
Chronická B-lymfatická leukemie a jí podobné stavy
B-cell chronic lymphocytic leukaemia and the similar states
B-cell chronic lymphocytic leukaemia and the similar diseases are seen predominantly in patients above the age 50 years, i.e. at the age when the patients also have other co-morbidities. The knowledge of these diseases on molecular level has improved significantly over the last decade. Molecular and biological prognostic factors are available in routine everyday practice. Assessment of these factors enables prediction of prognosis and, in some cases, also the response to therapy. The aim of the present review is to provide the medical community with the main information on this disease as patients with B-cell chronic lymphocytic leukaemia and similar disease states are of older age and very often suffer from a range of co-morbidities. Consequently, care for these patients involves physicians from various specialities. The aim of the following text is to present a clear overview of the basic information about this group of diseases that might be useful to all physicians who provide care to patients with B-cell chronic lymphocytic leukaemia and similar conditions. Since monoclonal immunoglobulin is sometimes identified in patients with these diseases, it is important to consider these conditions in the differential diagnosis of the states with the presence of monoclonal immunoglobulin.
Key words:
B-cell chronic lymphocytic leukaemia – monoclonal gammopathy – hairy cell leukaemia – prolymphocytic leukaemia – multiple myeloma
Authors:
M. Krejčí; Z. Adam; L. Pour; Y. Brychtová; J. Mayer; J. Vorlíček
Authors‘ workplace:
Interní hematoonkologická klinika Lékařské fakulty MU a FN Brno, pracoviště Bohunice, přednosta prof. MUDr Jiří Vorlíček, CSc.
Published in:
Vnitř Lék 2009; 55(9): 746-765
Category:
80th Birthday - prof. MUDr. Miloš Štejfa, DrSc., FESC
Overview
Chronická B-lymfatická leukemie a jí podobné stavy jsou dominantně nemoci věku nad 50 let, kdy obvykle nemocní mají již i další přítomné nemoci. Poznání těchto nemocí na molekulární úrovni se v posledním desetiletí výrazně prohloubilo. Molekulárně biologické prognostické faktory jsou již dostupné pro rutinní každodenní praxi. Jejich stanovení umožňuje předpovědět prognózu a někdy léčebnou odpověď. Cílem následujícího přehledu je předat informace o této nemoci široké lékařské veřejnosti, neboť pacienti s chronickou B-lymfatickou leukemií a s podobnými nemocemi jsou vyššího věku a mají velmi často četné další nemoci, takže na péči o jejich zdraví se podílejí lékaři vícero odborností. Cílem následujícího textu je přinést v přehledné formě základní informace o této skupině nemocí, které mohou být užitečné všem lékařům, kteří pečují o nemocné s chronickou B-lymfatickou leukemií a jí podobnými stavy. Vzhledem k tomu, že tyto nemoci někdy bývají provázeny přítomností monoklonálního imunoglobulinu, je nutné je vzít v potaz v diferenciální diagnostice přítomnosti monoklonálního imunoglobulinu.
Klíčová slova:
chronická B-lymfatická leukemie – monoklonální gamapatie – vlasatobuněčná leukemie – prolymfocytární leukemie – mnohočetný myelom
Chronická B-lymfatická leukemie
Definice
Chronická B-lymfatická leukemie je nízce agresivní (indolentní) lymfoproliferativní onemocnění, jehož podstatou je proliferace a akumulace klonálních, maligně transformovaných vyzrálých B-lymfocytů s charakteristickým imunofenotypem. Tyto lymfocyty nepodléhají apoptotické smrti tak rychle jako jejich fyziologické protějšky, takže postupný vzrůst počtu lymfocytů je způsoben nejen nekontrolovanou klonální proliferací, ale také poruchou jejich zániku (apoptózy). Na této skutečnosti se zřejmě spolupodílí zvýšená exprese genu bcl-2.
Plurální termín „chronické lymfatické leukemie“ se používá pro širší skupinu nemocí, které se projevují vyplavováním malých lymfocytů do periferní krve. Mimo B-lymfocytární leukemii patří do této skupiny ještě B - a T-prolymfocytární leukemie, vlasatobuněčná leukemie, splenický lymfom s vilózními lymfocyty a chronická lymfatická leukemie s velkými granulárními lymfocyty.
Chronická lymfatická leukemie (B-CLL) je nejčastější leukemií dospělých v Evropě a Severní Americe, kde tvoří 25–30 % všech leukemií. Její incidence je v Evropě 3/100 000. B-CLL se jen zřídka objevuje u lidí mladších 50 let. Medián věku stanovení diagnózy je 65 let, ale 20–30 % pacientů je mladších 60 let a 5–10 % pacientů je mladších 50 let. Obvyklá je až u starší populace, 81 % případů je diagnostikováno u osob starších 60 let.
Etiologické faktory a patogeneze
Nevíme, proč vzniká B-CLL tak často. Víme jen, že incidence B-CLL je vyšší u pracovníků v zemědělství a u lidí, kteří jsou v profesionálním kontaktu s azbestem a chemickými rozpouštědly. Na rozdíl od jiných typů leukemií není incidence B-CLL zvýšena u lidí vystavených radioaktivnímu záření a působení alkylačních cytostatik. Ačkoliv lymfoidní maligní choroby jsou časté u lidí s imunodeficiencí, u B-CLL tato souvislost není. Souvislost vzniku B-CLL s infekcí některým ze známých virů nebyla rovněž prokázána. Epidemiologické studie ukazují, že riziko vzniku CLL nebo jiných nádorů je vyšší u pokrevních příbuzných pacientů s CLL a jen některé epidemiologické studie prokázaly, že u potomků nemocných CLL se případná CLL manifestuje o 10–20 let dříve, než vznikla u jejich rodičů. S tím zřejmě souvisí nález monoklonální B-lymfocytózy, která byla popsána u 15–57 % zdravých potomků první linie, z toho u 13–18 % z nich měla monoklonální B-lymfocytóza imunofenotyp shodný s B-CLL.
V patogenezi choroby se uplatňuje opět gen bcl-2, jehož zvýšená exprese je typická pro folikulární lymfomy. Patologické buňky B-CLL běžně nemívají uvedenou translokaci 14;18 jako folikulární lymfomy. Ke zvýšené expresi genu bcl-2 dochází jiným mechanizmem. Výsledkem této zvýšené exprese genu bcl-2 je inhibice apoptotické smrti patologických lymfocytů, a tedy jejich dlouhověkost. Vzrůstající počet lymfocytů je zapříčiněn neschopností těchto buněk podstoupit fyziologickou cestu apoptotického zániku, a nikoliv rychlou proliferací maligních buněk.
Do nedávné doby se původ těchto patologických buněk odvozoval od naivních pregerminálních CD5+ buněk. Analýza mutace variabilní části imunoglobulinového genu prokázala, že asi polovina případů B-CLL má přítomnu somatickou mutaci, což znamená, že tyto buňky mají původ v paměťových B buňkách (memory B cells), které po kontaktu s antigenem prošly germinálním centrem. Přítomnost či nepřítomnost somatické mutace je spojená s určitými genetickými změnami, s expresí znaku CD38 a s prognózou.
Prognóza pacientů s buňkami B-CLL typu paměťových buněk majících často 13q14 deleci a nižší procento CD38+ je příznivá.
Naproti tomu prognóza pacientů s buňkami B-CLL typu naivních B buněk (nemutovaná variabilní část imunoglobulinového genu) majících často trizomii chromozomu 12 a vyšší zastoupení CD38+ buněk je horší, jejich choroba obvykle rychle progreduje.
Příznaky a průběh nemoci
Tato diagnóza je stanovována často (asi v 50 %) zcela náhodně při vyšetření krevního obrazu z jiné indikace než pro podezření na maligní krevní onemocnění, většinou v rámci vyšetření pro jinou chorobu či v rámci preventivní prohlídky. Některé formy nemoci mohou mít totiž dlouhé, roky trvající, bezpříznakové období, kdy jediným znakem nemoci je lymfocytóza, jak v periferním krevním obraze, tak i v kostní dřeni. Ojediněle byly popsány i dlouhodobé spontánní remise.
Jiné formy naopak mohou mít průběh poměrně agresivní, kde se stav nemocného rychle zhoršuje i přes odpovídající léčbu, proto některé nemocné přivede ke stanovení diagnózy vyšetření pro B symptomy (noční pocení, úbytek hmotnosti, teploty).
Klinické příznaky jsou shodné s příznaky maligních lymfomů. Asi 15 % nemocných má při stanovení diagnózy klasické B symptomy.
Lymfadenopatie
Pro B-CLL je typické generalizované zvětšení uzlin a sleziny, v pokročilých stadiích pak infiltrace nelymfatických orgánů (prostaty, jater, pohrudnice). Lymfadenopatie je obvykle již zpočátku generalizovaná, postihuje uzliny krku, axil i třísel. Jejich velikost však nebývá zpočátku nijak extrémní, pohybuje se kolem 2–3 cm, nápadná je však jejich mnohočetnost. Výrazná lymfadenopatie v mediastinu není pro B-CLL typická, stejně není pro počátečné formu B-CLL typická velká abdominální lymfadenopatie, ačkoliv postižení retroperitoneálních uzlin je prokazatelné u 50 % pacientů.
Masivní izolované zvětšení uzlin v kterékoliv lokalizaci je vždy podezřelé z transformace v lymfom vyššího stupně malignity (Richterův syndrom).
Splenomegalie a hepatosplenomegalie
Někteří pacienti mají splenomegalickou formu, u níž je nápadná velmi zvětšená slezina při žádném nebo jen nepatrném zvětšení uzlin. Mírně zvětšená slezina je však nalézána minimálně u 50 % nemocných již v počátku nemoci. Splenomegalie může někdy způsobovat abdominální dyskomfort, pocit sytosti po malé porci jídla a může vyústit v hypersplenizmus, a přispívat tak k anémii. Hepatomegalie je méně častá.
Infiltrace mimouzlinových tkání
Extranodální infiltrace může být odhalena histologickým vyšetřením tkání, málokdy však vyvolává symptomy. Nejčastěji se udává infiltrace kůže a tonzil, zatímco infiltrace zažívacího traktu, plíce, pleury, CNS, ledvin či kostí je vzácné.
Může vzniknout infiltrace prostaty s typickými močovými příznaky. Byla popsána i infiltrace ledvin, která však nemívá souvislost s nefrotickým syndromem pozorovaným u některých pacientů. Infiltrace gastrointestinálního traktu nebo kůže je výjimečná. Vznik infiltrace v CNS je zcela raritní. U některých pacientů s B-CLL byla však pozorována exfoliativní dermatitida nejasného původu.
Celkové klinické příznaky
Celkové klinické příznaky se objevují až při pokročilém onemocnění, při větší mase maligních buněk. Jsou způsobeny cytokiny, produkovanými buňkami B-CLL nebo autoimunitním mechanizmem. Typickými příznaky jsou: teploty neinfekčního původu, zvýšené (noční) pocení, úbytek na váze až kachektizace, anémie a trombocytopenie s krvácivými příznaky. Pokud neinfekční teplota přesahuje 38 °C nebo je výrazné noční pocení s nutností výměny prádla a případně váhový úbytek přesáhl za 6 měsíců 10 % původní váhy, označujeme tyto projevy jako B příznaky, podobně jako u Hodgkinovy nemoci. Pacienti s B-CLL mají dále zvýšenou náchylnost k infekcím, obzvláště k infekcím plic.
Anémie a další cytopenie
Chudokrevnost je snad nejčastější komplikací chronické lymfatické leukemie. Před zahájením její léčby je nutno pečlivě analyzovat její etiologii.
Autoimunní hemolytická anémie: protilátky proti erytrocytům (pozitivní Coombsův test) se objeví během nemoci u 10–25 % pacientů, hemolytická anémie se manifestuje méně často. Ikterus je tedy nejčastějším projevem autoimunitní hemolytické anémie.
V literatuře lze nalézt zprávy o současném výskytu B-CLL a systémového lupusu, revmatoidní artritidy, Sjögrenova syndromu, ulcerózní kolitidy, alergické vaskulitidy, perniciózní anémie a bulózního pemfigoidu. Výskyt těchto komplikujících autoimunitních nemocí se považuje za čistě náhodný na rozdíl od hemolytické anémie, která s touto nemocí souvisí.
Čistá aplázie červené řady (pure red cell aplasia): tato příčina anémie postihuje asi 6 % pacientů s B-CLL. Hematokrit klesá pod 21 %, retikulocyty pod 0,5 ‰, je normální počet trombocytů a neutrofilů v periferním krevním obraze. V kostní dřeni chybí normoblasty a přitom je normální počet megakaryocytů a myeloidních prekurzorů. Laboratorní zkoušky in vitro prokázaly, že T-lymfocyty těchto pacientů inhibují proliferaci erytroidních prekurzorů CFU-E a BFU-E normální kostní dřeně. Léčebným přístupem u těchto nemocných může být jak cytostatická léčba, redukující i T-lymfocyty, tak monoterapie cyklosporinem A, zaměřená jen na T-lymfocyty.
Anémie způsobená infekcí parvovirem B19: anémie způsobená parvovirem je vzácnější než „pure red cell aplasia“. Klinické a laboratorní příznaky v periferní krvi i kostní dřeni jsou shodné s výše uvedenou diagnózou. Diagnózu lze určit jedině detekcí parvoviru B19 metodou polymerázové řetězové reakce.
Anémie jako projev pokročilého onemocnění: anémie může být také projevem pokročilé choroby. Je způsobena jak cytokiny produkovanými maligními buňkami B-CLL, tak i mechanickým útlakem kostní dřeně maligními buňkami. Přispívá k ní i redukce kmenových krvetvorných buněk předchozí léčbou. U těchto nemocných je obvykle absolutně či relativně nedostatečná tvorba endogenního erytropoetinu.
Trombocytopenie: etiologie trombocytopenie kopíruje etiologii anémie. V podstatě je nutno se rozlišit:
- autoimunitní trombocytopenii s pro-tilátkami proti trombocytům a dostatečným počtem megakaryocytů v kostní dřeni
- trombocytopenii způsobenou tlumivým vlivem masivní lymfocytární infiltrace kostní dřeně na tvorbu trombocytů s velmi malým počtem megakaryocytů v kostní dřeni
Incidence autoimunitní trombocytopenie u B-CLL se pohybuje kolem 2 %. Pokud jsou přítomny protilátky proti trombocytům, jsou opět lékem volby steroidy či cytostatika, při neúspěchu splenektomie. V druhém případě lze zkusit opět cytostatika a steroidy k redukci choroby. Jediný rozdíl je v tom, že v případě autoimunitní etiologie se podávají steroidy kontinuálně, v případě trombocytopenie způsobené útlakem fyziologické krvetvorby masou patologických lymfocytů se steroidy podávají jen nárazově.
Imunosuprese
Pokles aktivity protilátkové imunity – hypogamaglobulinemie
Buňky B-CLL produkují cytokin TGFb (tumour growth factor b), který inhibuje proliferaci fyziologických B-lymfocytů. Proto pro nemocné s pokročilou chorobou je v klasickém elektroforetickém vyšetření typická nízká frakce gamaglobulinů a při kvantitativním vyšetření nízká koncentrace jednotlivých imunoglobulinů. První klesá koncentrace imunoglobulinu (Ig) A, následně pak IgM a IgG. Nízká koncentrace imunoglobulinů souvisí s nízkými hodnotami sedimentace erytrocytů. Plazmatická hladina imunoglobulinů odráží vývoj nemoci a je východiskem pro rozhodování o jejich léčebném podání při infekci.
Pokles aktivity T-buněčné imunity
Pacienti s B-CLL mají i při počátečním dostatečném počtu cirkulujících T-lymfocytů deficit T-buněčné imunity, neboť funkce všech T-lymfocytů je porušena. Je snížena aktivita NK (natural killer) buněk, LAK (lymphokine activated killer) buněk a také je snížená cytotoxická aktivita závislá na protilátkách. Pokles buněčné aktivity je příčinou častých virových chorob. Incidence manifestního onemocnění viry herpes simplex a herpes zoster dosahuje u těchto pacientů 30 %. Je důležité informovat pacienty o nutnosti okamžitého podání protivirových léků při prvních známkách infekce.
Snížený absolutní počet neutrofilů
Při progresi nemoci klesá absolutní počet neutrofilů, což spolu s hypogamaglobulinemií snižuje obranyschopnost proti bakteriálním infekcím.
Infekční komplikace
Vzhledem k výše uvedeným poruchám imunity je nyní pochopitelné, proč pacienti s touto nemocí často získávají běžné (pásový opar, pneumonie) i oportunní infekce (Pneumocystis carinii) a proč právě infekce jsou nejčastější příčinou smrti těchto nemocných.
Zvýšení viskozity krve vlivem lymfocytů
Počet lymfocytů má vliv na viskozitu krve. Poruchy prokrvení se však objevují až při značně vysokých počtech lymfocytů (500–1 000 × 109/l), zatímco např. u chronické myeloidní leukemie se kvůli větší velikosti krvinek klinické příznaky zvýšené viskozity objeví již při počtu leukocytů přesahujícím 100 × 109/l.
Transformace v lymfoproliferativní onemocnění vyšší malignity
Přibližně u 3–5 % nemocných dojde v průběhu nemoci k transformaci v maligní lymfom vyšší malignity, obvykle difuzní velkobuněčný lymfom. Klinickými příznaky této změny jsou: vzestup teplot, vzestup aktivity laktátdehydrogenázy (LDH), asymetrická lymfadenopatie a hyperkalcemie. Pro uvedenou transformaci se někdy používá název Richterův syndrom.
Možná je i transformace v prolymfocytární leukemii se změnou morfologie lymfocytů a agresivnějším průběhem choroby.
Přítomnost monoklonálního imunoglobulinu
Monoklonální imunoglobulin (M-Ig) bývá nalézán asi u 5–10 % pacientů s B-CLL. Četnost jeho záchytu závisí na citlivosti metody. Přítomný M-Ig může poškozovat organizmus. Např. může vyvolat glomerulonefritidu a nefrotický syndrom či rušivě zasahovat do koagulační kaskády a do funkce trombocytů. Léčili jsme však i nemocnou, která měla v kostní dřeni souběh dvou maligních klonů odpovídajících CLL a druhá mnohočetnému myelomu. Tato paní měla četné osteolytické fraktury odpovídající mnohočetnému myelomu.
Projevy progrese nemoci do terminálního stadia
V terminálních fázích nemoci se objevuje výrazný úbytek hmotnosti, hlavně svalové hmoty. Příčinou je jednak snížená chuť k jídlu, ale také katabolizmus navozený maligní chorobou, který vede k úbytku svalové hmoty se všemi důsledky, které s tím souvisejí.
Diagnóza a diferenciální diagnóza
Diagnóza chronické lymfatické B-leu-kemie se dříve odvíjela od početního a morfologického nálezu v periferní krvi a kostní dřeni. Tato kritéria lze i nynípoužít, pokud není k dispozici imunofenotypizace, která diagnostický proces podstatně zpřesňuje.
Chronická B-lymfocytární leukemie je definována imunofenotypem lymfocytů. Pro stanovení diagnózy B-CLL je požadováno nejméně 5 × 109/l lymfocytů v periferní krvi uniformní morfologie malých vyzrálých lymfocytů. Pokud se imunofenotyp a morfologie těchto lymfocytů shoduje s diagnózou B-CLL, není nutné vyšetření kostní dřeně pro potvrzení diagnózy (schéma 1).

Morfologická subtypizace. Běžné je, že část lymfocytů bývá atypických, větších, s naštípnutým jádrem, a dále lymfocytů morfologie prolymfocytů. Počet typických lymfocytů však musí být větší než 55 %. Přítomnost více než 55 % prolymfocytů je již kritériem prolymfocytární leukemie (PL).
Histologie uzliny infiltrované buňkami B-CLL je shodná s nozologickou jednotkou lymfocytární lymfom. Na B-CLL lze také pohlížet jako na lymfocytární lymfom s vyplavováním do periferní krve a infiltrací kostní dřeně. Pokud je histologicky potvrzená diagnóza lymfocytárního lymfomu a pacient má patologickou lymfocytózu v periferní krvi, jde o B-CLL. I v tomto případě je vhodné imunofenotypizační potvrzení diagnózy.
Imunofenotyp B-CLL
Pro CLL je diagnostická lymfocytóza v periferní krvi přesahující počet 5 × 109/l s přítomností typického fenotypu (CD5+ CD19+ CD23+ FMC7 – se slabou expresí CD 22 a CD79b) a s přítomností slabě exprimovaného povrchového imunoglobulinu.
Stanovení diagnózy je tedy závislé na použití průtokové cytometrie. Vyšetření kostní dřeně v případě stanovení diagnózy z periferní krve není nezbytné, je však možné imuhohistochemickým vyšetřením kostní dřeně potvrdit diagnózu CLL. Odběr kostní dřeně umožní vyšetřit další prognostické faktory. Podrobně jsou diagnostická kritéria uvedena v tab. 1.
![Diagnostická kritéria pro stanovení diagnózy B-CLL a základní vyšetření prováděná u této choroby [32,33].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/a031d441ab7756f30c928c66a6423bb4.png)
V rámci diferenciální diagnózy lymfocytózy je nutno zvažovat další jednotky chronicky probíhající lymfoproliferace, které mohou vyplavovat lymfocyty do cirkulace. Diferenciálně diagnosticky je nutné odlišit vzácnější typy chronických lymfatických leukemií, T - a B-prolymfocytární leukemii, leukemii z velkých granulárních lymfocytů a vlasatobuněčnou leukemii (tab. 2). Důležité je také odlišit leukemizovanou formu lymfomu z buněk plášťových buněk (mantle cell lymphoma), jehož buňky by neměly exprimovat CD23, a naopak by měly vykazovat zvýšenou expresi genu Bcl-1 a proteinu, který kóduje.
![Diferenciální diagnostika indolentních lymfoproliferativních chorob [34].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/fcd68bc64d01c50b81b0fd6edd830ceb.png)
Pro buňky B-CLL je typická přítomnost povrchového imunoglobulinu (sIg), intenzita jeho exprese je ale slabá. Nejčastěji jde o sIgM nebo sIgD. Cytoplazmatický imunoglobulin (cIg) bývá nalézán nepravidelně. Pravidelně jsou nacházeny společné znaky B lymfocytární řady: CD19, CD20, CD24, CD37, CD40, CD45RA a znaky II. třídy HLA systému. Antigen CD10, přítomný během pre-B vývoje a během folikulární fáze, nebývá u B-CLL nalézán, stejně jako antigen CD34, typický pro pluripotentní buňky. Antigen CD9, přítomný na normálních pre-B lymfocytech a subpopulaci aktivovaných B buněk, je nalézán jen u < 20 % případů. Antigeny plazmatických buněk CD38 a CD138 (Syndekan 1, BB4) se na buňkách B-CLL nenacházejí. Pro buňky B-CLL je typická přítomnost CD5 antigenu, který se fyziologicky nachází jen na T-lymfocytech a na tymocytech. Mimo buňky B-CLL je antigen CD5 nalézán i na buňkách lymfomu plášťové zóny (mantle cell lymphoma). Zcela vzácné jsou formy B-CLL s negativitou CD5. U nich se popisuje mírně odlišný klinický průběh, dominance velké splenomegalie bez lymfadenopatie a kratší přežití.
Druhým typickým znakem buněk B-CLL je přítomnost antigenu CD23, což je nízkoafinitní Fc-IgE receptor. Téměř vždy je přítomen na buňkách B-CLL a jen výjimečně je nalézán na jiných maligních B-lymfocytech. Antigen CD22 se v případě buněk B-CLL nachází převážně intracelulárně a jeho přítomnost na jejich povrchu je slabá nebo žádná. U dalších B-lymfatických maligních chorob se CD22 nachází jak na povrchu, tak v cytoplazmě.
Antigen FMC7 je přítomen výjimečně na buňkách B-CLL (< 4 %), ale je pravidelně nacházen na buňkách prolymfocytární nebo vlasatobuněčné leukemie. Antigen CD1c exprimuje 50 % fyziologických B-lymfocytů, u buněk CLL je výjimečný. Častěji je nacházen u dalších maligních lymfoproliferací. Antigen CD21 je přítomen na povrchu buněk B-CLL ve velmi nízké denzitě. Antigen CD35 (receptor pro komplement) je přítomný na normálních B-lymfocytech, ale není na buňkách B-CLL. Antigen CD79b (SN8) je u B-CLL obvykle negativní, u ostatních lymfocytárních leukemií je pozitivní.
Diferenciální diagnóza
Některé virové infekce mohou způsobit vzestup počtu lymfocytů na hodnoty shodné s počínající formou B-CLL. Lymfocytóza může provázet EB virózu, infekci cytomegalovirem, HIV, rubeolu, varicelu, adenovirózu, infekční lymfocytózu, hepatitidy a některé bakteriální infekce, brucelózu, černý kašel nebo také infekci Toxoplazma gondii.
Nejen B-CLL, i jiné maligní B-lymfoproliferace mohou přejít do leukemické fáze se zvýšeným počtem patologických lymfocytů v periferní krvi. Jedná se o následující jednotky: prolymfocytární leukemie, vlasatobuněčná leukemie, splenický lymfom s vilózními lymfocyty, folikulární lymfomy, lymfom plášťové zóny (mantle cell lymphoma). Diskutovanou a některými autory zpochybňovanou jednotkou je CD5 negativní B-CLL. Morfologicky se mohou buňkám B-CLL podobat buňky některé formy T-lymfatické leukemie: T-prolymfocytární leukemie, vlasatobuněčná leukemie, T-leukemie z velkých granulárních lymfocytů (large granular lymphocytic leukeamia). V diferenciální diagnóze pomůže morfologie s odpovídajícím barvením a s hodnocením ve světelném, imunofluorescenčním nebo elektronovém mikroskopu a imunofenotypizační vyšetření.
Prognostické faktory
Pro klasifikaci stupně pokročilosti choroby a velikosti masy maligních buněk se používají kritéria, která definovali Rai a Binet. Stanovení klinického stadia dle uvedených kritérií má prognostický význam (tab. 3, 4).

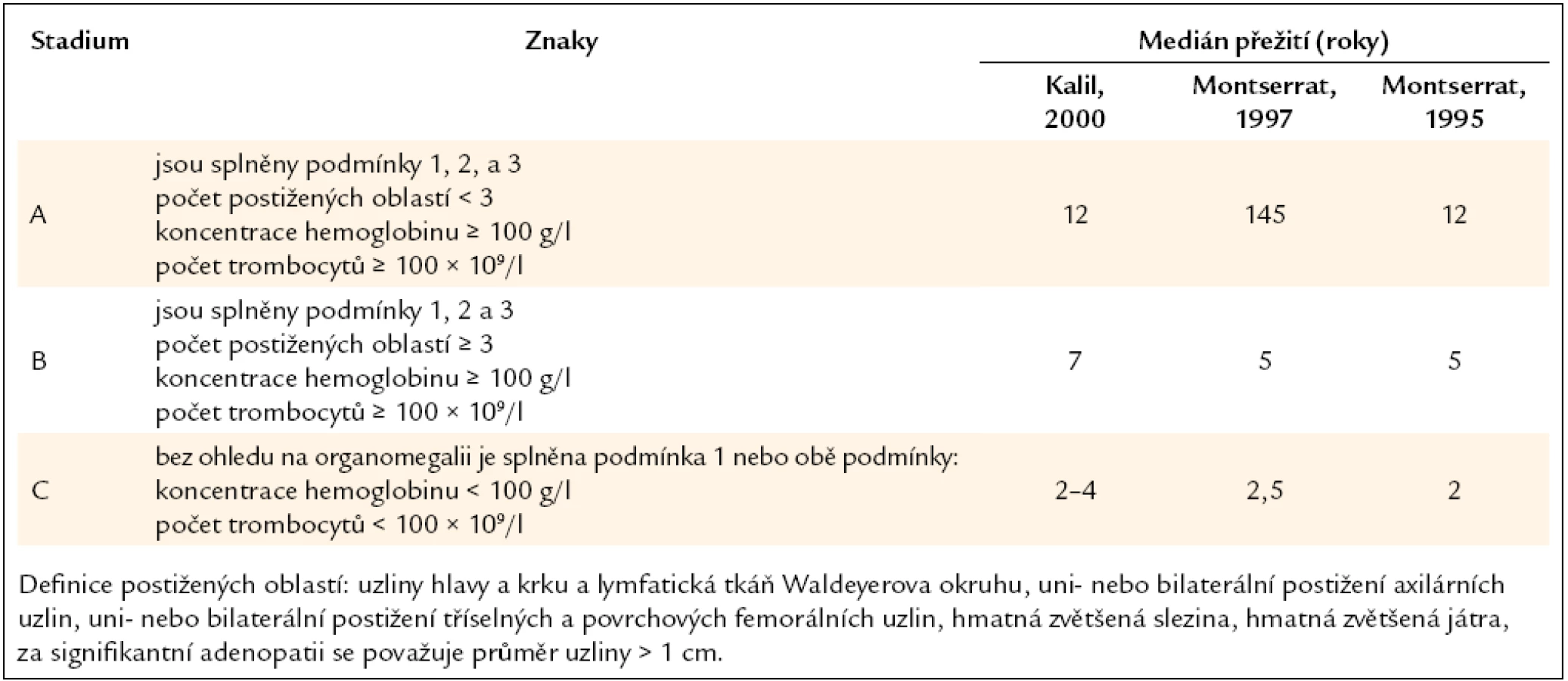
Ke starším a stále platným prognostickým parametrům patří zdvojovací čas počtu lymfocytů kratší než 12 měsíců, zvýšená koncentrace b2-mikroglobulinu, zvýšená aktivita sérové tymidinkinázy, zvýšená sérová hladina solubilního CD23 a masivní infiltrace kostní dřeně.
V roce 2007 jsou standardně používány následující prognostické markery:
- sérové markery: aktivita tymidinkinázy, solubilní sCD23, b2-mikroglobulin
- FISH cytogenetické vyšetření
- IgVH mutační status
- V3.21 exprese
- CD38 exprese
- ZAP-70 exprese
Mutační status – přestavba genetické informace pro variabilní části těžkého řetězce imunoglobulinu IgVH
V roce 1999 bylo 2 nezávislými týmy zjištěno, že B-CLL lze v podstatě rozdělit na dva typy, na B-CLL vzniklou z lymfocytů prošlých zárodečným (germinálním) centrem s příznivým průběhem a na B-CLL vzniklou z naivních (pregerminálních) B-lymfocytů, která probíhá obvykle velmi agresivně.
Tzv. naivní lymfocyty, které neprošlygerminálním centrem, mají nemutova-nou informaci pro těžký řetězec imunoglobulinu, zatímco lymfocyty prošlé zárodečním centrem již mají tuto genetickou informaci mutovanou. Prognózu v závislosti na mutačním statusu znázorňuje graf 1. Zjištění mutačního stavu má signifikantní prognostickou informaci nezávislou na cytogenetice a klinickém stadiu. Výjimkou je však exprese genu z variabilní části označeného jako V3-21, který signalizuje nepříznivý průběh.
![Délka přežití v závislosti na mutované či nemutované genetické informaci pro těžký řetězec imunoglobuloinu (IgV<sub>H</sub> genu) [37].](https://pl-master.mdcdn.cz/media/image/fb22ef69a611a8d986a702361981af17.png?version=1537790346)
Průkaz mutace je však relativně obtížný, a tak se hledají náhradní, snáze prokazatelné prognostické parametry, které by korelovaly s mutačním stavem genetické informace pro těžký řetězec imunoglobulinu.
Hledání zástupných ukazatelů mutačního stavu – ZAP-70 a CD38 antigenu
Přítomnost zeta asociovaného proteinu (ZAP-70) je spojena s agresivnějším průběhem nemoci, je tedy nepříznivým prognostickým faktorem. Protein ZAP-70 byl detekován u většiny nemutovaných B-CLL. Neplatí to však absolutně, v 6–29 % je exprese proteinu ZAP diskordantní k IgVH mutačnímu stavu (graf 2).
![Kaplan-Meierova křivka přežití v závislosti na expresi znaku ZAP-70 [38].](https://pl-master.mdcdn.cz/media/image/f04d4146c654e3607877a77f04f0c323.png?version=1537790346)
Vysoká exprese antigenu CD38 signalizuje agresivnější průběh a je také častěji detekována u nemutovaných forem B-CLL (graf 3, 4). Exprese CD38 se mění v průběhu nemoci a je nutné opakovat její vyšetření při nové progresi.
![Přežití v závislosti na mutaci IgV<sub>H</sub> genu a na CD38 expresi [39].](https://pl-master.mdcdn.cz/media/image/553f9d1e37de5c5e57f468b80bd75f3b.png?version=1537790346)
![Vliv mutace genu p53 na přežití u pacientů s B-CLL [40].](https://pl-master.mdcdn.cz/media/image/b7e387eb705b5dd4071cec54662bbd02.png?version=1537790346)
Cytogenetické vyšetření
Cytogenetické vyšetření přináší také důležité informace o prognóze nemocných a o jejich odpovědi na léčbu. Kromě klasického vyšetření karyotypu je nutné vyšetření metodou fluorescenční in situ hybridizace (FISH), které má potenciál detekovat cytogenetickou abnormalitu u 90 % vyšetřených.
Medián přežití pacientů s delecí (17p), delecí (11q), trisomií (12q), dále s normálním karyotyopem a delecí (13q) jako jedinou aberací byl 32, 79, 114, 111 a 133 měsíců.
Incidence cytogenetických změn, kteréjsou dnes považovány za prognosticky signifikantní, se pohybuje v následujících číslech:
- delece (13q) (solitární abnormalita) je nalézána ve 14–40 %
- delece (11q) je popisována v 10–32 %
- delece (12q+ ) v 11–18 %
- delece (17p) ve 3–27 %
- delece (6q) ve 2–9 %
Vliv cytogenetického nálezu na délku přežití ukazuje graf 5.
- Pacienti s delecí (13q), jako jedinou genetickou abnormalitou, mají excelentní prognózu s mediánem přežití 133 měsíců.
- Trisomie 12 je spojena s atypickou morfologií a imunofenotypem.
- Delece 6q je častější u případů, jejichž lymfocyty mají plazmocytoidní morfologii a koreluje s intermediární prognózou.
- Delece 11q, související se ztrátou ná-dorového supresoru ATM, se častějivyskytuje u mužů, bývá provázena ma-sivní lymfadenopatií (bulky disease), medián přežití je 79 měsíců.
- Nález (17p–), související se ztrátou nádorového supresoru p53, a pacienti s nálezem (17p–) mají nízký počet léčebných odpovědí a krátké přežití, medián 32 měsíců, proto je u nich na zvážení jiná iniciální léčba (alemtuzumab, alogenní transplantace), než je podávána ostatním pacientům.
![Prognostický význam cytogenetiky u B-CLL [41].](https://pl-master.mdcdn.cz/media/image/b4276232680e4a2dbf1972bbc1461505.png?version=1537790346)
Velmi nepříznivou prognostickou in-formaci přináší průkaz mutace a/neboztráty genu p53. Mutace genu p53 jsou recesivní, a proto musí většinou dojít k inaktivaci obou alel genu p53. Nejčastějším mechanizmem inaktivace první alely jsou bodové mutace, které lze vyšetřit sekvenací DNA genu p53 nebo funkční analýzou FASAY. Druhá alela p53 bývá nejčastěji kompletně deletována, a proto je vhodnou metodou FISH analyzující ztrátu p53 specifického lokusu 17p13. Cytogenetický nález a funkční stav p53 se mohou měnit v průběhu onemocnění, a je proto vhodné jejich opakované vyšetření při každé progresi nemoci nebo při vzniku rezistence v průběhu terapie.
Education textbook Americké hematologické společnosti uvádí dvě zásadní kategorie pacientů dle cytogenetických nálezů:
- skupina s nízkým rizikem: normální karyotyp, nebo izolovaná delece 13q
- skupina se vysokým rizikem del(17p) nebo del(11q) nebo trisomie 12; tito pacienti mívají agresivní průběh nemoci, avšak na rozdíl od pacientů s del(17p) a del(11q) buňky CLL obsahující trisomii 12 obvykle reagují na léčbu fludarbinovým režimem a jejich přežití je delší než v případě předchozích dvou skupin
Dle multivariantních analýz představuje nezávislý nepříznivý prognostický faktor:
- nemutovaný stav těžkého řetězce imunoglobulinu
- delece (17p)
- delece (11q)
- vyšší biologický věk
- vysoký počet lymfocytů
- zvýšená hodnota LD
Pacienti s těmito nepříznivými faktory mají podstatně kratší celkové trvání léčebné odpovědi i celkové přežití.
Uvedená prognostická vyšetření majípraktický význam pouze u mladších pa-cientů v dobrém stavu, u nichž je možnérozhodování mezi agresivní léčbou zahrnující alogenní transplantaci a klasickou chemoterapii.
Léčba
Indikace k zahájení léčby
Pro zahájení léčby jsou přijata přesná pravidla, která lze obecně vyjádřit – nemoc se léčí, až se začne projevovat klinickými příznaky, podobně jako folikulární lymfom.
Za indikaci k léčbě lze považovat:
- B symptomy
- progresivně se zvětšující lymfatické uzliny nebo hepatosplenomegalie způsobující klinické potíže
- pokles hemoglobinu pod 100 g/l anebo pokles trombocytů pod 100 × 109/l v důsledku narůstající infiltrace kostnídřeně
- autoimunitní trombocytopenie či ané-mie nereagující na glukokortikoidy
Podrobněji jsou indikace k léčbě shrnuty v tab. 5.

Cílem léčby je vždy navodit kompletní remisi, to znamená normalizaci hodnot krevního obrazu a normalizaci velikosti lymfatických uzlin, jater a sleziny. Po dosažení remise se léčba přeruší a pacient je dále jenom pravidelně kontrolován řádově v měsíčních intervalech. Cílem kontrol je však rozpoznat případný relaps nemoci, který je důvodem k obnovení léčby této nemoci s cílem dosáhnout další remise.
Délka dosažených remisí se postupně zkracuje. V průběhu nemoci může dojít k transformaci v agresivnější typ lymfomu nebo se nemoc postupně stane rezistentní na léčbu a pacienta zahubí.
V případě mladších nemocných s nepříznivou formou B-CLL je možné zvážit provedení alogenní transplantace již jako první léčbu této nemoci nebo jako léčbu časného relapsu. V ostatních případech se používá medikamentózní léčba.
Terminální stavy pacientů s B-CLL jsou charakterické výraznou kachexií se svalovou atrofií a četnými atakami běžných i oportunních infekcí, z nichž jedna se nakonec stane příčinou úmrtí. Proto při zřetelném selhání protinádorové léčby by měla dominovat paliativní a symptomatická léčba.
Význam glukokortikodů
Glukokortikoidy indukují v jádře některých fyziologických, ale i patologických lymfocytů apoptotickou smrt buňky. Účinnost glukokortikoidů je však u B-CLL omezená. Monoterapie glukokortikoidy dosahuje jen 10 % remisí, což je podstatně slabší léčebný účinek, než jaký mají glukokortikoidy např. u mnohočetného myelomu nebo u akutní lymfatické leukemie. Prednison přidaný k monoterapii alkylačními cytostatiky zvýší počet léčebných odpovědí, neprodlouží však přežití. Obvykle se proto při základní léčbě chlorambucilem podává prednison v dávce 1–2 mg/kg váhy po dobu 1 týdne v měsíčních intervalech.
Vysoké dávky glukokortikoidů jsou používány v terminálních fázích nemoci, kdy je fyziologická krvetvorba značně utlačena lymfoidní infiltrací. Omezená krvetvorná kapacita limituje další chemoterapii a vysoké dávky glukokortikoidů zde představují alternativu léčby, která nepoškodí zbývající krvetvorbu a která může redukovat počet patologických lymfocytů. Používají se vysoké dávky metylprednisolonu (250 mg/m2 po 5 dní ve 14–30denních intervalech) nebo dexametazonu (40 mg/den, 1.–4., 10.–13. a 20.–23. den v 28–30denních cyklech).
Léčba autoimunitní cytopenie
Kontinuální dlouhodobé podávání prednisonu je oprávněné pouze u nemocných s hemolytickou anémií. Zde má tato léčba za cíl nejen brzdit rozvoj chronické lymfatické leukemie a tvorbu protilátek, ale také inhibovat destrukci erytrocytů potažených protilátkou buňkami retikuloendoteliálního systému. V této indikaci se obvykle používá prednison 40 mg/m2 po dobu 14 dní a pak se dávka postupně snižuje a hledá se nejnižší, dostatečně účinná dávka pro dlouhodobou léčbu. Při neúspěchu se prednison kombinuje s cyklofosfamidem nebo azatioprinem. Lze zkusit i velké dávky gamaglobulinů, 0,4 g/kg/den po 5 dnů. Při neúčinnosti medikamentózní léčby přichází na řadu splenektomie. Někteří autoři v této indikaci uvádí i cyklosporin A.
Alkylační cytostatika a kombinovaná chemoterapie obsahující antracykliny
Alkylační cytostatika byla prvním léčebným prostředkem s potenciálem navodit remisi nemoci. Nejčastěji je používán chlorambucil, lze však použít i jiná alkylační cytostatika. Chlorambucil byl podáván buď dlouhodobě v nižších dávkách, které se upravovaly dle kontrol krevního obrazu, nebo ve vyšších dávkách podávaných v určitých časových intervalech.
Alkylační cytostatika v monoterapii mají pomalu nastupující účinek, takže léčba perorálními alkylačními cytostatiky trvala nezřídka více než 1 rok. Perorální léčba alkylačními cytostatiky dosáhne kolem 10 % kompletních remisí a celkový počet léčebných odpovědí se pohybuje mezi 50 a 60 %. Medián přežití od zahájení léčby alkylačními cytostatiky byl 50–70 měsíců.
Chlorambucil je stále považován za důležitý lék pro tuto skupinu pacientů, je lékem volby pro starší polymorbidní nemocné, u nichž by agresivnější léčba vedla k neakceptované toxicitě.
Rychlejší nástup účinku umožňují kombinovaná chemoterapie, nejčastěji používaná je zřejmě stejná kombinace jako u lymfomů, vinkristin, cyklofosfamid a prednison (COP) nebo vinkristin, adriamycin, cyklofosfamid a prednison s akronymem CHOP. Vzhledem k vyššímu věku těchto pacientů používají v některých zemím modifikaci zvanou CLL-CHOP s 50% dávkou adriamycinu. Tato léčba dosahuje léčebné odpovědi rychleji než monoterapie chlorambucilem, není však prokázáno, že by léčba chemoterapií CHOP dosahovala delší přežití než léčba samotnými alkylačními cytostatiky.
Purinová analoga
Purinová analoga, fludarabin, ale i 2-chlordeoxyadenosin, mají potenciál dosáhnout u CLL výraznější léčebné odpovědi než monoterapie alkylační cytostatikem, tedy vyšší počet kompletních remisí a celkových léčebných odpovědí, ale také delšího trvání remisí.
Na otázku, zda zavedení fludarabinu či 2-chlordeoxyadenosinu do léčebných schémat prodloužilo délku celkového přežití, není jednoznačná odpověď, protože v prospektivních randomizovaných studiích obvykle pacienti, kteří byli randomizováni do ramene s chlorambucilem, byli později při relapsu léčeni fludarabinem, takže nakonec obě skupiny nemocných se s tímto lékem setkali.
V roce 2007 je však jednoznačně prokázáno, že fludarabin či 2-chlordeoxyadenosin dosahují podstatně vyššího počtu léčebných odpovědí, pokud jsou podávány v kombinaci s alkylačními cytostatikem cyklofosfamidem (režim fludarabin cyklofosfamid), případně ještě s mitoxantronem (fludarabin, cyklofosfamid, mitoxantron). To bylo prokázáno četnými prospektivními randomizovanými studiemi.
V současnosti je kombinace fludarabinu a cyklofosfamidu považována za standardní léčbu první linie.
Přehled studií uvádí tab. 6. Léčba fludarabinem v kombinaci s alkylačními cytostatiky má však své nežádoucí dopady.
![Randomizované klinické studie srovnávající monoterapii purinovým analogem a kombinovanou léčbu purinovým analogem a alkylačním cytostatikem v rámci iniciální léčby [35].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/0d1d1027aeb2751affbe235499602936.png)
Monoklonální protilátka rituximab
Při léčbě B-CLL je možné použít protilátku antiCD20 (rituximab) nebo monoklonální protilátku antiCD52 (alemtuzumab).
Protilátka antiCD20, rituximab, je velmi dobře tolerována. Rituximab lze použít v monoterapii, podobně jako u jiných nízce agresivních maligních B-lymfoproliferací. Kompletní remise lze rituximabem v monoterapii získat jen výjimečně, údaje z klinických studií se pohybují od 0 do 9 % CR, počet celkových léčebných odpovědí se pohybuje mezi 13 a 58 %, interval do progrese pak mezi 5 a 19 měsíci.
Antigen CD20 je na buňkách B-CLL exprimován méně než u jiných neoplastických B-buněk. Navíc v plazmě cirkuluje solubilní CD20, který může inhibovat kapacitu rituximabu vázat se na leukemické buňky. Solubilní antigen CD20 má za následek rychlou clearence rituximabu a negativně ovlivňuje jeho farmakodynamiku. Proto účinek standardní dávky rituximabu 375 mg/m2 i.v. 4krát v týdenním intervalu lze zlepšit navýšením dávky (dose intensive regimen) a zkrácením intervalů mezi podáváním (dose dense regimen), tak jak dokumentuje tab. 7.
![Rituximab v monoterapii, výjimkou je poslední studie, v níž se jednalo o pacienty již dříve léčené chemoterapií [35].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/c4f2028e9f49781aad655f92645ea9bb.png)
Rituximab však výrazně zvyšuje aktivitu léčebných režimů založených na fludarabinu, a byl proto inkorporován v četných studiích do kombinované léčby. Rituximab snižuje expresi antiapoptotického proteinu bcl-2, a tím se buňky CLL stávající senzitivnější k apoptotickému efektu fludarabinu a dalších cytostatik z této skupiny.
Navíc fludarabin snižuje expresi proteinů, které jsou zodpovědné za rezistenci na komplement, CD46, CD55 a CD59, a tím zcitlivuje tyto leukemické buňky na cytotoxické působení komplementu, který se aktivuje navázáním rituximabu na CD20 antigen.
Protože FC je považován za optimální cytostatický režim první volby, tak se zcela logicky očekávalo jeho další zlepšení po začlenění rituximabu. Výsledky klinických studií fáze 2 byly opravu velmi působivé, ale zde uvedeme pouze výsledky klinické studie fáze III German CLL Study Group (tab. 7).
![Výsledky klinické randomizované studi e fáze III srovnávající dvojkombinaci fludarabin a cyklofosfamid s trojkombinací rituximab + fludarabin a cyklofosfamid (fludarabin 25 mg/ m<sup>2</sup> i.v. 1.– 3. den, cyklofosfamid 250 mg/ m<sup>2</sup> i.v. 1.– 3. den, rituximab 375 mg/ m<sup>2</sup> i.v. v 1. cyklu a 500 mg/ m<sup>2</sup> v každém dalším cyklu, celkem 6 cyklů léčby [36].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/f40bf3e948e6af15f818eec09a79450b.png)
Monoklonální protilátka alemtuzumab
Alemtuzumab je monoklonální protilátka proti antigenu CD52, který je hojně exprimován na buňkách CLL, ale také na normálních B - a T-lymfocytech, ale i monocytech. Proto po aplikaci alemtuzumabu dochází jak k ničení buněk patologického klonu B-CLL, tak i dalších fyziologických imunokompetentních buněk (B-, T-lymfocytů), zatímco po aplikaci protilátky antiCD20nedochází k eliminace buněk zodpovědných za buněčnou imunitu T - a NK-lymfocytů. Proto je podávání alemtuzumabu spojeno se zvýšeným rizikem závažných oportunních infekcí. Běžně se tedy při léčbě alemtuzumabem podává profylaxe oportunních infekcí.
První studie prokázaly vysokou účinnost u pacientů s recidivující nemocí a po nich následovaly studie prokazující účinnost této látky podávané v monoterapii také v rámci primoléčby. Alemtuzumab aplikovaný v dávce 30 mg/m2 3krát týdně po dobu 18 měsíců dosáhl ve skupině dříve neléčených nemocných 41 pacientů 19 % kompletních remisí a 87 % celkových léčebných odpovědí. Následovalo randomizované srovnání alemtuzumabu (148 pacientů) a chlorambucil (149 pacientů). Počet kompletních remisí byl v první skupině 22 %, ve druhé 2 % a počet celkových léčebných odpovědí byl 85 vs 56. Reaktivace cytomegalové infekce postihla 11 % nemocných léčených alemtuzumabem.
Alemtuzumab je zatím jediným FDA schváleným lékem, který má potenciál působit i na leukemické buňky s narušenou funkcí p53. Pacienti s delecí 17p mají narušenou aktivitu p53 a jsou rezistentní na léčbu ostatními, standardně používanými léky u této nemoci.
Podobně jako rituximab je i alemtuzumab testován v kombinaci s purinovými cytostatiky a dalšími léky.
U pacientů, u nichž dříve předcházela jakákoliv léčba, dosahuje alemtuzumab v monoterapii 33–53 % léčebných odpovědí s mediánem trvání 8,7 až15,4 měsíců.
Léčba reziduální nemoci
Potentní účinek alemtuzumabu nabízí ideu podat jej jako konsolidaci např. po ukončené cytostatické léčbě fludarabinem a cyklofosfamidem. Alemtuzumab má sice potenciál dosáhnout molekulární remise, je však podáván osobám s deficitem imunity po předchozí léčbě, kterou ještě dále oslabí. Německá CLL skupina proto studii testující konsolidační léčbu alemtuzumabem pozastavila pro vysoký počet infekčních komplikací. To však neznamená opuštění ideje dosažení molekulární remise a totální eradikace nemoci, je však nutné najít vhodný léčebný postup s akceptovatelnými nežádoucími účinky.
Radioterapie
Část nemocných má potíže způsobené splenomegalií, které jsou buď mechanického (útlak okolních orgánů), nebo funkčního rázu (pancytopenie). Standardním postupem k odstranění těchto potíží je cílená radioterapie na slezinu v dávce 20–30 Gy.
Cílené ozáření lze použít i na oblast značně zvětšených lymfatických uzlin, které činí svému nositeli potíže, obvyklá dávka je 5krát týdně 2 Gy do celkové dávky 24 Gy.
Další indikací radioterapie je případná osteolýza na pokladě infiltrace skeletu, ke které dochází obvykle až po dlouhodobějším průběhu nemoci.
Splenektomie
Méně často používanou alternativou ozařování sleziny je splenektomie. Za indikovanou se považuje u medikamentózně obtížně zvládnutelné hemolytické anémie anebo u autoimunitní trombocytopenie, dále při hypersplenizmu a také při recidivujících bolestivých infarktech sleziny.
Při hypersplenizmu (u pacientů s cytopenií v periferní krvi a se zvýšenou aktivitou erytropoézy a trombopoézy v kostní dřeni) zlepší splenektomie situaci u 70 % operovaných. Operační mortalita se udává kolem 4 %.
Alogenní transplantace
Alogenní transplantace je vyhrazena pro mladší nemocné. Lze ji nabídnout jako léčbu volby druhé linie v případě nepříznivých prognostických faktorů (17p–) při selhání fludarabinového režimu, podaného v rámci iniciální léčby. Podmínkou je dobrý stav transplantovaného a dostupnost dárce. V posledních letech jsou používány režimy s redukovanou intenzitou přípravného chemoterapeutického režimu. Léčba je zobrazena na schématu 2.

Komplikace chronické B-lymfatické leukemie
Autoimunitní poruchy
Chronická lymfatické leukemie je spojena s vyšší frekvencí výskytu autoimunitních komplikací, hemolytické anémie, autoimunitní trombocytopenie a vzácně s aplazií červené řady (pure red aplasia).
Hemopoetická insuficience
B-CLL je nemoc s relativně dlouhým přežitím, komplikovaná postupnou neschopností kostní dřeně vytvořit dostatečný počet neutrofilů, trombocytů a erytrocytů a také postupně se zhoršující imunitní odpovědní na infekt. V rámci podpůrné léčby je tedy nutno substituovat krvinky, případně aplikovat erytropoetin.
Infekční komplikace
CLL je spojená s defektní B i T imunitní odpovědí a zvýšeným počtem infekcí.
Pro B-CLL je typická hypogamaglobulinemie, tedy nedostatečná protilátková imunita. Nedostatečná opsonizace bakterií imunoglobuliny brzdíjejich odstraňování neutrofily. Často mají pacienti velmi nízké počty neutrofilů. Proto mají bakteriální infekce u těchto nemocných mnohem agresivnější průběh a při léčbě závažných infekcí je ke zvážení substituce gamaglobulinů.
Pacienti s B-CLL mají i při počátečním dostatečném počtu cirkulujících lymfocytů deficit T - a NK-buněčné imunity. Pravidelně bývá snížena funkce všech T-lymfocytů a také aktivita NK (natural killer) buněk, LAK (lymphokine activated killer) buněk. Snížena je také cytotoxická aktivita závislá na protilátkách. To je příčinou častých virových infekcí.
Vzhledem k výše uvedeným poruchám imunity je nyní pochopitelné, proč pacienti s touto nemocí mají často běžné infekce (pásový opar, pneumonie) a infekce oportunní (Pneumocystis jiroveci, mykotické infekce) a proč právě infekce jsou nejčastější příčinou smrti těchto nemocných. Incidence manifestního onemocnění viry herpes simplex a herpes zoster dosahuje u těchto pacientů 30 %.
Snížení frekvence infekcí, zvláště infekcí dýchacích cest lze dosáhnout profylaktickou substitucí gamaglobulinů, obvykle v dávce 10 g/měsíc. Tato léčba sníží četnost infekcí, nevede však ke zlepšení celkového přežití. Vzhledem k nákladům na tuto léčbu je podávána pouze nemocných s prokázaným deficitem imunoglobulinů (IgG < 4 g/l, případně IgG < 4–6 g/l) a velmi časté recidivující bakteriální infekce (často sinobronchiální).
Při závažných život ohrožujících infekcích se doporučuje podávat gamaglobuliny v léčebné dávce 0,4 g/kg.
Profylaktické podání acykloviru je doporučováno jen pacientům s nízkými CD4+ lymfocyty a dále u pacientů léčených antiCD52 protilátkou. U těchto imunokompromitovaných nemocných se může také vyskytnout cytomegalovirová infekce. Proto se vyšetřuje replikace CMV viru PCR metodou.
Ani mykotické infekce (aspergilóza, kandidóza a jiné) nejsou výjimkou a je třeba po nich cíleně pátrat (průkaz aspergilového galaktomananu a jiné).
Vzhledem k četným bakteriálním in-fekcím doporučují někteří autoři pneu-mokokovou vakcínu jednou za pět let a roční očkování proti chřipce, i když je imunologická odpověď na vakcinaci u těchto pacientů oslabená.
Kachektizace
V případě velkého dlouhodobého nechutenství může pomoci megestrolacetát (Megace), který zvyšuje chuť k jídlu.
Sekundární maligní nemoci
Průběh nemoci mohou komplikovat sekundární malignity, jejich četnost dosahuje 11 %. Jiné zprávy hodnotící četnost dalších malignit udávají, že jejich výskyt je 3krát častější než u stejně staré průměrné populace. Proto se pacientům doporučuje maximálně eliminovat kancerogeny z denního života a dále při kontrolách rozšířit prohlídky s cílem detekovat včas druhotné maligní choroby (karcinom prsu, děložního čípku, střeva, prostaty, ale i kožní tumory a tumory CSN). Tumory CNS jsou velmi záludné, jejich rozpoznání je často velmi pozdní. Jejich vyšší četnost se přisuzuje narušení imunitní obrany, která je obligátně spjata s touto nemocí.
Jako příklad sekundárních maligních nemocí můžeme uvést muže s příznivým průběhem B-CLL, u něhož byl při sledování B-CLL diagnostikován karcinom prostaty a vyléčen, avšak pozdě byl rozpoznán meningeom v oblasti frontálního laloku, který při pomalém růstu utlačil tkáň frontálního laloku a způsobil změnu osobnosti, která byla mylně až do prvního CT interpretována jako arteriosklerotické změny osobnosti. Takže i změna osobnosti u těchto pacientů by mohla být signálem k postavení otázky, zda příčinou není maligní expanze v CNS.
Zvýšená frekvence myelodysplastického syndromu je pozorována u nemocných léčebných fludarabinem v kombinaci s alkylačním cytostatikem.
Souběh více lymfoproliferativních nemocí
V praxi jsme se setkali s pacienty, u nichž v průběhu B-CLL byl diagnostikován mnohočetný myelom s vysokou hodnotou monoklonálního gamaglobulinu a osteolytickými projevy. Nové bolesti kostí je tedy třeba v průběhu této nemoci vždy vyšetřit a včas zahájit léčbu případného souběhu B-CLL a mnohočetného myelomu.
Transformace nemoci do agresivnějšího lymfoproliferativního onemocnění (Richterův syndrom)
Přibližně u 10 % nemocných se B-CLL transformuje v agresivnější typ nehodgkinského lymfomu – difuzní velkobuněčný B-lymfom.
Příznaky transformace jsou rychle se zvětšující uzliny či hepatosplenomegalie, anémie, trombocytopenie, postižení mimouzlinových oblastí (mozek, dýchací trakt, kůže, zažívací trakt). U některých osob se transformace do agresivnější histologické formy projeví vícečetným osteolytickým postižením skeletu a případně i hyperkalcemií.
Laboratorně odpovídá této transformaci vzestup aktivity LD.
Transformace do výše agresivního typu nehodgkinského lymfomu je spojena s malou léčebnou odpovědí, medián přežití po transformaci bývá kratší než 6 měsíců. Ani nejnovější léčebné postupy zásadně nezlepšily prognózu u pacientů s transformací této indolentní choroby do agresivnějšího histologického typu lymfoproliferace.
Další možností je transformace do prolymfocytární leukemie. Tato změna je spojená s výraznou lymfocytózou nad 100 × 109/l, splenomegalií, hematomegalií a lymfadenopatií a často infiltrací CNS, vznikem pleurálního výpotku, ascitu, a periorbitálních otoků. I v těchto případech bývá pozorována hyperkalcemie se vznikem osteolytických destrukcí skeletu.
Medián přežití po transformaci do prolymfocytární leukemie je krátký, obvykle méně než šest měsíců.
Prognóza
Medián přežití všech nemocných je 8–10 let. Asi 1/3 nemocných s B-CLL má dlouhé přežití a nemoc u těchto pacientů do konce jejich života nepřejde do progredující formy, která vyžaduje léčbu. U 1/3 zpočátku indolentní CLL začne progredovat a vyžadovat léčbu, takže dalším osudem těchto osob jsou relapsy a remise nemoci. U poslední 1/3 má CLL agresivní průběh vyžadující záhy po stanovení diagnózy její léčbu.
Dle výše uvedených prognostických faktorů je možné nemocné začlenit do jedné ze 3 prognostických skupin:
- CLL nízkého rizika: mutovaný IgVH, negativní ZAP 70, CD38–, del RB1 a normální cytogenetický nález
- CLL středního rizika: nemutovaný IgVH, pozitivní ZAP 70, CD38+, +12, del ATM
- CLL vysokého rizika: mutace/delece p53
Existuje samozřejmě více prognostických stratifikací, další z nich znázorňuje schéma 3.
![Schéma 3. Jedno z možných prognostických členění [31].](https://pl-master.mdcdn.cz/media/image/7712962a7cf10829c9e84c00b1957444.png?version=1537790346)
Vlasatobuněčná leukemie
Vlasatobuněčná leukemie (hairy cell leukemia dle WHO klasifikace) je vzácnou chorobou, podobně jako další typy chronických lymfatických leukemií z této kapitoly.
Podstatou nemoci je proliferace patologických lymfoidních buněk, majících četné cytoplazmatické výběžky (vlásky), které jsou nejzřetelněji patrné jenom v elektronovém mikroskopu. Od těchto leukemických buněk s jemnými výběžky cytoplazmatické membrány (tricholymfocytů) je odvozen název nemoci.
Tyto buňky infiltrují kostní dřeň, slezinu a játra. V nevelkém počtu cirkulují v krvi, jejich průkaz v cirkulující krvi se však vždy nemusí podařit. Splenomegalie je přítomna v 90 % případů, masivní splenomegalie je v době stanovení diagnózy přítomna nejméně u 20 % nemocných. Ve slezině je postižena červená pulpa a atrofuje bílá pulpa sleziny, což je rozdíl od CLL, kde je postižena dominantně bílá pulpa sleziny. Lymfadenopatie je přítomna jen u pokročilých případů. Hepatomegalie je u této nemoci neobvyklá, ačkoliv játra jsou pravidelně infiltrována.
V kostní dřeni stimulují patologické lymfoidní buňky tvorbu retikulinových vláken, což může být příčinou nezdařené aspirace kostní dřeně.
Patologické vlasaté lymfoidní buňky produkují četné cytokiny, jejichž tlumivý vliv na krvetvorbu je hlavní příčinou pancytopenie, i když se na pancytopenii také podílí mechanický útlak kostní dřeně leukemickými buňkami. Podíl na cytopenii má také velká slezina.
Incidence vlasatobuněčné leukemie není velká, u dospělých představuje 2 % z celkového počtu maligních lymfoproliferativních nemocí a také tvoří 2 % ze všech leukemií. Postihuje v 75–80 % muže. Medián věku pacientů s nově diagnostikovanou chorobou se pohybuje mezi 50 a 55 roky.
Nemoc bývá často asociována s au-toimunitními chorobami.
Zatím stále není jednoznačně objasněno, který buněčný typ stojí na počátku vývoje vlasatobuněčné leukemie. Dřívější práce naznačovaly, že vlasatobuněčná leukemie vzniká expanzí zralých B buněk, které exprimují na svém povrchu exkluzivní lehké řetězce, ale násobné izotopy těžkých imunoglubulinových řetězců s převahou IgG3. Současnější práce poukazují na existenci klonálně příbuzných izotypů v jednotlivých vlasatých buňkách. To naznačuje možnost, že tyto izotypy vznikají na úrovni sestřihu RNA. Předpokládá se proto, že vlasaté buňky jsou zablokovány ve fázi izotypového přepnutí, kdy sestřih RNA může předcházet deleční rekombinaci.
Příznaky, s nimiž pacienti přicházejí k lékaři
Dominujícím příznakem je splenomegalie a pancytopenie a z ní odvoditelné příznaky (slabost, únava, krvácivé projevy). Hepatomegalie provází jen asi 1/3 pacientů, lymfadenopatie nemusí být zpočátku zřetelná, při progresi nemoci je pak lymfadenopatie (periferní, intraabdominální, retroperitoneální a mediastinální) patrná u nadpoloviční většiny pacientů.
Choroba je provázena závažným snížením intenzity imunitní odpovědi (neutropenií, monocytopenií a porušením funkce T - i B-lymfocytů), a proto jsou pacienti s vlasatobuněčnou leu-kemií ohroženi častými a závažnými infekcemi včetně oportunních infekcí.
Podezření na diagnózu je nutné získat z výše uvedených klinických projevů,splenomegalie a cytopenie, anémie (85 %), trombocytopenie (60–80 %) a leukopenie (60 %) a indikovat speciální vyšetření, neboť běžný diferenciální krevní obraz diagnózu neprokáže. Diagnózu lze prokázat na základě:
- cytologického vyšetření periferní krve a kostní dřeně ve světelném mikroskopu se speciálním cytochemickým barvením k průkazu vlasatobuněčné leukemie (tartarát rezistentní kyselá fosfatáza), požadavek na provedení speciálního barvení k průkazu vlasatobuněčné leukemie je nutno napsat na žádanku
- histologie kostní dřeně s imunohistochemickým barvením (CD antigeny)a případně se znázorněním zmnoženíretikulinových vláken pomocí stříbření; toto vyšetření umožní odhalit infiltraci kostní dřeně vlasatobuněčnými lymfoidními buňkami
- imunofenotypizace (průtokové cytometrie) lymfocytů získaných aspirací kostní dřeně
- imunofenotypizace lymfocytů z periferní krve; stanovení vyžaduje erudovanou analýzu vzorku specialistou v oboru průtokové cytometrie, protože patologické buňky jsou domi-nantně lokalizované ve slezině a kostnídřeni a v cirkulaci je jich málo
- hodnocení lymfocytů periferní krve a kostní dřeně elektronovou mikroskopií patří díky imunofenotypizaci většinou již minulosti a není běžně prováděno
Vzhledem k fibrotizaci dřeně je vhodnější ihned provést trepanobiopsii, neboť sternální punkce s aspirací se ne vždy podaří. Podezření na vlasatobuněčnou leukemii, získané z vyšetření periferní krve, lze verifikovat trepanobiopsií s imunohistochemickým vyšetřením kostní dřeně. Diferenciálně diagnosticky nutno odlišit splenický lymfom s vilózními lymfocyty či jiné nízce agresivní lymfocytární onemocnění.
U vlasatobuněčné leukemie platí, že pouze lékař, který ví o této nosologické jednotce a je si vědom, že ji lze rozpoznat pouze speciálními vyšetřeními a že běžné mikroskopické vyšetření krevního nátěru (diferenciální krevní obraz) ji neodhalí, je schopen zorganizovat diagnostické kroky vedoucí ke stanovení této diagnózy.
Léčba a prognóza
Nemoc se léčí až ve stadiu klinických příznaků. Běžná alkylační cytostatika nelze u této nemoci použít. Metodou léčby před rokem 1980 byla splenektomie. Prvním úspěšným lékem pro tuto nemoc byl interferon a, po něm následoval 2-chlordeoxyadenosin, který je dnes lékem volby (nebo také 2-deoxycoformycin). Léčba 2-chlordeoxyadenosinem má excelentní výsledky, ilustrujeme je údaji Gooldmana, který sledoval 209 pacientů nejméně 7 let. Nemoc relabovala u 76 (36 %) pacientů v průběhu uvedených sedmi let, ostatní zůstávali v kompletní remisi. Relaps nemoci citlivé na 2-chlordeoxyadenosin je možno znovu léčit stejným preparátem.
Pro případy, kdy je nutná léčba druhé linie, je zde účinná monoklonální protilátka antiCD20 rituximab. Další monoklonální protilátkou použitou pro léčbu této nemoci je BL22, rekombinantní imunotoxin, tedy upravená protilátka proti antigenu CD22 s navázaným pseudomonádovým toxinem.
Na povrchu vlasatých buněk je však také navázán antigen CD52 a byly zveřejněny popisy případů účinnosti antiCD52 protilátky alemtuzumabu. V literatuře je popsán také velmi dobrý účinek pentostatinu.
Léčba 2-chlordeoxyadenosinem dosahuje excelentních výsledků, udává se, že 10leté celkové přežití dosáhne 84 % a 20leté 65 % pacientů, u nichž byla podána léčba 2-chlordeoxyadenosinem. Léčba je shrnuta ve schématu 4.

B-buněčná prolymfocytární lymfatická leukemie (B-PLL)
Definice a stanovení diagnózy
B-prolymfocytární leukemie je extrémněvzácnou nemocí, dle údajů WHO klasifikace tvoří 1 % všech případů chronických lymfatických leukemií. Většina pacientů je starší 60 let, medián věku pacientů se udává 70 let (rozptyl 41–92 let), poměr mužů a žen je 1,6 : 1. WHO klasifikace tuto jednotku (B-cell prolymphocytic leukemie) uvádí jako samostatnou nosologickou jednotku.
Klasickými příznaky nemoci jsou: splenomegalie (87 %), extrémně vysoký počet lymfocytů (38 %), lymfadenopatie (38 %), anémie (64 %), trombocytopenie (64 %). Postižení uzlin nebývá časté.
Diagnóza této nemoci se stanovuje na základně morfologického vyšetření – přítomnosti prolymfocytů. Kritériem pro stanovení diagnózy přítomnost více než 55 % prolymfocytů ze všech lymfoidních buněk.
Prolymfocyty se morfologicky odlišují od zralých lymfocytů, jsou to středně velké buňky, mají středně kondenzovaný chromatin a jeden výrazný nukleolus v jádře. Obvykle tyto prolymfocyty difuzně infiltrují kostní dřeň.
Pacienti počtem prolymfocytů v intervalu 10–55 % jsou řazení do kategorie CLL/B-PLL. U pacientů této skupiny je pravděpodobná pozdější transformace do prolymfocytární leukemie.
Imunofenotyp patologických prolymfocytů je odlišný od imunofenotypu buněk B-CLL. Buňky B-PLL exprimují pan-B znaky (CD19, CD20, CD22 FMC7 CDF79a a CD79b). Je výrazně přítomný povrchový imunoglobulin s restrikcí lehkých řetězců, těžké řetězce imunoglobulinu jsou nejčastěji typu IgM nebo IgD, menší část případů má těžké řetězce typu IgG nebo IgA.
Na rozdíl od klasické B-CLL exprese znaků CD5 a CD23 je slabá či zcela chybí. Naproti tomu znak CD20 je na buňkách B-PLL podstatně výraznější než u klasické CLL. K dalším důležitým znakům z hlediska diferenciální diagnostiky je negativita CD11c a CD103 a slabá či chybějící exprese CD10 a CD25.
Cytogenetickým vyšetřením lze prokázat četné abnormality. Většinou postihují chromozom 14 (14q32). Translokace t(11;14)(q13;q32), která je typicky popisována u lymfomu plášťové zóny, způsobující zvýšenou expresi cyklinu D1, byla zjištěna u 25 % případů prolymfocytární leukemie. Další popsané chromozomální aberace postihují chromozom 6q21, 11q23, 12p12 a 13q14 a také 17p. Trisomie 12, která je častá u B-CLL, je v případě prolymfocytární leukemie řídká.
Prognosticky důležitou informací je skutečnost, že abnormality (delece či mutace) postihují nejméně v 50 % gen p53, což pravděpodobně přispívá k rezistenci na běžnou léčbu.
Léčba a prognóza
Tradičními léčebnými postupy byly chemoterapeutické kombinace, nověji pak monoterapie či kombinace obsahující purinová analoga, fludarabin nebo 2-chlordeoxyadenosin. Silná exprese antigenu CD20 na povrchu těchto buněk je zřejmě důvodem úspěchu monoklonální protilátky antiCD20, rituximabu. Účinná je také léčba monoklonální protilátkou antiCD52, alemtuzumabem. Vzhledem k vzácnosti těchto případů jsou informace o léčbě odvozené dominantně z popisů případů z menších souborů nemocných.
Alogenní transplantace je vhodná pro mladší nemocné. U pacientů s velkou slezinou, která způsobovala klinické příznaky, lze provést splenektomii buď klasickou cestou, nebo endoskopicky.
Léčebné výsledky jsou méně příznivé, než je tomu u klasické B-CLL. Stav pacienta se obvykle progresivně zhoršuje a nemoc bývá rezistentní na chemoterapii.
Medián přežití těchto nemocných se uvádí kolem pěti let.
Nepříznivými prognostickými faktory jsou anémie, trombocytopenie a mutace genu p53.
T-buněčná prolymfocytární leukemie (T-PLL)
Definice a stanovení diagnózy
T-buněčná prolymfocytární leukemie (T-PPL – T-cell prolymfocytic leukemia WHO klasifikace) je charakteristická proliferací malých prolymfocytů postthymického imunofenotypu. Medián věku při stanovení diagnózy je 63 (33–91) let.
Pacienti při stanovení této diagnózy mívají obvykle splenomegalii (75 %), lymfadenopatii (50 %), kožní infiltráty (25 %), výpotky v serózních dutinách (15 %). Výjimečně může být tato nemoc zastižena v asymptomatické formě při náhodném vyšetření krevního obrazu.
U většiny postižených přesahuje počet lymfocytů 100 × 109/l. Anémie a trombocytytopenie je méně častá než u B-PLL. Na rozdíl od B-CLL jsou sérové koncentrace imunoglobulinů normální.
Diagnóza T-PLL se stanoví na základě morfologického nálezu periferní krve či kostní dřeně, typickým obrazem je infiltrace tvořená malými až středními lymfoidními buňkami s prominujícími nukleoly. Diagnózu je však nutné potvrdit imunofenotypizačním vyšetřením (CD2+, CD3+, CD4+, CD5+,CD7+, CD8 – a asi ve 1/4 případů CD8+). Imunofenotyp těchto patologických T-prolymfocytů odpovídá imunofenotypu thymických a postthymických T-lymfocytů. Cytogenetickým vyšetřením lze prokázat různé aberace, které však nejsou diagnostické pro tuto chorobu.
Léčba a prognóza
Jde o extrémně agresivní chorobu. Cel-kové přežití i při agresivní léčbě purinovými analogy (fludarabin, 2-chlor-deoxyadenonosin, pentostatin) je velmi krátké, pohybuje se kolem sedmi měsíců. Průměrné celkové přežití těch pacientů, kteří po úvodní léčbě dosáhnou kompletní remise, je pouze 14,8 měsíce. Za nejúčinnější dostupnou léčbu je považován alemtuzumab v monoterapii či v kombinaci. Alemtuzumab dosáhl v jedné studii 60 % CR a 16 % PR, medián bezpříznakového intervalu byl ale jen sedm měsíců. Pacienti, kteří dosáhli kompletní remise, měli delší přežití než nemocní bez léčebné odpovědi.
Podobně i četné další studie potvrdily vysoký počet léčebných odpovědí u této nemoci. Teoretickým vysvětlení excelentní účinnosti alemtuzumabu u této nemoci je vysoké exprese antigenu CD52 na T-prolymfocytech. Alemtuzuman v monoterapii, případně v kombinaci, je tedy zřejmě lékem vhodným pro iniciální léčbu, i když i při úspěšné léčbě je vysoký počet recidiv.
Jedině alogenní transplantace zde představuje potenciální naději na delší život.
T-buněčná leukemie z velkých granulárních lymfocytů
Patofyziologie nemoci
Velké granulární lymfocyty je popisné pojmenování velkých vyzrálých lymfocytů, majících četná jemná azurofilní granula s cytochemickým průkazem kyselé fosfatázy.
Tyto buňky jsou fyziologicky přítomny v periferní krvi přibližně v počtu 220 buněk na 1 µl, tvoří 10–15 % mononukleárních buněk. Morfologicky jsou tyto lymfocyty odlišné od B-lymfocytů, typické pro ně je nadbytek cytoplazmy s jemnými azurofilními granuly. Jsou to obvykle T - nebo NK-buňky. Zvýšený počet těchto buněk (nad 2 000/µl) může souviset s různými patologickými stavy. Jde o syndrom heterogenní povahy.
Dočasný vzestup jejich počtu se objevuje při virových infekcích, při infarktu myokardu a po transplantaci kostní dřeně. Zvýšený počet těchto buněk může doprovázet různé revmatoidní choroby. Tato reaktivní forma lymfocytózy z velkých granulárních lymfocytů má polyklonální charakter a trvá obvykle méně než 1/2 roku.
Na druhé straně existuje také leu-kemie z velkých granulárních lymfocytů, která má dlouhodobý, nízce agresivní průběh. Trvalý vzestup počtu těchto buněk představuje klonální proliferaci buď z CD3+, nebo CD3 – řady T-lymfocytů. Populace CD3+ představuje aktivní cytotoxické T-lymfocyty a CD3 – představuje NK buňky (natural killer cells). Klonální proliferací těchto buněk vznikají T-LGL (T-cell large granular lymphocyte leukemia – T-buněčná leukemie z velkých granulačních lymfocytů) nebo NK-LGL (NK-cell large granular lymphocyte leukemia – NK-buněčná leukemie z vel-kých granulárních lymfocytů) dle WHO klasifikace.
Klinické příznaky a průběh
Chronická lymfatická leukemie z velkých granulárních lymfocytů nezpůsobuje pacientům zpočátku vážnější problémy. Pokud ano, jedná se o celkovou slabost a náchylnost k bakteriálním infekcím často postihujícím paranazální dutiny, kůži, perianální krajinu nebo plíce.
Středně velká slezina je nacházena u 20–30 % pacientů. Hepatomegalie je vzácnější a lymfadenopatie je zcela výjimečná. U 25 % pacientů však bývají kožní infiltráty. Koincidence této chronické T-lymfatické leukemie se séropozitivní revmatoidní artritidou dosahuje 20–30 %. Vysoká je i koincidence s jinými sérologickými abnormalitami, jako jsou antinukleární protilátky nebo protilátky proti neutrofilům.
Leukocytóza vůbec není nápadná. Někteří autoři udávají, že počet leukocytů se pohybuje maximálně kolem hodnot 10–20 × 109/l. Častěji je však počet leukocytů nezvýšen. Počet lymfocytů přesahuje 4 × 109/l jen u 70 % pacientů. Počet velkých granulárních lymfocytů je přitom obvykle menší než 25 % z počtu lymfocytů. Medián počtu velkých granulárních lymfocytů při stanovení diagnózy činí 1,5 × 109/l, s rozmezím 0,1–10,0 × 109/l.
U 80 % pacientů je ihned při stanovení diagnózy nápadná neutropenie, u 40 % pacientů je počet neutrofilů menší než 0,5 × 109/l. Anémie je při stanovení diagnózy přítomna u 50 % a trombocytopenie u 20 % pacientů.
Patogeneze anémie a trombocytopenie, pokud se objeví, je vysvětlována inhibičním vlivem patologických buněk a jimi produkovaných cytokinů na krvetvorbu, a nikoliv excesivní infiltrací kostní dřeně.
Chronická granulární CD3+ lymfocytóza může být příčinou chronické neutropenie při koincidenci s revmatoidní artritidou a také může souviset s cyklickou neutropenií manifestující se v dospělém věku. Závažná anémie může být způsobena brzdivým vlivem velkých CD3+ granulárních lymfocytů na krvetvorbu.
Na rozdíl od B-CLL je infiltrace kostní dřeně méně výrazná, počet lymfocytů v kostní dřeni se pohybuje mezi 20 a 50 % a u některých nemocných se může blížit normálu. U 50 % pacientů chorobu provází polyklonální hypergamaglobulinemie.
Průběh T-LGL a NK-LGL se mírně liší. Obecně lze konstatovat, že pacienti s T-LGL mají chronický průběh nemoci, charakterizovaný neutropenií a dalšími cytopeniemi.
Naproti tomu pacienti s NK-LGL mají obvykle B symptomy (úbytek hmotnosti, noční poty, horečky), masivní splenohepatomegalii a obvykle agresivnější průběh nemoci. T-LGL je však podstatně častější, představuje zhruba 85 % všech případů LGL.
Stanovení diagnózy
Diagnóza této vzácné nemoci byla dříve stanovována na základně nalezení > 2 × 109/l buněk odpovídajících popisu T-LGL. Nověji je diagnostika založena na prokázání jejich klonality pomocí průkazu přestavby (přeskupení) genu TCR.
Typickým imunofenotypizačním nálezem je CD1–, CD2+, CD3+, CD4–, CD5–, CD7–, CD8+, CD16+/–, CD57+ a TCR α/β+. Pokud je na velkých granulárních lymfocytech přítomen znak CD56, je průběh nemoci obvykle zhoubnější.
S T-LGL bývají často asociované autoimunitní poruchy (pure red cell aplasia, revmatoidní artritida, Feltyho syndrom) a dále sérologické abnormality bez projevů autoimunitní choroby, např. polyklonální hypergamaglobulinemie nebo hypogamaglobulinemie, antineutrofilní a antitrombocytární protilátky, přítomnost revmatoidního faktoru anebo antinukleárních protilátek.
Léčba a průběh
Nemoc má indolentní průběh s mediá-nem přežití 13 let, jsou však možné spontánní remise stejně jako agresivní průběh nemoci.
Indikací k zahájení léčby je anémie, B příznaky či neutropenie. Léčba se pro nevelký počet případů nezakládá na výsledcích klinických studií, ale na zkušenostech získaných formou menších studií či popisů případů.
Byly popsány úspěchy následujících léčebných alternativ: nízké dávky metotrexátu, cyklosporin, nízké dávky cyklofosfamidu, neukleosidová analoga. Nejlepší výsledy byly zřejmě dosaženy kombinací cyklosporinu a nízké dávky metotrexátu. Kortikoidy v monoterapii se pro tuto nemoc nedoporučují. Pro úpravu cytopenie byly použity jak hemopoetické růstové faktory (G-CSF, erytropoetin), tak i splenektomie. Nejnovější alternativu představuje antiCD52 protilátka alemtuzumab.
T-buněčná leukemie dospělých
Výskyt T-buněčné leukemie dospělých (Adult T-cell leukemie dle WHO klasifikace) je omezen pouze na endemické oblasti, jihozápadní Japonsko, Karibskou oblast, jihovýchod USA a centrální a jižní Ameriku. Tento typ leukemie souvisí s infekcí virem HTLV-1. Je zajímavé, že protilátky proti tomuto viru má v endemických oblastech Japonska až 37 % obyvatel, ale pouze u 2–4 % nositelů propukne tato choroba.
Tato choroba má asi u 50 % postižených akutní průběh, méně časté jsou chronické formy nemoci či průběhy podobné lymfomům.
Vzhledem tomu, že lidská leukemie související prokazatelně s virovou infekcí se v ČR nevyskytuje, omezíme se pouze na informaci o jejím výskytu.
Tato práce vznikla a byla podporována v rámci projektu MŠMT: LC 06027 a VZ 0021622434.
MUDr. Marta Krejčí
www.fnbrno.cz
e-mail: mkrejci@fnbrno.cz
Doručeno do redakce: 15. 7. 2009
Sources
1. Adam Z, Doubek M. Chronické lymfatické leukémie. Postgrad Med 2000; 2 : 165–179.
2. Cmunt E, Trněný M, Karban J et al. Analýza rizikových faktorů u 248 pacientů s chronickou B-lymfocytární leukemií. Transfuze Hematol Dnes 2006; 12 : 232–239.
3. Dědič K, Žák P. Vlasatobuněčná leukémie. Čes Slov Patologie 2002; 38 : 69–74.
4. Doubek M, Ráčil Z, Mayer J. Klinické využití humanizované monoklonální protilátky alemtuzumab. Brno: Masarykova univerzita 2006.
5. Doubek M. Alemtuzumab v léčbě pacientů s chronickou lymfatickou leukémií. Postgrad Onkol 2005; 1 : 13–15.
6. Chrobák L. K českému názvu leukemie charakterizované vlasatými buňkami. Transfuze Hematol Dnes 2004; 10 : 106–111.
7. Jarošová M, Divoký V, Papajík T et al. Význam a přínos moderní cytogenetiky a molekulární cytogenetiky pro současnou hematoonkologii. Transfuze Hematol Dnes 2005; 11 : 84–86.
8. Kuhrová V, Francová H, Klimešová D et al. Molekulárně genetická charakteristika agresivity chronické lymfocytární leukemie u českých pacientů: nukleotidová variabilita genů kódujících syntézu těžkého řetězce imunoglobulinů. Čas Lék Čes 2006; 145 : 855–858.
9. Kuhrová V, Francová H, Klimešová D et al. Molekulárně-genetická charakterizace agresivity chronické lymfocytární leukemie u českých pacientů: nukleotidová variabilita genů kódujících syntézu těžkého řetězce imunoglobulinů. Čas Lék Čes 2006; 145 : 855–858.
10. Kuhrová V, Francová H, Klimešová D et al. Chronická lymfatická leukemie: mutační status těžkého řetězce imunoglobulinového genu je významné prognostické kritérium. Transfuze Hematol dnes 2004; 10 : 143–148.
11. Kvapil F, Doubek M, Brychtová Y et al. Splenektomie v diagnostice a léčbě hematologických onemocnění – indikace, komplikace a výsledky z jednoho pracoviště. Trans Hematol Dnes 2006; 12 : 146–152.
12. Mayer J, Doubek M, Brychtová Y et al. Využití rituximabu v léčbě chronické lymfatické leukemie. Klin Onkol 2003; 16 : 178–183.
13. Žák P. Vlasatobuněčná leukemie a přínos 2-chlorodeoxyadenisnu v léčbě. Hradec Králové: Nukleus 2006.
14. Papajík T, Jarošová M, Pikalová Z et al. Chronická B-lymfocytární leukemie. Část II. Diagnostická kritéria a význam stanovení individuální prognózy nemocného. Transfuze Hematol Dnes 2006; 12 : 132–139.
15. Papajík T, Jarošová M, Plachý R et al. Chronická B-lymfocytární leukemie: Část I. Pohled na původ, biologii a genetické změny leukemických buněk. Transfuze Hematol Dnes 2006; 12 : 53–61.
16. Papajík T, Urbanová R, Kubová Z et al. Chronická B-lymfocytární leukemie. Část IV. Možnosti léčby s použitím monoklonálních protilátek alemtuzumabu a rituximabu. Transfuze Hematol dnes 2007; 13 : 48–55.
17. Papajík T, Urbanová R, Procházka V et al. Chronická B-lymfocytární leukemie: část III. Současné konvenční možnosti primární léčby. Transfuze Hematol Dnes 2006; 12 : 249–256.
18. Raida L, Papajík T, Pikalová Z. Terapeutická účinnost cladribinu a buněčný imunodeficit – spojené nádoby u leukemie s vlasatými buňkami. Vnitř Lék 2002; 48 : 384–389.
19. Singlová Z, Cetkovský P. Alogenní transplantace krvetvorných buněk po přípravném režimu redukované intenzity v léčbě lymfoproliferací. Výsledky ÚHKT 1999–2005. Transfuze Hematol Dnes 2006; 12 : 67–72.
20. Smolej L, Andrýs C, Belada D et al. Plazmatické koncentrace solubilního endoglinu u nemocných s lymfoidními malignitami. Transfuze Hematol Dnes 2006; 12 : 37–39.
21. Smolej L, Kašparová-Benešová P. Význam angiogeneze u maligních nádorů. Acta Medica (Hradec Králové) 2005; 48 : 69–72.
22. Smolej L, Saudková L, Špaček M et al ZAP-70 u chronické B-lymfocytární leukemie: klinický význam a metody detekce. Vnitř Lék 2006; 52 : 1194–1199.
23. Smolej L. Kombinační režim fludarabin + cyklofosfamid u chronické B-lymfocytární leukemie. Vnitř Lék 2005; 51 : 1142–1143.
24. Suková V, Klabusay M, Čoupek P et al. Denzita exprese antigenu CD20 na populacích maligních buněk u pacientů s chronickými lymfoproliferacemi. Čas Lék Čes 2006; 145 : 712–716.
25. Trbušek M, Malčíková J, Šmardová Jet al. Starý známý neznámý TP53, tentokráte u B-CLL. Čas Lék Čes 2005; 144 : 350–351.
26. Trbušek M, Mayer J. Molekulární patogeneze chronické lymfocytární leukemie se zaměřením na regulaci buněčného cyklu a apoptózy. Čas Lék Čes 2004; 143 : 84–89.
27. Žák P, Chrobák L, Dědič K. The degree of bone marrow infiltration in patients with hairy cell leukemia treated with splenectomy compatible with long-term hematological remission. Neoplasma 2001; 48 : 72–75.
28. Žák P, Dědič K, Chrobák L et al. Imunohistochemická detekce a kvantifikace minimální reziduální choroby protilátkou DBA.44 v trepanobioptických vzorcích u pacientů s vlasatobuněčnou leukemií (popis metody). Hematol Transfuziol (Bratislava) 1999; 4 : 40–45.
29. Žák P, Chrobák L, Podzimek K et al. Neobvyklý průběh leukemie s vlasatými buňkami s výraznou abdominální lymfadenopatií, leukemickými infiltráty oční rohovky a kožními změnami. Vnitř Lék 1996; 42 : 463–466.
30. Žák P, Chrobák L, Dědič K et al. Sledování minimální reziduální nemoci u nemocných s vlasatobuněčnou leukemií v kompletní remisi po terapii 2-chlorodeoxyadenosinem. Vnitř Lék 2000; 46 : 697–703.
31. Shanafelt TD, Byrd JC, Call TG et al. Narrative review: Initial management of newly diagnosed early stage chronic lymphocytic leukemia. Ann Intern Med 2006; 145 : 435–447.
32. Binet JL, Caligaris–Cappio F, Catovsky D et al. International Workshop od Chronic lymphocytic leukemie – IWCLL. Perspectives on the use of new diagnostic tools in the treatment of chronic lymphocytic leukemia. Blood 2006; 107 : 859–861.
33. Cheson BD, Bennett JM, Grever M et al. National Cancer Institute sponsored working Group guidelines for chronic lymphocytic leukemia: revised guidelines for diagnosis and treatment. Blood 1996; 87 : 4990–4997.
34. Yee KW, O’Brien SM. Chronic lymphocytic leukemia: diagnosis and treatment. Mayo Clin Proc 2006; 81 : 1105–1129.
35. Wiera WG. Current and invetigational therapies for patients with CLL. American Society of Hematology Education Program Book 2006 : 285–294.
36. Hallek et al. Blood 2008; 112 (Suppl): Abstract 325.
37. Hamblin TJ, Davis Z, Gardiner A et al. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood 1999; 94 : 1848–1854.
38. Crespo M, Bosch F, Villamor N et al. ZAP-70 expression as a surrogate for immunoglobulin-variable-region mutations in chronic lymphocytic leukemia. N Engl J Med 2003; 348 : 1764–1775.
39. Damle RN, Wasil T, Fais F et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood 1999; 94 : 1840–1847.
40. Sturm I, Bosanquet AG, Hermann S et al. Mutation of p53 and consecutive selective drug resistance in B-CLL occurs as a consequence of prior DNA-damaging chemotherapy. Cell Death Differ 2003; 10 : 477–484.
41. Döhner H, Stilgenbauer S, Benner Aet al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med 2000; 343 : 1910–1916.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2009 Issue 9
Most read in this issue
- Hladiny cholesterolu v závislosti na věku
- Chronická B-lymfatická leukemie a jí podobné stavy
- Jaterní cirhóza a její léčba
- Transplantace krvetvorných buněk