
Leukemie z dendritických buněk CD4+56+, typ DC2
CD4+56+ leukemia from dendritic cells type DC2
CD4+CD56+ malignancies are rare hematological tumours with poor prognosis affecting primarily the skin; despite good initial response to chemotherapy, they result in early relapse and rapid progression and dissemination of the disease into the bone marrow, peripheral blood and lymphatic nodes. Even though the origin of tumorous cells was initially being associated with NK cells because of the CD56 expression, recent studies suggest the disease is derived from precursor plasmocytoid dendritic cells. It is the co - expression of CD4 and CD56 and an absence of line - specific markers that defines this new entity within the last WHO - EORCT classification for cutaneous lymphomas. Patients with this immunophenotype have common clinical features and morphological findings. The specific genetic anomaly is not known. Immunohistochemical and flow cytometric analyses have an exclusive place in the diagnostics. We present two cases of CD4+CD56+ malignancies with different clinical course. In an 18 years old female, the disease presented as an acute leukemia without the typical cutaneous lesions, was chemoresistant and the patient died 12 months following diagnosis for relapse of the disease after allogeneic hematopoietic stem cell transplantation. In a 64 years old male, the disease manifested as cutaneous lymphoma only. Chemotherapy resulted in an 8 months lasting first complete remission. Treatment of the first relapse with disease dissemination resulted in a short-term 2nd complete remission. The second relapse followed and the patient died 2 years following diagnosis.
Key words:
CD4+CD56+ haematodermic neoplasm – DC2 cells – DC2 malignancy
Authors:
M. Pevná 1; J. Kissová 2; M. Doubek 1; Z. Adam 1; M. Klabusay 1
Authors‘ workplace:
Interní hematoonkologická klinika Lékařské fakulty MU a FN Brno, pracoviště Bohunice, přednosta prof. MU Dr. Jiří Mayer, CSc.
1; Oddělení klinické hematologie FN Brno, pracoviště Bohunice, přednosta prof. MU Dr. Miroslav Penka, CSc.
2
Published in:
Vnitř Lék 2010; 56(Supplementum 2): 183-187
Category:
Langerhans cell histiocytosis and some other Hematology rare diseases
Overview
CD4+CD56+ malignity jsou vzácné hematologické nádory primárně postihující kůži se špatnou prognózou. I přes dobrou iniciální odpověď na chemoterapii dochází k časnému relapsu a prudké progresi a diseminaci onemocnění do kostní dřeně, periferní krve a lymfatických uzlin. Ačkoli byl zprvu původ nádorových buněk vzhledem k expresi znaku CD56 vztahován k NK buňkám, nové studie svědčí o odvození z prekurzorové plazmocytoidní dendritické buňky. Právě vzácná koexprese znaků CD4 a CD56 a nepřítomnost markerů specifických pro linie definuje tuto novou jednotku v poslední WHO - EORTC klasifikaci pro kožní lymfomy. Skupina s tímto imunofenotypem sleduje společné klinické rysy a morfologický nález. Specifická genetická anomálie není známa. Své výhradní místo v diagnostice zde má imunohistochemická a flowcytometrická analýza. Uvádíme 2 případy CD4+CD56+ malignit s odlišným klinickým průběhem. U 18leté ženy se onemocnění prezentuje jako akutní leukemie bez typických kožních lézí, onemocnění je chemorezistentní a pacientka umírá na relaps po alogenní transplantaci krvetvorných buněk 12 měsíců od stanovení diagnózy. U 64letého muže se nemoc prezentovala pouze jako kožní lymfom. Chemoterapií bylo dosaženo 8 měsíců trvající první kompletní remise. První relaps s diseminací onemocnění byl léčen s odpovědí krátce trvající 2. kompletní remise. Poté nastal 2. relaps a pacient umírá 2 roky od stanovení diagnózy.
Klíčová slova:
CD4+CD56+ hematodermické neoplazma – DC2 buňky – DC2 malignita
Úvod
CD4+CD56+ hematodermické neo-plazma [1] nebo také leukemie z časných dendritických buněk DC2 je nově stanovenou jednotkou v poslední WHO - EORTC klasifikaci pro kožní lymfomy z roku 2005 [2], která je založena především na koexpresi CD4 a CD56 a na chybění znaků specifických pro linie (B lymfoidních, T lymfoidních nebo myeloidních buněčných markerů). Onemocnění se obvykle prezentuje na kůži solitárními nebo mnohočetnými noduly nebo tumory. U 1/ 2 pacientů jsou současně postiženy uzliny nebo kostní dřeň. U většiny pacientů pouze s kožními lézemi se rychle rozvíjí postižení kostní dřeně, periferní krve, lymfatických uzlin a extranodálních míst. Počáteční odpověď na léčbu je vysoká bez ohledu na její intenzitu [3], ale u většiny pacientů nastává časný relaps, leukemizace a diseminace onemocnění. Jedinou známou kurativní léčbou u této nově stanovené jednotky je alogenní transplantace kostní dřeně.
Jedná se o velmi vzácné onemocnění [4] s neobvyklou současnou expresí znaků CD4 a CD56, která byla prvně popsána v literatuře v roce 1995 [5]. V roce 2001 byl zjištěn in vitro testy a srovnáním imunofenotypu ontogenetický původ nádorových buněk v prekurzoru plazmocytoidní dendritické buňky (DC2) [6]. DC2 buňky nesou znaky CD4, BDCA - 2, BDCA - 4, CD45RA, CD68, CD123, a HLA-DR a jsou negativní na znaky specifické pro linie (CD3, CD11c, CD14, CD16, CD19 a CD56). Rozdílná mezi nádorovými a DC2 buňkami je exprese znaku CD56, která dříve vedla k připisování příslušnosti nádorových buněk k NK linii [7]. Znak CD56 přitom může být vedle DC2 malignit také aberantně exprimován např. u akutní myeloidní leukemie [8] nebo u mnohočetného myelomu [9].
Kazuistiky
Případ 1
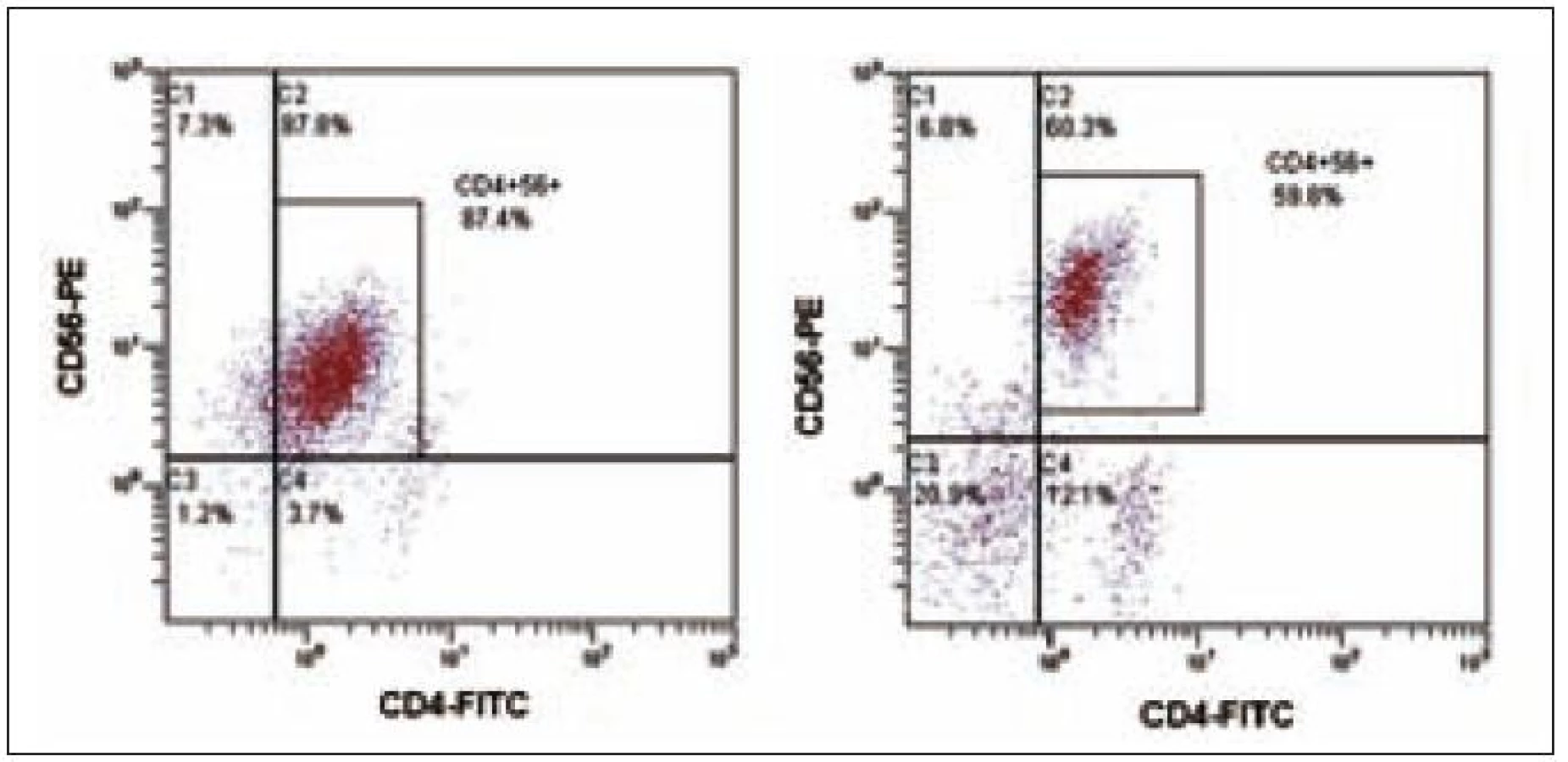
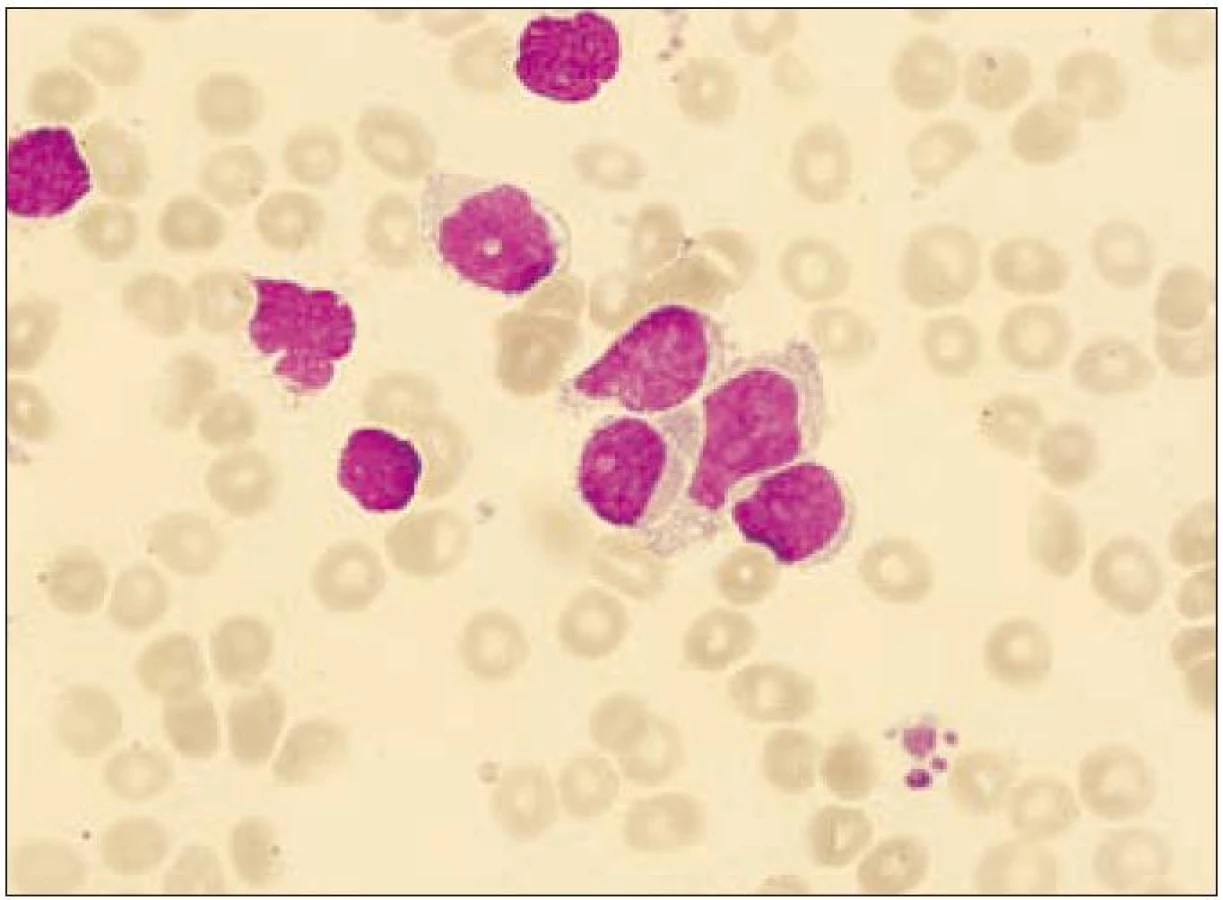
64letý pacient s anamnézou ischemické choroby srdeční, stavem po dvojnásobném aortokoronárním bypassu, hypertenzí gr. III, se prezentoval 3 blízko uloženými kožními ložisky v dubnu roku 2003, ze kterých byla histologicky stanovena diagnóza kožní lymfom z blastických NK buněk. Biopsie kožního ložiska ukázala infiltraci buňkami střední velikosti v celé retikulární dermis a přilehlém podkožním tuku, bez epidermo-tropizmu. Mitotická aktivita byla značná. Buňky vykazovaly pozitivitu CD56, CD43, slabě CD4, CD79a a zcela negativní byly znaky CD3, CD5, CD7, CD10, CD20, CD23, CD30, CD45RO, cyklin D1, TIA a TdT. Staging neprokázal diseminaci onemocnění. Pacient podstoupil 6 cyklů chemoterapie CHOP s efektem kompletní remise, ale v červnu roku 2004 nastal relaps základního onemocnění s postižením kůže. Dle počítačové tomografie krku, hrudníku a břicha nebyla prokázána patologická lymfadenopatie. Začátkem září roku 2004 byl pacient přijat na našem pracovišti ve fulminantní progresi kožních ložisek. Kůže byla postižena téměř v celém rozsahu splývajícími ložisky lividní barvy, ojediněle velikosti až 4 × 5 cm, některými exulcerovanými a barvy červené, fialové až černé. Zvětšené mízní uzliny byly lokalizovány bilaterálně na krku, supraklavikulárně a oboustranně pakety uzlin v axilách a tříslech. Tumor postihl i paranazální dutiny a pravou orbitu. Cytologie likvoru neprokázala infiltraci patologickými buňkami. Cytogeneticky nebyla shledána žádná anomálie. Vyšetření periferní krve při přijetí ukázalo WBC 28,9 × 109/ l (neutrofily 10 %, lymfocyty 41 %, monocyty 1 %, myelocyty 2 %, blasty 46 %), hematokrit 0,11, Hb 41 g/l, PLT 16 × 109/ l, kreatinin 149 µmol/ l, urea 10,7 mmol/ l, hodnoty jaterních enzymů v mezích normy, LD 9,45 µkat/ l, bilirubin 39,5 µmol/ l, PT 1,36 R, aPTT 38,4 s. Flowcytometrickou analýzou byla prokázána v periferní krvi i v kostní dřeni výrazná lymfocytóza (92 %), granulocytopenie a dominující patologická populace buněk fenotypu CD4+CD56+ (90 % lymfocytů) bez koexprese jiných specifických antigenů (tab. 1, obr. 1 a 2). Po léčbě septického stavu (etiologie Escherichia coli) byla v září roku 2004 zahájena chemoterapie MINE, která byla komplikována rozvojem tumor lysis syndromu. V říjnu roku 2004 následoval 1 cyklus chemoterapie ESHAP. Pacient dosáhl 2. kompletní remise. Vzhledem k vysokému věku a komorbiditám nebyla indikována alogenní transplantace kostní dřeně. Koncem ledna roku 2005 došlo k lokálnímu relapsu na levém bérci a pacient byl léčen radioterapií. V polovině února roku 2005 se však připojil relaps v kostní dřeni a CNS a pacient umírá v progresi nemoci přes záchrannou léčbu metotrexátem (MTX) 22 měsíců od stanovení diagnózy.

Případ 2
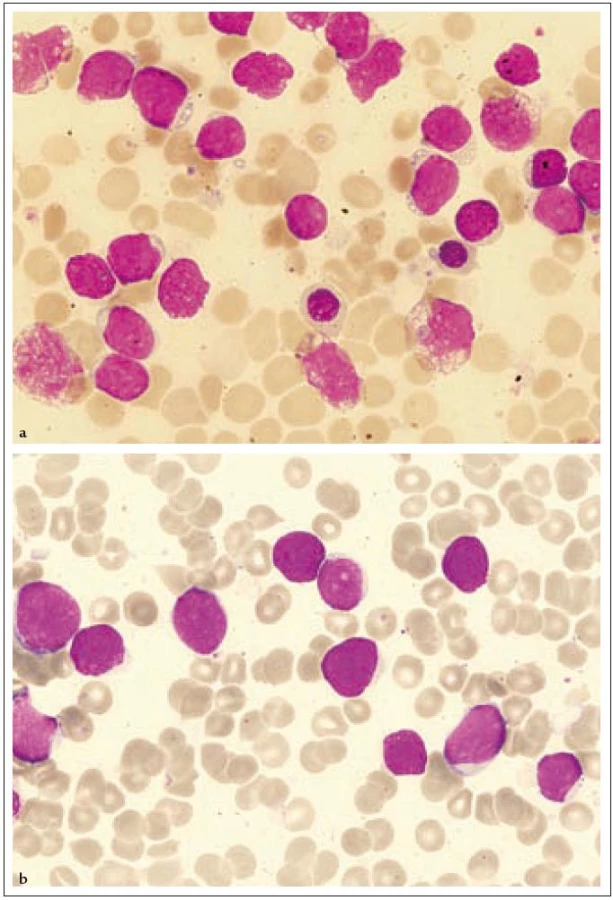
18letá pacientka byla poprvé vyšetřena na jiném pracovišti v březnu roku 2007 pro měsíc trvající subfebrilie, kašel, únavu a slabost. Pro nález nediferencovaných blastů v aspirátu kostní dřeně byla přeložena na naše pracoviště. Fyzikální vyšetření neprokázalo hepatomegalii nebo splenomegalii a lymfadenopatii a rovněž žádné kožní léze. Vyšetření periferní krve ukázalo WBC 3,83 × 109/ l, hematokrit 0,24, Hb 88 g/ l, PLT 37,3 × 109/ l, kreatinin 58 µmol/ l, LD 5,19 µkat/ l, CRP 2,4 mg/ l. Počítačová tomografie hrudníku a břicha byla bez patologie. Lumbální punkce neprokázala infiltraci nádorovými buňkami. Diagnóza CD4+56+ leukemie byla stanovena koncem března flowcytometrickým vyšetřením periferní krve, kde byla prokázána relativní lymfocytóza (61 %) a patologická populace buněk fenotypu CD4+56+ (22 % lymfocytů) bez koexprese jiných pro linie specifických antigenů (tab. 1, obr. 1). Biopsie kostní dřeně ukázala hypercelulární dřeň s extenzivní difuzní infiltrací malými okrouhlými a vřetenitými buňkami, z nichž 85 % buněk neslo imunofenotyp CD3–4+56+79a−HLA-DRdim+ TdT− granzym B− perforin− . Cytogenetické vyšetření neodhalilo žádnou anomálii. Začátkem dubna roku 2007 byla zahájena chemoterapie CHOP s následnou intratekální aplikací MTX, cytarabinu a dexamethasonu. Po 1. cyklu CHOP flowcytometrie prokazuje v kostní dřeni perzistující populaci 63 % buněk imunofenotypu CD4+56+ (obr. 3). Vzhledem k vysoké expresi CD52 byla zahájena v polovině května roku 2007 terapie alemtuzumabem (monoklonální protilátkou anti-CD52), která byla 2krát přerušena pro febrilní neutropenii a trombocytopenii. Týden po ukončení terapie protilátkou byla z histologie kostní dřeně zjištěna opět progrese, 100 % tvořily patologické buňky. Po léčbě sepse (etiologie Escherichia coli) byla nemocná indikována k příbuzenské alogenní transplantaci s přípravným režimem FLAMSA + TBI (total body irradiation) + cyklofosfamid a s imunosupresí cyklosporin A + my-kofenolát mofetil. Sedm měsíců po alogenní transplantaci trvala kompletní remise, ale u pacientky v polovině února roku 2008 došlo k otoku dásní a z histologie byl diagnostikován relaps nádoru. V aspirátu kostní dřeně byla nalezena 62% infiltrace nádorovými buňkami. Přes záchrannou léčbu mitoxantronem a cytarabinem pacientka umírá v progresi 12 měsíců od stanovení diagnózy.
Etiologie a klinické znaky
Etiologické a patogenetické faktory DC2 malignit jsou neznámé, vliv latentní virové infekce (EBV, HSV - 6, HTLV - 1) nebyl prokázán [3,10].
CD4+CD56+ malignity jsou jednotnou skupinou z pohledu klinického průběhu. Postihují starší pacienty (průměrný věk 67 let) a muže 3krát častěji než ženy. Z původně kožních lézí se nemoc rychle šíří do extrakutánních míst, nejčastěji do kostní dřeně, lymfatických uzlin, sleziny a jater. Ostatní orgány jako nazofarynx, tonsily, CNS, slzné kanálky, svaly nebo gynekologický trakt mohou být postiženy sporadicky [3].
Byla zaznamenána transformace DC2 malignit v myelomonocytární leukemii [11,12]. Diagnózu DC2 malignit může předcházet anamnéza myelodysplastického syndromu anebo mohou být nalezeny myelodysplastické rysy v kostní dřeni v době diagnózy [13,14].
Morfologie, imunofenotyp a cytogenetika
V kožních lézích maligní buňky difuzně postihují dermis bez epidermotropizmu a angiotropizmu [1,13]. Cytoplazma obsahuje prominující vakuoly a je myeloperoxidáza a butyrát - esteráza negativní [1,5,13].
Maligní CD4+CD56+ buňky se podobají blastům akutní leukemie a rovněž při vyšetření flowcytometrií ukazují nízkou expresi znaku CD45 (CD45dim+). Progenitorové markery CD10, CD34 a CD117 zcela chybí [6]. Znaky B linie nejsou přítomny a z markerů T linie mohou být v některých případech pozitivní CD2, CD5 a CD7, ale T buněčný znak CD3 je negativní [3,6]. Obecně, myeloidní markery nejsou přítomny, ačkoli ani exprese myeloidního znaku CD33 nemusí vylučovat diagnózu DC2 malignity [15]. Monocytární znaky CD68 a CD36 a dále HLA-DR a CD38 bývají exprimovány [1,3,6,13,16 – 18]. Původ CD4+CD56+ malignit v DC2 buňkách podporují exprese znaků CD4, CD45RA, CD101, CD123, BDCA - 2 (blood dendritic cell antigen - 2) a negativita znaků CD45RO a CD11c (11c+ jsou myeloidní dendritické buňky neboli DC1) [6,19 – 21]. Pro odvození z této linie svědčí u nádorových buněk in vitro produkce IFN α v odpověď na stimulaci virem, přežití a diferenciace ve funkční dendritické buňky pod vlivem IL-3 a indukce Th2 polarizace při aktivaci IL-3 [6].
Neexistuje žádná konzistentní chromozomální odchylka, byly ale zaznamenány reprodukovatelné ztráty chromozomální hmoty na chromozomech 5q, 6q, 9, 12p, 13q a 15q [22].
Léčba
Vzhledem k neznámé optimální terapii vzácných DC2 malignit, léčebné přístupy se u publikovaných případů značně lišily. Jejich efekt shrnuje práce autorů Reimer et al [3]. Iniciální odpověď na léčbu je vysoká, až 70 % pacientů dosahuje kompletní remise bez ohledu na to, zda byly užity pouze lokální terapie nebo systémové terapie různé intenzity (od „menší než CHOP“ až po myeloablativní terapii s transplantací kostní dřeně). U většiny pacientů ale nastává relaps a progrese onemocnění, přičemž medián celkového přežití je 13 měsíců. Udržení kompletní remise je lepší jen s agresivnějšími protokoly léčby (protokoly pro akutní leukemie) a výrazný pozitivní vliv na celkové přežití má jedině myeloablativní režim s transplantací kostní dřeně (průměrné přežití 31,5 měsíce).
Diskuze
64letý pacient svým věkem při diagnóze, klinickým průběhem, morfologickým nálezem i imunofenotypem naplňuje kritéria této nově definované jednotky. Nebyla shledána žádná cytogenetická anomálie, a ačkoliv byla popsána cílová místa genetických alterací [22], nejsou pravidlem a není známa specifická genetická anomálie. Pacient měl závažná přidružená onemocnění, proto nemohlo být přistoupeno k intenzivním protokolům ani k alogenní transplantaci kostní dřeně, zatím jediné léčebné modalitě schopné u této diagnózy navodit dlouhotrvající kompletní remisi. Chemoterapií CHOP byla navozena první kompletní remise (1. CR), ale po 8 měsících nastal relaps kožních ložisek a fulminantní progrese onemocnění s diseminací do uzlin, kostní dřeně, sleziny i paranazálních dutin a pravé orbity. Chemoterapií ESHAP a MINE se podařilo dosáhnout krátce trvající 2. CR, nastal však 2. relaps opět zpočátku s lokálním postižením kůže a záhy s diseminací do kostní dřeně a CNS.
U 18leté pacientky se nemoc prezentovala ve velmi mladém věku, ačkoliv ani dětský věk u této diagnózy není výjimkou [13], jako akutní leukemie bez kožních ložisek pro tuto diagnózu typických, která se neobjevila ani v dalším průběhu onemocnění. Onemocnění bylo od počátku chemorezistentní, bez odpovědi na chemoterapii CHOP, která byla nejužívanější léčbou u této nové jednotky a byla schopna navodit kompletní remisi u 55 % pacientů [3]. Byla započata imunoterapie alemtuzumabem vzhledem k vysoké expresi antigenu CD52, ale vzhledem k časnému přerušení pro nežádoucí účinky její efekt nemohl být pozorován. Pacientka podstoupila příbuzenskou transplantaci kostní dřeně, kterou se podařilo navodit efekt první kompletní remise. Po 7 měsících ale nemoc relabovala a pacientka umírá v progresi choroby.
CD4+56+ DC2 leukemie je vzácné nádorové onemocnění vykazující se nejčastěji infiltrací kůže, ale jak dokazuje 2. případ, nemusí ani to být pravidlem. Lze se pouze domnívat, že čistě leukemizující forma onemocnění může být prognosticky nepříznivým faktorem. Prognóza onemocnění je velmi závažná. V léčbě lze doporučit režimy kombinované intenzivní chemoterapie s intratekální profylaxí leukemické infiltrace CNS s cílem navození remise a indikaci alogenní transplantace pokud možno časně v 1. CR, což je v současnosti dle literárních údajů jediná kurativní alternativa tohoto onemocnění. Suverénní diagnostickou metodou je zde flowcytometrie.
Seznam zkratek
- CHOP – cyklofosfamid, adriamycin, vinkristin a prednison
- MINE – mitoxantron, ifosfamid, etoposid a mesna
- ESHAP – etoposid, methylprednisolon, cytosinarabinosid a cisplatina
- MTX – metotrexát
- FLAMSA – fludarabin, cytosinarabinosid a amsakrin
- TBI – celotělové ozáření, total body irradiation
- HSV - 6 – lidský herpervirus - 6
- HTLV - 1 – lidský lymfotropní virus T buněk typu I
- 1. CR – první kompletní remise
Tato práce byla podpořena grantem Interní grantové agentury Ministerstva zdravotnictví ČR číslo NS 9671 - 4.
doc. MUDr. Martin Klabusay, Ph.D.
www.fnbrno.cz
e-mail: mklabus@fnbrno.cz
Doručeno do redakce: 20. 9. 2010
Sources
1. Petrella T, Dalac S, Maynadié M et al. CD4+ CD56+ cutaneous neoplasms: a distinct hematological entity? Am J Surg Pathol 1999; 23 : 137 – 146.
2. Willemze R, Jaffe ES, Burg G et al. WHO - EORTC classification for cutaneous lymphomas. Blood 2005; 105 : 3768 – 3785.
3. Reimer P, Rüdiger T, Kraemer D et al. What is CD4+CD56+ malignancy and how should it be treated? Bone Marrow Transplant 2003; 32 : 637 – 646.
4. Bueno C, Almeida J, Lucio P et al. Incidence and characteristics of CD4+/ HLA DRhi dendritic cell malignancies. Haematologica 2004; 89 : 58 – 69.
5. Brody JP, Allen S, Schulman P et al. Acute agranular CD4 – positive natural killer cell leukemia. Comprehensive clinicopathologic studies including virologic and in vitro culture with inducing agents. Cancer 1995; 75 : 2474 – 2483.
6. Chaperot L, Bendriss N, Manches O et al. Identification of a leukemic counterpart of the plasmacytoid dendritic cells. Blood 2001; 97 : 3210 – 3217.
7. DiGiuseppe JA, Louie DC, Williams JE et al. Blastic natural killer cell leukemia/ lymphoma: a clincopathologic study. Am J Surg Pathol 1997; 21 : 1223 – 1230.
8. Scott AA, Head DR, Kopecky KJ et al. HLA-DR – , CD33+, CD56+, CD16 – myeloid/ natural killer cell acute leukemia: a previously unrecognized form of acute leukemia potentially misdiagnosed as French - American - British acute myeloid leukemia M3. Blood 1994; 84 : 244 – 255.
9. Van Camp B, Durie BG, Spier C et al. Plasma cells in multiple myeloma express a natural killer cell-associated antigen: CD56(NKH - 1; Leu - 19). Blood 1990; 76 : 377 – 382.
10. Savoia P, Fierro MT, Novelli M et al. CD56 - positive cutaneous lymphoma: a poorly recognized entity in the spectrum of primary cutaneous disease. Br J Dermatol 1997; 137 : 966 – 971.
11. Khoury JD, Medeiros LJ, Manning JT et al. CD56+ TdT+ blastic natural killer cell tumor of the skin: a primitive systemic malignancy related to myelomonocytic leukemia. Cancer 2002; 94 : 2401 – 2408.
12. Herling M, Teitell MA, Shen RR et al. TCL1 expression in plasmacytoid dendritic cells (DC2s) and the related CD4+ CD56+ blastic tumors of skin. Blood 2003; 101 : 5007 – 5009.
13. Feuillard J, Jacob MC, Valensi F et al. Clinical and biologic features of CD4+CD56+ malignancies. Blood 2002; 99 : 556 – 1563.
14. Kazakov DV, Mentzel T, Burg G et al. Blastic natural killer - cell lymphoma of the skin associated with myelodysplastic syndrome or myelogenous leukaemia: a coincidence or more? Br J Dermatol 2003; 149 : 869 – 876.
15. Garnache - Ottou F, Chaperot L, Biichle Set al. Expression of the myeloid-associated marker CD33 is not an exclusive factor for leukemic plasmacytoid dendritic cells. Blood 2005; 105 : 1256 – 1264.
16. Petrella T, Comeau MR, Maynadié M et al. “Agranular CD4+ CD56+ hematodermic neoplasm “ (blastic NK - Cell lymphoma) originates from a population of CD56+ precursor cells related to plasmacytoid monocytes. Am J Surg Pathol 2002; 26 : 852 – 862.
17. Herling M, Teitell MA, Shen RR et al. TCL1 expression in plasmacytoid dendritic cells (DC2s) and the related CD4+ CD56+ blastic tumors of skin. Blood 2003; 101 : 5007 – 5009.
18. Ko YH, Kim SH, Park K et al. CD4+CD56+CD68+ hematopoietic tumor of probable plasmacytoid monocyte derivation with weak expression of cytoplasmic CD3. J Korean Med Sci 2002; 17 : 833 – 839.
19. Trimoreau F, Donnard M, Turlure P et al. The CD4+ CD56+ CD116 – CD123+ CD45RA+ CD45RO – profile is specific to DC2 malignancies. Haematologica 2003; 88: ELT10.
20. Meyer N, Petrella T, Poszepczynska - Guigné E et al. CD4+ CD56+ blastic tumor cells express CD101 molecules. J Invest Dermatol 2005; 124 : 668 – 669.
21. Urosevic M, Conrad C, Kamarashev J et al. CD4+ CD56+ hematodermic neoplasms bear a plasmacytoid dendritic cell phenotype. Hum Pathol 2005; 36 : 1020 – 1024.
22. Leroux D, Mugneret F, Callanan M et al. CD4+, CD56+ DC2 acute leukemia is characterized by recurrent clonal chromosomal changes affecting 6 major targets: a study of 21 cases by the Groupe Francais de Cytogénétique Hématologique. Blood 2002; 99 : 4154 – 4159.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2010 Issue Supplementum 2
Most read in this issue
- Hemofagocytující lymfohistiocytóza
- Erdheimova-Chesterova nemoc v obrazech
- Systémová mastocytóza
- Histiocytóza z Langerhansových buněk u dětí a dospívajících