypertrofických, umírajících, případně mrtvých adipocytů.
Důležitou úlohu v indukci buněčné smrti hypertrofovaných adipocytů zaujímá fenomén nazývaný jako „stres endoplazmatického retikula“. Tato nitrobuněčná organela má primární funkci v syntéze proteinů a jejich úpravě pro sekreci vně buněk (zde získávají proteiny řadou modifikací svou sekundární a terciární strukturu). Přebytek nutrientů je jedním z faktorů, který vyvolává zvýšené nároky na endoplazmatické retikulum v adipocytech [18]. Součástí reakce na tento stres jsou rozsáhlé změny v buněčném metabolizmu, jejichž cílem je omezení proteosyntetické aktivity. Pokud ovšem tyto obranné mechanizmy nejsou dostatečné nebo pokud stresový faktor přesahuje adaptační mechanizmy, je výsledkem „stresu endoplazmatického retikula“ zahájení řízené buněčné smrti – apoptózy [36]. Na molekulární úrovni reakce provázející „stres endoplazmatického retikula“ zahrnují aktivaci serinových kináz IKK (IκB kináza, kináza inhibitoru κ B) a JNK (c-Jun NH2‑terminální kináza). Tyto enzymy hrají klíčovou úlohu při regulaci zánětlivé odpovědi, diferenciaci a apoptóze buněk (včetně adipocytů). Mezi dominantní účinky těchto kináz patří omezení přenosu signálu z inzulinového receptoru do nitra buněk (vzniká inzulinová rezistence), jakož i zvýšení sekrece již zmíněných prozánětlivých cytokinů [10,31]. Celý circulus vitiosus se uzavírá zjištěním, že JNK i IKK mohou být aktivovány přímo zvýšenou hladinou saturovaných mastných kyselin, jejichž zdrojem je často vysokotuková dieta [31].
Při dnešním stupni poznání by se dala etiopatogeneze inzulinové rezistence ve vztahu k prozánětlivému stavu organizmu shrnout do tohoto sledu pochodů: přebytek nutrientů a nedostatečná pohybová aktivita vede k hypertrofii adipocytů a poruchám v inzulinové citlivosti cílových tkání, zejména svalové. Hypertrofované adipocyty a takto pozměněná tuková tkáň dále zapříčiňují mírný prozánětlivý stav, a to produkcí zánětlivých cytokinů a vyšší hladinou volných mastných kyselin uvolňovaných při lipolýze v adipocytech. S dalším rozvojem stavu dochází k vyšší infiltraci tukové tkáně makrofágy, které jsou do tukové tkáně aktivně rekrutovány a digestují odumírající adipocyty. Tyto makrofágy jsou dominantním zdrojem prozánětlivých cytokinů. Na úrovni periferních inzulin‑dependentních tkání, zejména ve svalové a jaterní buňce, vede vyšší hladina prozánělivých cytokinů k aktivaci vnitrobuněčných signalizačních drah, jejichž součástí jsou i regulační enzymy (JNK, PKC, mTOR) schopné díky své aktivitě výrazně snížit signalizaci inzulinovým receptorem na dané buňce. Vzniká tak inzulinová rezistence. Díky zvýšeným nárokům na pankreatickou sekreci inzulinu dochází ke vzniku stresu endoplazmatického retikula v pankreatických β buňkách, který je dále potencován zánětlivými cytokiny a aktivací nitrobuněčných signalizačních kaskád, v nichž opět hlavní roli hraje JNK. Tím jsou vytvořeny obě základní podmínky pro vznik diabetes mellitus 2. typu.
Infekce, zánět a ateroskleróza
Pohlížíme‑li dnes na aterosklerózu jako na chronický fibroproliferativní zánět [2,3], není překvapivé, že se v centru názorů na patogenezi tohoto onemocnění objevily rizikové faktory související s aktivací imunitního systému, tedy infekce a chronická zánětlivá onemocnění obecně včetně autoimunitních chorob. Lokální či systémový zánět přirozeně vede k produkci imunokinů a cytokinů (např. IL‑6, IL‑10, IL‑18, TNF‑α, solubilní CD40L a dalších), které dále aktivují buňky cévní stěny účastnící se aterosklerotického procesu, zejména monocyty a z nich odvozené pěnové buňky, T‑lymfocyty, buňky hladké svaloviny medie, endotelové buňky a trombocyty (k trombogenezi může dále přispívat i zvýšená produkce tkáňového faktoru a aktivace f. VII koagulační kaskády a zvýšený poměr PAI‑1/TPA v endoteliích). Nejedná se však zdaleka o názor nový. Hypotézu, že infekce může způsobovat aterosklerózu, vyslovil již v roce 1921 Ophulsi. Byla založena na nálezu zánětlivých infiltrátů ve stěně cév, které tvořily převážně makrofágy a pěnové buňky. Na dlouhou dobu poté však tato hypotéza ustoupila do pozadí a dostalo se jí zvýšené pozornosti teprve o 50 let později. V roce 1978 Fabricant et al publikovali nález arteriálních lézí u kuřat infikovaných ptačím herpesvirem, které byly identické s aterosklerotickými změnami u člověka. V českém písemnictví se objevil v roce 1958 článek profesora Františka Bláhy, prvního děkana pražské vinohradské lékařské fakulty, který popsal svá pozorování, kdy za 2. světové války jako vězeň jednoho koncentračního tábora byl přítomen sekcím některých zemřelých spoluvězňů. Zejména u mladých osob byly překvapivě nalezeny na tepnách výrazné aterosklerotické změny, které neodpovídaly věku a nemohly být za daných okolností ani dávány do souvislosti s tradičními rizikovými faktory, jako je obezita, zvýšená konzumace živočišných tuků či kouření. Profesor Bláha však tehdy vyslovil myšlenku, že popsané změny v tepenném řečišti mohly být v souvislosti s chronickými infekcemi, kterými tyto osoby v průběhu svého věznění trpěly [8]. Z klasiků české medicíny však již kladl aterosklerózu do vztahu k infekčním onemocněním, zejména diftérii a tyfu, Thomayer v roce 1909 [52]. Od konce 90. let 20. století byla identifikována řada infekčních agens, u kterých se předpokládá vliv na vznik a akceleraci aterosklerózy u člověka. Na základě přímého průkazu v plátech, častěji však podle sérologie a na základě experimentálních studií na zvířecích modelech se nejčastěji uváděly Chlamydia pneumoniae, Cytomegalovirus, Herpes simplex virus, Virus hepatitidy A, Helicobacter pylori, Porphyromonas gingivalis a další. Byly vysloveny následující mechanizmy, kterými se infekce může podílet na patogenezi aterosklerotického procesu:
- Přímá infekce cévní stěny – patogen iniciuje nebo infiltruje již existující poškození cévní stěny v daném místě, kde stimuluje a akceleruje zánětlivou odpověď. Pro tuto teorii svědčí právě studie prokazující genom příslušného infekčního agens, nejčastěji Chlamydia pneumoniae, v ateromovém plátu. My sami jsme v recentně publikované studii demonstrovali poměrně vysokou prevalenci chlamydiové DNA (47,4 %) v ateromových plátech ze vzorků získaných invazivně angiologickou metodou od pacientů s obliterující aterosklerózou periferních tepen [29].
- Indukce nebo akcelerace lokálního inflamatorního procesu v arteriální stěně systémovým zánětem způsobeným chronickou orofaciální, respirační, gastrointestinální či urologickou infekcí [14]. Často se v této souvislosti hovoří o tzv. náloži patogenů (pathogen burden), kdy souhra působení více infekčních agens vedla k rozsáhlejší zánětlivé odpovědi reflektované vyššími plazmatickými koncentracemi CRP a vyšším rizikem koronární aterosklerózy [62]. Dále se ukázalo, že některé bakteriální toxiny mohou poškozovat cévní stěnu. Zejména endotoxin produkovaný gramnegativními bakteriemi může přímo poškodit endotelové buňky, zvyšovat oxidační stres a aktivovat syntézu cytoadhezivních molekul a proinflamatorních cytokinů [5].
- Akcelerace aterosklerózy je dále přisuzována inflamatornímu stavu provázejícímu onemocnění, jako je obezita, diabetes mellitus [20], hypertenze, chronická renální insuficience [48], systémový lupus erythematodes, revmatoidní artritida, antifosfolipidový syndrom [4,34]. Autoimunitní charakter může být přisuzován i zkřížené imunitní reakci proti bakteriálnímu proteinu a jemu strukturálně podobnému antigenu exprimovanému na povrchu poškozených endotelových buněk (např. hsp-60) [27].
Souhrnně můžeme konstatovat, že se názor na úlohu zánětu v patogenezi aterosklerózy za posledních 15 let měnil. Teorie, že proces může být spuštěn specifickým infekčním agens, začala ztrácet na síle poté, co se ukázalo, že např. Chlamydia pneumoniae je ubikviterní bakterie a antichlamydiové protilátky lze detekovat u velké části obecné populace. Dnes již víme, že ani ne tak konkrétní infekce jako spíše zánětlivá imunitní odpověď obecně akceleruje pochody v cévní stěně vedoucí k tvorbě plátu. Hovoříme‑li však o chronickém zánětu v souvislosti s manifestací např. ischemické choroby srdeční, nemáme na mysli pouze chronické systémové infekce, ale i autoimunitní onemocnění pojiva. K nim dále přistupují nosologické jednotky, jako jsou obezita, diabetes mellitus či chronická renální insuficience, u kterých je spojitost se zánětem jednoznačně zřejmá až v posledních letech. Ve studiích in vitro na kultuře pupečníkových endotelových buněk bylo opakovaně prokázáno, že proinflamatorní cytokin produkovaný např. tukovou tkání, ale též IL‑1 či IL‑8 zvyšují expresi cytoadhezivních molekul (VCAM‑1, ICAM‑1) na povrchu těchto buněk [24]. Jak jsme však ukázali v naší práci z roku 2004, kultivované endotelie exprimují hojně VCAM‑1, ICAM‑1 a ELAM-1 i po hyperglykemické expozici, a to mechanizmem, který účinek TNF‑α potencuje, působí však i na TNF‑α nezávisle [1] (obr. 1, 2).
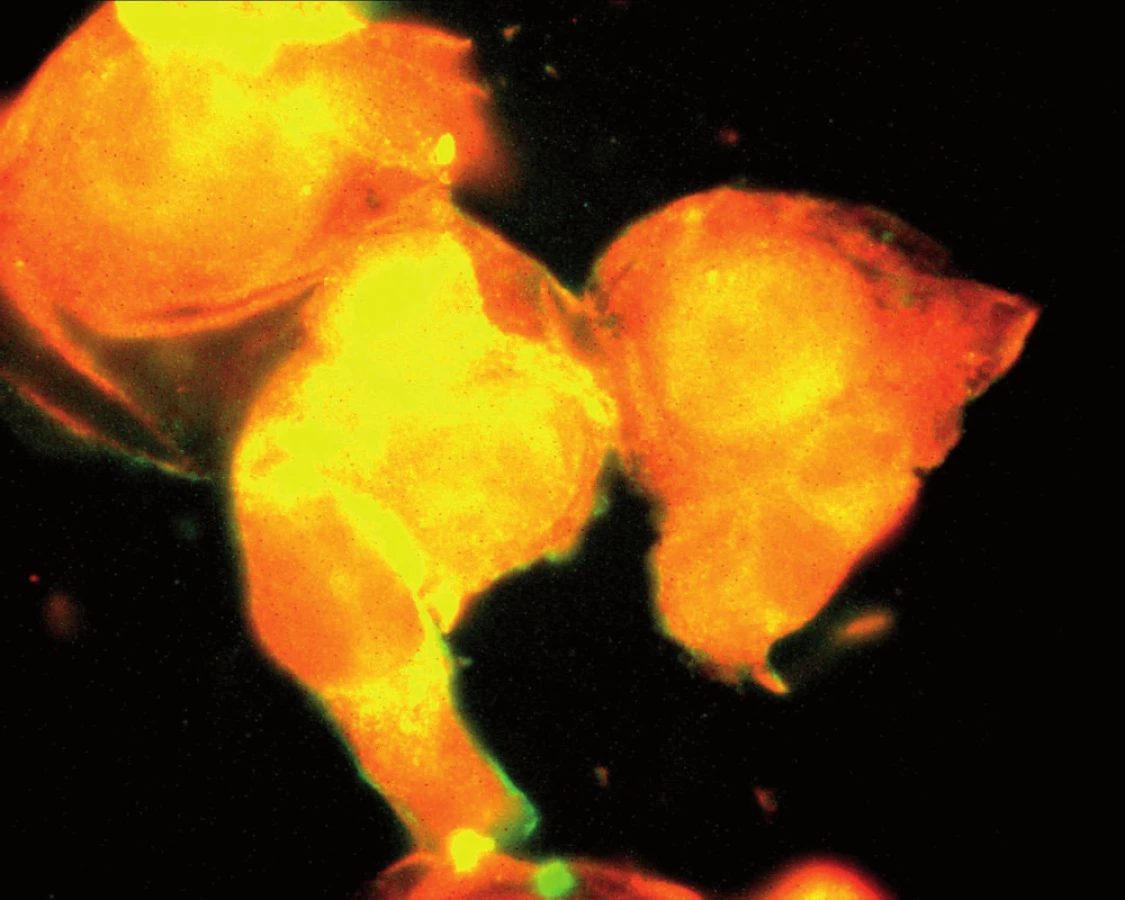
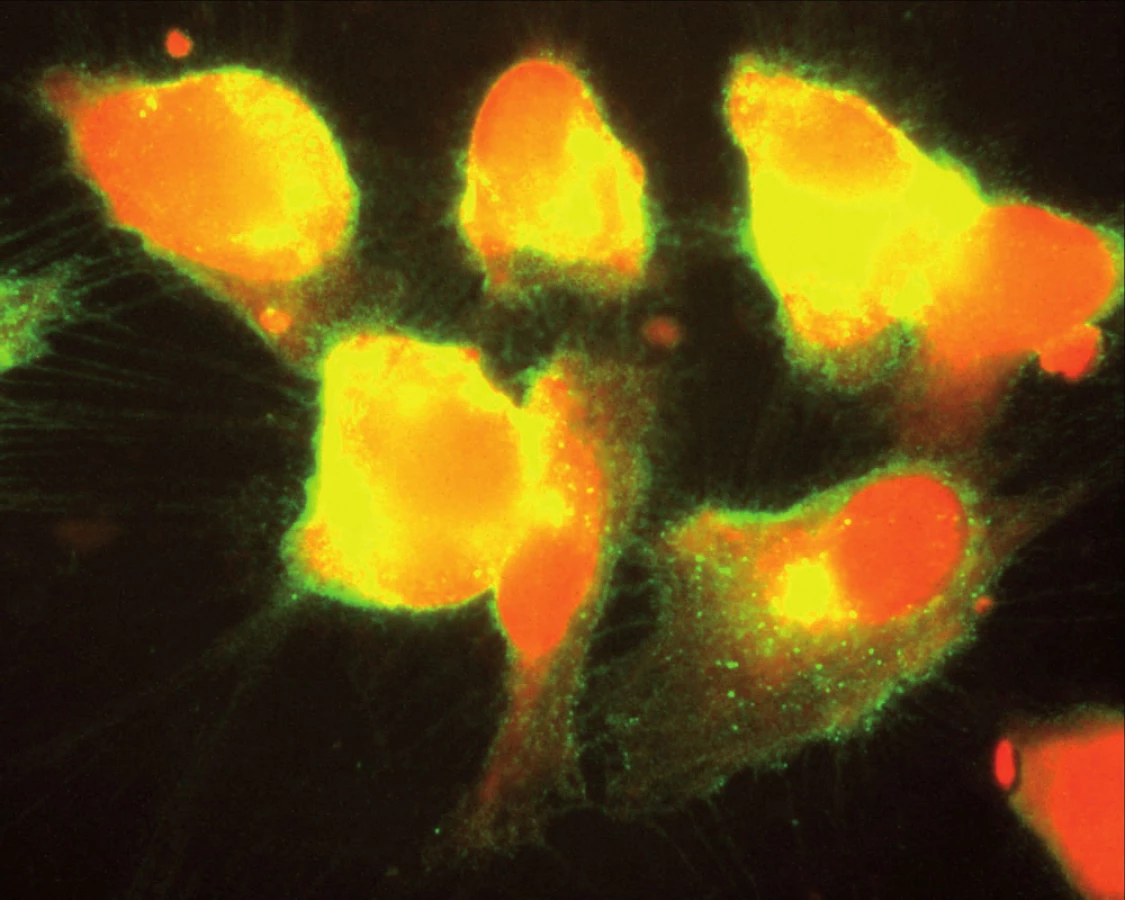
Protože chronický mírný zánět je jedním z aspektů abdominální obezity, je možné, že chronická expozice zánětlivým cytokinům může představovat vedle metabolické spojnice další důležité pojítko mezi těmito spolu provázanými jednotkami.
Diabetes mellitus 2. typu a chronický mírný zánět
V posledních letech je zřetelné, že cesta od inzulinové rezistence provázené hyperinzulinemií a ve značné většině případů také nadváhou či obezitou k diabetu 2. typu je spojena s poklesem původně zvýšené sekrece a také hladiny plazmatického inzulinu. Přitom může být absolutní hladina inzulinu ještě zvýšená nebo normální, relativně je však vzhledem k dané úrovni inzulinové rezistence nedostatečná. Za poklesem hladiny inzulinu v těchto případech stojí snížení počtu β buněk v důsledku jejich zániku. Na tomto zániku se může podílet zvýšená glukotoxicita, lipotioxicita, amyloidoza či zánět [56]. Chronická subklinická aktivizace imunitního systému může tvořit spojnici mezi obezitou, inzulinovou rezistencí a diabetem [22,38,55].
Chronický zánět zhoršuje i inzulinovou rezistenci. K inzulinové rezistenci přispívají jak TNF‑α, tak interleukin‑6, který v játrech zvyšuje produkci CRP, což může vést na jedné straně k indukci či akceleraci aterosklerózy [6], na straně druhé i k poškození β buněk. CRP je nejčastěji měřený cirkulující marker subklinického zánětu, běžně dosažitelný a široce užívaný [30]. V současné době existuje již na tucet prací spojujících CRP a diabetes 2. typu, v menší části těchto prací, po adjustaci na obezitu a inzulinovu rezistenci tento vztah nebyl bezpečně prokázán [30].
TNF‑α, interleukin 1β, IL‑6 a interferon - γ snižují o desítky procent sekreci adiponektinu z tukových buněk, aniž by snižovaly jeho oligomeraci [50]. Snížení hladin adiponektinu je faktorem zhoršení citlivosti k inzulinu. Infuze TNF‑α indukuje inzulinovou rezistenci cestou inhibice Akt metabolických cest v kosterním svalu [40]. Z tohoto hlediska tak zánětlivé cytokiny představují další spojnici mezi abdominální depozicí tuku a vznikem diabetu. Zánět pak představuje jeden z univerzálních patogenetických mechanizmů, který má vliv i na patogenezi diabetu 2. typu [17].
V patologii ostrůvků u diabetu 2. typu hraje zřejmě zánět významnou roli: nalezena byla infiltrace ostrůvků imunitními buňkami, cytokiny, apoptotické buňky, depozita amyloidu a fibróza. V β buňkách nemocných diabetem 2. typu byla nalezena vyšší exprese IL‑1β. Dále byly u těchto nemocných a u většiny experimentálních zvířecích modelů diabetu 2. typu nalezeny s ostrůvky asociované makrofágy. Zvýšené množství makrofágů v ostrůvcích bylo nalezeno již u obézních prediabetických myší. Tyto imunitní buňky jsou pravděpodobně atrahovány chemokiny ostrůvků produkovaných v odpovědi na metabolický stres a pod kontrolou IL‑1β. Modulace v ostrůvcích vznikajících zánětlivých mediátorů, zejména interleukinu-1β, by mohlo být prevencí zánětu u diabetu 2. typu a představovat slibný terapeutický potenciál [13]. Vlastní zánětlivý proces β buněk je zřejmě důsledkem součinného působení hyperglykemie, hyperlipidemie a cirkulujících cytokinů [11]. Prozánětlivý cytokin interleukin‑1β (IL‑1β) byl nalezen zvýšený u obézních pacientů i u obézních hlodavců a má se za to, že zodpovídá za zhoršení sekrece inzulinu, snížení proliferace β buněk a zvyšuje apoptózu pankreatických β buněk [35]. V experimentu vedlo podávání protilátky proti interleukinu-1β u obézních myší k výraznému zlepšení hladin glykemie [35].
Faktor inhibující migraci mikrofágů (MIF), prozánětlivý cytokin podílející se na řadě zánětlivých procesů a reakcí ovlivňuje homeostázu glukózy. MIF je produkován β buňkami a může zvyšovat sekreci inzulinu. Moduluje také odběr glukózy, glykolýzu a inzulinovou rezistenci v tkáních inzulin‑dependentních, jakými jsou adipocyty, myocyty a kardiomyocyty. MIF má jak imunologické, tak endokrinní vlastnosti a mohl by hrát roli při rozvoji diabetu 2. i 1. typu [53].
Steatohepatitida vede k portálnímu zánětu. Podobně také byl nalezen vztah mezi mírnou portální endotoxemií, jaterním zánětem a zánětlivým postižením β buněk [25]. Podle některých recentních názorů by vysoce senzitivní CRP měl být vedle metabolického sledování (BMI, HbA1c, glykemie, cholesterolemie, HDL‑cholesterolemie, triacylglycerolemie, hladina adiponektinu) součástí monitorování diabetiků [37].
Poznatky o roli chronického mírného zánětu v patogenezi diabetu 2. typu již nyní vyústily v pokusy podávat protizánětlivou terapii při jeho léčbě. O salicylátech však bylo již od konce 19. století známo, že u diabetiků vedou k poklesu hyperglykemie [12]. Na začátku 20. století byl publikován podobný článek v anglosaském písemnictví [59]. Podobný názor publikovali Reid et al v polovině 50. let minulého století [45]. Salicyláty nejenže vedou k poklesu ranní glykemie, snižují i postprandiální hladinu glukózy a glykemii po i.v. podání glukózy [42]. K těmto efektům dochází nejen u diabetiků, ale zlepšení metabolického profilu včetně zlepšení inzulinové rezistence bylo nalezeno i u obézních po salsalátu, dimeru salicylové kyseliny [16]. Vliv salsalátu na snižování glykemie je více založen na zvýšení koncentrace inzulinemie než na zlepšení účinku inzulinu [28].
Další nálezy ve vztahu k zánětu v tukové tkáni
U obézních nemocných byly nalezeny vyšší hladiny cirkulujícího amyloidu [41]. Přestože tento amyloid není identický s amyloidem deponovaným v nervových buňkách, někteří autoři dávají obezitu, diabetes a Alzheimerovu chorobu přinejmenším do nepřímého vztahu [26]. Recentně publikovaná švédská studie potvrdila zvýšení rizika Alzheimerovy choroby u pacientů se špatně kontrolovaným diabetem [61].
Samozřejmě je zcela zásadní otázkou, jak se na zvýšeném výskytu Alzheimerovy choroby podílí zánětlivé a jak metabolické faktory. Ty jsou ostatně nepominutelné. Ve výše uvedené studii Irie et al představoval diabetes stejné riziko jako nosičství APOE-ε4, dobře známého metabolického rizika.
Závěr
Důkazy, které přináší teorie chronického mírného zánětu, představují další způsob interpretace vztahů mezi obezitou, metabolizmem a imunitními funkcemi či mezi adipocyty a buňkami zodpovědnými za imunitní funkce. Důsledky pro další buňky, zejména pro β buňky Langerhansových ostrůvků, endotelie a hepatocyty, snad i pro buňky nervové a pro orgány z těchto buněk složené, mohou významně posunout poznatky o patofyziologii metabolického syndromu včetně jejich důsledků, tedy aterosklerózy, jaterní steatózy a diabetu 2. typu. Současně tyto poznatky dávají naději na další principy cílené prevence i léčby nejrozšířenějších onemocnění v populaci rozvinutých zemí.
Práce vznikla díky výzkumnému záměru Univerzity Karlovy podporovanému MŠMT MSM 00216 208 14.
Doručeno do redakce: 18. 6. 2009
prof. MUDr. Michal Anděl, CSc.
www.lf3.cuni.cz
e‑mail: michal.andel@lf3.cuni.cz
Sources
1. Altannavch TS, Roubalová K, Kucera P et al. Effect of high glucose concentrations on expression of ELAM-1, VCAM‑1 and ICAM‑1 in HUVEC with and without cytokine activation. Physiol Res 2004; 53 : 77–82.
2. Anděl M, Kraml P, Dlouhý P et al. Patogeneze aterosklerózy – od metabolických změn k chronickému zánětu s aspekty střádavého onemocnění. Prak Lékař 1997; 77 : 266–274.
3. Anděl M, Tsevegjav A, Roubalová K et al. Infekční a zánětlivé faktory v patogenezi aterosklerózy. Vnitř Lék 2003; 49 : 960–966.
4. Avalos I, Rho YH, Chung CP et al. Atherosclerosis in rheumatoid arthritis and systemic lupus erythematosus. Clin Exp Rheumatol 2008; 26 (5 Suppl 51): S5–S13.
5. Bannerman DD, Goldblum SE. Direct effects of endotoxin on the endothelium: barrier function and injury. Lab Invest 1999; 79 : 1181–1199.
6. Bastard JP, Maachi M, Lagathu C et al. Recent advances in the relationship between obesity, inflammation, and insulin resistance. Eur Cytokine Netw 2006; 17 : 4–12.
7. Bednář B. Úvod do patologie. Praha: Avicenum 1982.
8. Bláha F. O patogenezi aterosklerózy. Čas Lék Čes 1958; 97 : 85–89.
9. Cinti S, Mitchell G, Barbatelli G et al. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J Lipid Res 2005; 46 : 2347–2355.
10. de Luca C, Olefsky JM. Inflammation and insulin resistance. FEBS Lett 2008; 582 : 97–105.
11. Donath MY, Schumann DM, Faulenbach M et al. Islet inflammation in type 2 diabetes: from metabolic stress to therapy. Diabetes Care 2008; 31 (Suppl 2): S161–S164.
12. Ebstein W. Therapie des Diabetes mellitus, insbesondere über die Anwendung des salicylsauren Natron bei demselben. Berl Klin Wochenschr 1897; 13 : 337–340.
13. Ehses JA, Böni-Schnetzler M, Faulenbach M et al. Macrophages, cytokines and beta‑cell death in Type 2 diabetes. Biochem Soc Trans 2008; 36 : 340–342.
14. Epstein SE, Zhou YF, Zhu J. Infection and atherosclerosis: emerging mechanistic paradigms. Circulation 1999; 100: e20–e28.
15. Farmer SR. Transcriptional control of adipocyte formation. Cell Metab 2006; 4 : 263–273.
16. Fleischman A, Shoelson SE, Bernier R et al. Salsalate improves glycaemia and inflammatory parameters in obese young adults. Diabetes care 2008; 31 : 289–294.
17. Festa A, Hanley AJ, Tracy RP et al. Inflammation in the prediabetic state is related to increased insulin resistance rather than decreased insulin secretion. Circulation 2003; 108 : 1822–1830.
18. Gregor MF, Hotamisligil GS. Thematic review series: Adipocyte Biology. Adipocyte stress: the endoplasmic reticulum and metabolic disease. J Lipid Res 2007; 48 : 1905–1914.
19. Haque WA, Shimomura I, Matsuzawa Y et al. Serum adiponectin and leptin levels in patients with lipodystrophies. J Clin Endocrinol Metab 2002; 87 : 2395.
20. Heilbronn LK, Campbell LV. Adipose tissue macrophages, low grade inflammation and insulin resistance in human obesity. Curr Pharm Des 2008; 14 : 1225–1230.
21. Heilbronn L, Smith SR, Ravussin E. Failure of fat cell proliferation, mitochondrial function and fat oxidation results in ectopic fat storage, insulin resistance and type II diabetes mellitus. Int J Obes Relat Metab Disord 2004; 28 (Suppl 4): S12–S21.
22. Hotamisligil GS. Inflammation and metabolic disorders. Nature 2006; 444 : 860–867.
23. Hotta K, Funahashi T, Arita Y et al. Plasma concentrations of a novel, adipose‑specific protein, adiponectin, in type 2 diabetic patients. Arterioscler Thromb Vasc Biol 2000; 20 : 1595–1599.
24. Hung CF, Huang TF, Chen BH et al. Lycopene inhibits TNF‑alpha‑induced endothelial ICAM‑1 expression and monocyte-endothelial adhesion. Eur J Pharmacol 2008; 586 : 275–282.
25. Hsieh PS, Chan JY, Shyu JF et al. Mild portal endotoxaemia induces subacute hepatic inflammation and pancreatic beta‑cell dysfunction in rats. Eur J Clin Invest 2008; 38 : 640–648.
26. Irie F, Fitzpatrick AL, Lopez OL et al. Enhanced risk for Alzheimer disease in persons with type 2 diabetes and APOE epsilon4: the Cardiovascular Health Study Cognition Study. Arch Neurol 2008; 65 : 89–93.
27. Kol A, Sukhova GK, Lichtman AH et al. Chlamydial heat shock protein 60 localizes in human atheroma and regulates macrophage tumor necrosis factor‑alpha and matrix metalloproteinase expression. Circulation 1998; 98 : 300–307.
28. Koska J, Ortega E, Bunt JC et al. The effect of salsalate on insulin action and glucose tolerance in obese non‑diabetic patients: result of a randomised double-blind placebo-controlled study. Diabetologia 2009; 52 : 385–393.
29. Kraml PJ, Roubalová K, Bulvas M. Markers of Chlamydia pneumoniae and human cytomegalovirus infection in patients with chronic peripheral vascular disease and their relation to inflammation, endothelial dysfunction and changes in lipid metabolism. Folia Microbiol (Praha) 2008; 53 : 551–557.
30. Lee CC, Adler AI, Sandhu MS et al. Association of C – reactive protein with type 2 diabetes: prospective analysis and meta‑analysis. Diabetologia 2009; 52 : 1040–1047.
31. Lee JY, Hwang DH. The modulation of inflammatory gene expression by lipids: mediation through Toll‑like receptors. Mol Cells 2006; 21 : 174–185.
32. Levine JA, Jensen MD, Eberhardt NL et al. Adipocyte macrophage colony-stimulating factor is a mediator of adipose tissue growth. J Clin Invest 1998; 101 : 1557–1564.
33. Maixner E. Patologie a terapie vnitřních nemocí, část prvá Nemoci srdce a cévstva. Praha: Bursík a Kohout 1912.
34. Matsuura E, Kobayashi K, Lopez LR. Atherosclerosis in autoimmune disease. Curr Rheumatol Rep 2009; 11 : 61–69.
35. Osborn O, Brownell SE, Sanchez-Alavez M et al. Treatment with an Interleukin 1 beta antibody improves glycemic control in diet‑induced obesity. Cytokine 2008; 44 : 141–148.
36. Ozcan U, Cao Q, Yilmaz E et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004; 306 : 457–461.
37. Pfützner A, Weber MM, Forst T. A biomarker concept for assessment of insulin resistance, beta‑cell function and chronic systemic inflammation in type 2 diabetes mellitus. Clin Lab 2008; 54 : 485–490.
38. Pickup JC, Crook MA. Is type II diabetes mellitus a disease of the innate immune system? Diabetologia 1998; 41 : 1241–1248.
39. Pittenger MF, Mackay AM, Beck SC et al. Multilineage potential of adult human mesenchymal stem cells. Science 1999; 284 : 143–147.
40. Plomgaard P, Bouzakri K, Krogh-Madsen R et al. Tumor necrosis factor‑alpha induces skeletal muscle insulin resistance in healthy human subjects via inhibition of Akt substrate 160 phosphorylation. Diabetes 2005; 54 : 2939–2945.
41. Poitou C, Viguerie N, Cancello R et al. Serum amyloid A: production by human white adipocyte and regulation by obesity and nutrition. Diabetologia 2005; 48 : 519–528.
42. Prince RL, Larkins RG, Alford FP. The effect of acetylsalicylic acid on plasma glucose and the response of glucose regulatory hormones to intravenous glucose and arginine in insulin treated diabetics and normal subjects. Metabolism 1981; 30 : 293–298.
43. Randle PJ, Garland PB, Hales CN et al. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1963; 13 : 785–789.
44. Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes 1988; 37 : 1595–1607.
45. Reid J, Macdougall AI, Andrews MM. Aspirin and diabetes mellitus. Br Med J 1957; 2 : 1071–1074.
46. Robertson RP, Harmon J, Tran PO et al. Beta‑cell glucose toxicity, lipotoxicity, and chronic oxidative stress in type 2 diabetes. Diabetes 2004; 53 (Suppl 1): S119–S124.
47. Rosen ED, MacDougald OA. Adipocyte differentiation from the inside out. Nat Rev Mol Cell Biol 2006; 7 : 885–896.
48. Saran AM, DuBose TD Jr. Cardiovascular disease in chronic kidney disease. Ther Adv Cardiovasc Dis 2008; 2 : 425–434.
49. Schrauwen P, Hesselink MK. Oxidative capacity, lipotoxicity, and mitochondrial damage in type 2 diabetes. Diabetes 2004; 53 : 1412–1417.
50. Simons PJ, van den Pangaart PS, Aerts JM et al. Pro‑inflammatory delipidizing cytokines reduce adiponectin secretion from human adipocytes without affecting adiponectin oligomerization. J Endokrinol 2007; 192 : 289–299.
51. Skurk T, Herder C, Kräft I et al. Production and release of macrophage migration inhibitory factor from human adipocytes. Endocrinology 2005; 146 : 1006–1011.
52. Thomayer J. Pathologie a a terapie nemocí vnitřních. 3. vydání. Praha: Bursík a Kohout 1909.
53. Toso C, Emamaullee JA, Merani S et al. The role of macrophage migration inhibitory factor on glucose metabolism and diabetes. Diabetologia 2008; 51 : 1937–1946.
54. van Harmelen V, Skurk T, Röhrig K et al. Effect of BMI and age on adipose tissue cellularity and differentiation capacity in women. Int J Obes Relat Metab Disord 2003; 27 : 889–895.
55. Vozarova B, Weyer C, Lindsay RS et al. High white blood cell count is associates with a worsening of insulin sensitivity and predicts the development of type 2 diabetes. Diabetes 2002; 51 : 455–461.
56. Wajchenberg BL. Beta‑cell failure in diabetes and preservation by clinical treatment. Endocr Rev 2007; 28 : 187–218.
57. Wang P, Mariman E, Renes J et al. The secretory function of adipocytes in the physiology of white adipose tissue. J Cell Physiol 2008; 216 : 3–13.
58. Weisberg SP, McCann D, Desai M et al. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 2003; 112 : 1796–1808.
59. Williamson RT. On the treatment of glycosuria and diabetes mellitus with sodium salicylate. Br Med J 1901; 1 : 760–762.
60. Wititsuwannakul D, Kim KH. Mechanism of palmityl coenzyme A inhibition of liver glycogen synthase. J Biol Chem 1977; 252 : 7812–7817.
61. Xu WL, von Strauss EW, Qiu CX et al. Uncontrolled diabetes increases the risk of Alzheimer disease: a population‑based cohort study. Diabetologia 2009; 52 : 1031–1039.
62. Zhu J, Quyyumi AA, Norman JE et al. Effects of total pathogen burden on coronary artery disease risk and C‑reactive protein levels. Am J Cardiol 2000; 85 : 140–146.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2009 Issue 7-8
Most read in this issue
- Metabolický syndrom a játra (NAFLD/NASH)
- Anxiózně depresivní poruchy a metabolický syndrom
- Chronický mírný zánět spojuje obezitu, metabolický syndrom, aterosklerózu a diabetes
- Laboratorní markery metabolického syndromu v praxi