
Postižení CNS histiocytózou z Langerhansových buněk a Erdheimovou-Chesterovou nemocí. Přínos PET-CT vyšetření pro diagnostiku a vyhodnocení léčebné odpovědi
CNS sequelae in Langerhans cell histiocytosis and Erdheim-Chester disease. The importance of PET-CT for the diagnostics and evaluation of treatment response
Our centre monitors 23 patients with Langerhans cell histiocytosis (LCH) and 2 patients with Erdheim-Chester disease. Of this group of 25, 8 patients have some form of histiocytosis-associated CNS involvement. Four of the 8 patients had been referred to our centre for diabetes insipidus that developed in adulthood. In these patients, PET-CT was performed to detect potential extracranial signs of the disease that had induced diabetes insipidus. PET-CT revealed extracranial pathological changes; histological examination was performed on biopsies from these lesions. LCH was confirmed in two patients and Erdheim-Chester disease in the other two. In the fifth patient, an intracranial expansion from the occipital bone was detected that compressed the brain in the area of visual cortex and caused visual field failure. A follow-up MR and PET-CT were performed after a last cycle of cladribine treatment and confirmed complete remission. The sixth patient with headache had suggestive yet unclear MR finding in the area of temporal lobe. The MR signal changes could have been interpreted as delayed post-radiation changes or as LCH infiltrations of this part of the brain. PET-CT imaging confirmed pathological accumulation of fludeoxyglucose in this area, corresponding to malignant infiltration. As detected during a follow up examination, fludeoxyglucose accumulation declined from SUV 12 to SUV 5 and confirmed sensitivity of the disease to the 2 administered cycles of cladribine. The seventh and eighth patient had LCH diagnosed in childhood but the neurological sequelae, such as ataxia and dysarthria, did not occur before they reached adulthood. PET-CT examination showed reduced accumulation of fludeoxyglucose in cerebellum and in basal ganglia, corresponding to an MR depiction of atrophy in this area, and, at the same time, excluded a relapse of the disease.
Conclusion:
The whole body PET-CT examination might be useful in identification of the causes of diabetes insipidus and it may confirm active foci of LCH in the brain, while decreased fludeoxyglucose accumulation is a typical sign of delayed neurodegenerative changes rarely occurring following long-term course of LCH.
Key words:
PET-CT – Langerhans cell histiocytosis – Erdheim-Chester disease – diabetes insipidus – cladribine – 2-chlorodeoxyadenosine
Authors:
Z. Adam 1; Z. Řehák 2; R. Koukalová 2; P. Szturz 1; L. Pour 1; M. Krejčí 1; T. Nebeský 3; J. Vaníček 4; R. Hájek 1; J. Mayer 1
Authors‘ workplace:
Interní hematoonkologická klinika Lékařské fakulty MU a FN Brno, pracoviště Bohunice, přednosta prof. MUDr. Jiří Mayer, CSc.
1; Oddělení nukleární medicíny a pozitronové emisní tomografie Masarykova onkologického ústavu Brno, přednosta prim. MUDr. Karol Bolčák
2; Radiologická klinika Lékařské fakulty MU a FN Brno, pracoviště Bohunice, přednosta prof. MUDr. Vlastimil A. Válek, CSc.
3; Klinika zobrazovacích metod Lékařské fakulty MU a FN u sv. Anny Brno, přednosta doc. MUDr. Petr Krupa, CSc.
4
Published in:
Vnitř Lék 2010; 56(Supplementum 2): 94-104
Category:
Langerhans cell histiocytosis and some other Hematology rare diseases
Overview
Na našem pracovišti registrujeme celkem 23 pacientů s histiocytózou z Langerhansových buněk (LCH) a 2 pacienty s Erdheimovou-Chesterovou nemocí. Z tohoto 25členného souboru má 8 pacientů některou z forem postižení CNS histiocytárním onemocněním. Čtyři pacienti z této osmičlenné skupiny byli na našem pracovišti konzultováni pro diabetes insipidus vzniklý v dospělosti. Provedli jsme u nich PET-CT vyšetření s cílem detekovat extrakraniální projevy té nemoci, která způsobila také diabetes insipidus. PET-CT vyšetření u všech těchto pacientů odhalilo extrakraniální patologické změny, z nichž byl odebrán materiál pro histologické vyšetření. Ve dvou případech byla prokázána LCH a v dalších dvou případech Erdheimova-Chesterova nemoc. U pátého pacienta jsme pomocí MR vyšetření detekovali intrakraniální expanzi z okcipitální kosti, která komprimovala mozek v oblasti zrakového centra a způsobovala výpady zorného pole. Pomocí kontrolního MR a PET-CT vyšetření po ukončené léčbě kladribinem jsme potvrdili kompletní remisi. U šesté nemocné s bolestmi hlavy byl suspektní, ale nejasný MR nález z oblasti temporálního laloku. Změnu MR signálu bylo možné interpretovat jako pozdní poradiační změny anebo jako LCH infiltraci této části laloku. PET-CT zobrazení prokázalo patologicky zvýšenou akumulaci fluorodeoxyglukózy v této oblasti, odpovídající maligní infiltraci. Pokles akumulace fluorodeoxyglukózy při kontrolním vyšetření po 2 cyklech kladribinu z SUV 12 na 5 potvrdil senzitivitu nemoci na aplikovanou léčbu. U sedmého a osmého pacienta byla diagnóza LCH stanovena již v dětství a teprve v dospělosti vznikly neurologické problémy typu ataxie a dysatrie. PET-CT vyšetření prokázalo sníženou akumulaci fluorodeoxyglukózy v cerebellu a v bazálních gangliích, odpovídající MR obrazu atrofie této oblasti a zároveň vyloučilo recidivu nemoci.
Závěr:
Celotělové PET-CT vyšetření může být přínosem pro rozpoznání příčiny diabetes insipidus, může potvrdit aktivní ložiska LCH v mozkové tkáni, a naopak snížení akumulace fluorodeoxyglukózy je typickým projevem pozdních neurodegenerativních změn, které vzácně vznikají po mnohaletém průběhu LCH.
Klíčová slova:
PET-CT – histiocytóza z Langerhansových buněk – Erdheimova-Chesterova nemoc – diabetes insipidus – kladribin – 2-chlorodeoxyadenosin
Úvod
Histiocytóza z Langerhansových buněk je vzácné onemocnění. V dětském věku má tato nemoc agresivnější průběh, než je tomu v dospělosti. U dospělých pacientů má nemoc ve většině případů neagresivní průběh a nejsou výjimkou ani spontánní remise. Z hlediska rozsahu nemoci se rozlišují následující formy LCH:
- unifokální forma LCH: v těle je přítomno pouze jedno izolované ložisko. Obvykle to bývá solitární kostní eozinofilní granulom;
- multifokální forma LCH: v těle je více ložisek v rámci určité tkáně či orgánu. U dospělých to bývají nejčastěji vícečetná kostní ložiska nebo difuzní postižení plicního parenchymu;
- multisystémová (synonymem multiorgánová) forma LCH: nemoc postihuje více orgánů a tkání.
V dospělosti nemoc postihuje nejčastěji kosti ve formě eozinofilního kostního granulomu, o něco méně často pak kůži a plíce, postižení ostatních orgánů je vzácnější. Postižení centrálního nervového systému v dospělosti je mnohem vzácnější, než je tomu v dětském věku, ale má podobné formy jako v dětském věku:
- postižení hypotalamu a hypofýzy. Buňky LCH mají nevysvětlenou afinitu právě k hypotalamu a hypofýze. Klinicky se to nejčastěji projeví deficitem antidiuretického hormonu, klinické projevy deficitu ostatních hormonů jsou méně časté, ale je nutno s nimi počítat. Na MR zobrazení mozku je nejobvyklejším projevem infiltrát v oblasti stopky infundibula.
- infiltráty CNS mimo hypotalamus a hypofýzu. Tyto infiltráty mohou být buď důsledkem extraoseální expanze Langerhansových buněk z primárního kostního ložiska do CNS, nebo mohou vznikat v CNS bez souvislosti s kostmi kalvy.
- pozdní neurodegenerativní změny, obligátně počínající v oblasti cerebella a bazálních ganglií, způsobující ataxii a poruchu řeči. Později se neurodegenerace rozšiřuje i na obě hemisféry.
Postižení CNS lze detekovat metodou CT nebo MR. PET-CT vyšetření je nová metoda, s jejíž pomocí lze excelentně detektovat extrakraniální rozsah nemoci a po léčbě pak vyhodnocovat léčebnou odpověď u pacientů s LCH. Pouze nečetné publikace se věnují přínosu PET-CT pro postižení mozku touto chorobou, a proto v následujícím příspěvku chceme upozornit na přínos PET-CT vyšetření pro postižení CNS histiocytózou z Langerhansových buněk
Metody zobrazení a soubor pacientů
PET a PET-CT vyšetření, metodika
U pacientů souboru jsme prováděli PET a později PET-CT po 6hodinovém lačnění v euglykemii. Aplikovaná aktivita byla v rozmezí 312–409 MBq 18F-FDG i.v. Akumulační fáze byla 60 min. Akvizice byla prováděna do roku 2008 na PET skeneru ECAT ACCEL SIEMENS ve 3D modu (3 vyšetření u 1 nemocného v letech 2004–2007) a později od roku 2008 na hybridním PET-CT skeneru True Point PET-CT Biograph 64 SIEMENS, a to vždy v rozsahu proximální třetiny stehen – baze lební, ve 4 případech i se snímáním hlavy. U Erdheimovy-Chesterovy choroby byly snímány i končetiny. Snímána byla emisní a také transmisní data s korekcí absorpce a iterativní rekonstrukcí dat. Při akvizici na PET-CT skeneru je možné použít režim low dose CT (LD-CT) nebo high dose CT (HD-CT). Vzhledem k opakovaným kontrolám byl ve většině případů zvolen low dose CT protokol, díky kterému jsme výrazně zredukovali radiační zátěž. CT data byla rekonstruována v poli o šíři 500 nebo 700 mm dle habitu pacienta. Parametry LD-CT protokolu byly tyto: slice 5 mm, kolimace 24 × 1,2 mm, faktor stoupání (pitch) 0,8 mm. Parametry HD-CT protokolu byly zvoleny tyto: slice 5 mm, kolimace 24 × 1,2 mm, pitch 0,8 mm. Pro zvýraznění struktur plicního parenchymu byl použit rekonstrukční algoritmus: slice 1,5–2,0 mm, kernel B 80 f (ultra sharp), recon. increment 0,4 mm, window – lung.
Míru metabolické aktivity jsme stanovovali semikvantitativní analýzou SUVmax (maximum standardized uptake value) v měřitelné lézi (např. v lymfatické uzlině, ložisku).
Soubor pacientů
Na našem pracovišti registrujeme celkem 23 pacientů s LCH a 2 pacienty s Erdheimovou-Chesterovou nemocí [1,2]. Postižení CNS bylo prokázáno u 8 z celkového počtu 25 nemocných. Jedná se o 7 mužů a 1 ženu, medián věku, v němž bylo prokázáno postižení CNS histiocytárním onemocněním, je 34,4 (25–57) let. V dalším textu uvedeme popis jednotlivých případů a soubor pacientů rozdělíme dle typu poškození CNS.
Diferenciální diagnostika diabetes insipidus vzniklého v dospělosti
Na naše pracoviště byli odesláni celkem 4 pacienti s diabetes insipidus, zjištěným v dospělosti. Biopsie stopky hypofýzy je spojena s rizikem jejího poškození a minimum odebrané tkáně nemusí přitom vést k histologickému ověření diagnózy. Proto jsme u těchto osob provedli PET-CT vyšetření s cílem detekovat extrakraniální známky choroby, která vedla k poškození hypofýzy. Ve všech námi popsaných případech PET-CT vyšetření prokázalo extrakraniální patologická ložiska, z nichž jsme odebrali vzorky pro histologické vyšetření. Uvedeme stručný popis těchto případů a shrneme je do tabulky.
Pacient s diabetes insipidus a kožními projevy LCH
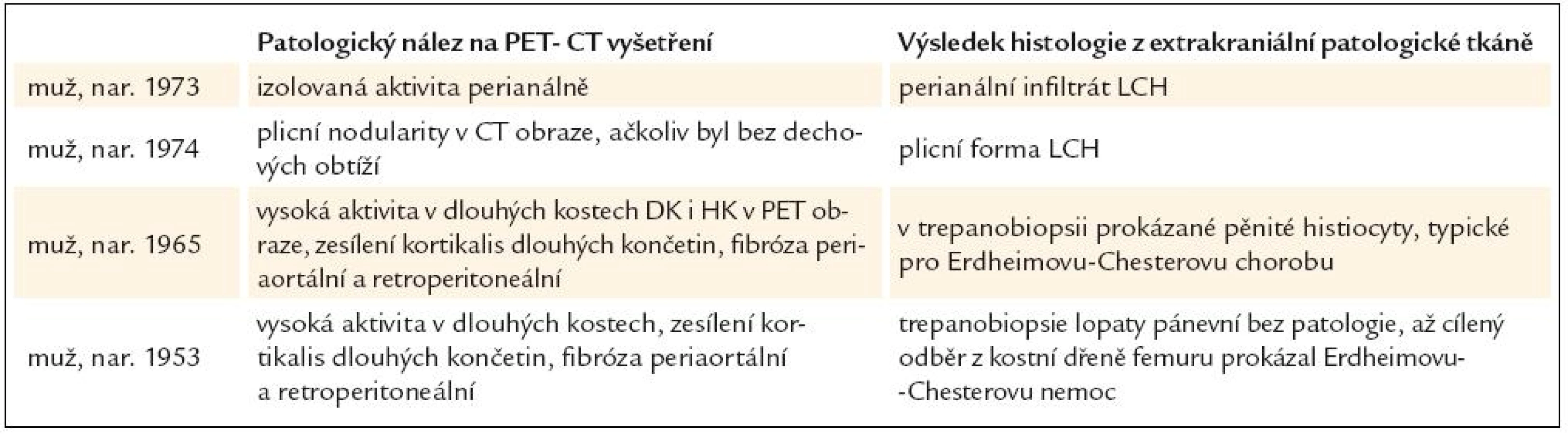
U 1. pacienta, narozeného 1973, byl dia-betes insipidus diagnostikován v roce 2005, ve 32 letech věku. MR mozku prokázala infiltrát v infundibulu hypofýzy. S tímto nálezem byl sledován bez dalšího zákroku.
V roce 2007 bylo provedeno PET vyšetření, které sice v oblasti CNS nezachytilo aktivitu odpovídající maligní tkáni, zatímco v oblasti perianální bylo jasné solitární ložisko patologicky zvýšené akumulace fluorodeoxyglukózy. Histologické vyšetření materiálu z verukózního periananálního infiltrátu prokázalo LCH.
Z nálezu kožní formy LCH jsme pak dedukovali, že infiltrát ve stopce hypofýzy bude také způsoben stejnou nemocí, protože u LCH je známa vysoká afinita této nemoci právě ke stopce hypofýzy a k hypotalamu.
Na základě PET vyšetření byla dia-gnóza uzavřena jako multisystémová forma postihující CNS a kůži, nebylo prokázáno postižení jiné tkáně či jiného orgánu, což bylo prognosticky příznivé. Po aplikaci 2-chlorodeoxyadenosinu vymizela perianální infiltrace a také vymizel infiltrát stopky hypofýzy.
Kontrolní PET-CT i MR mozku bylo provedeno v roce 2010 a bylo bez recidivy nemoci, infundibulum bylo nadále bez patologického infiltrátu.
Pacient s diabetes insipidus a asymptomatickou plicní formou LCH
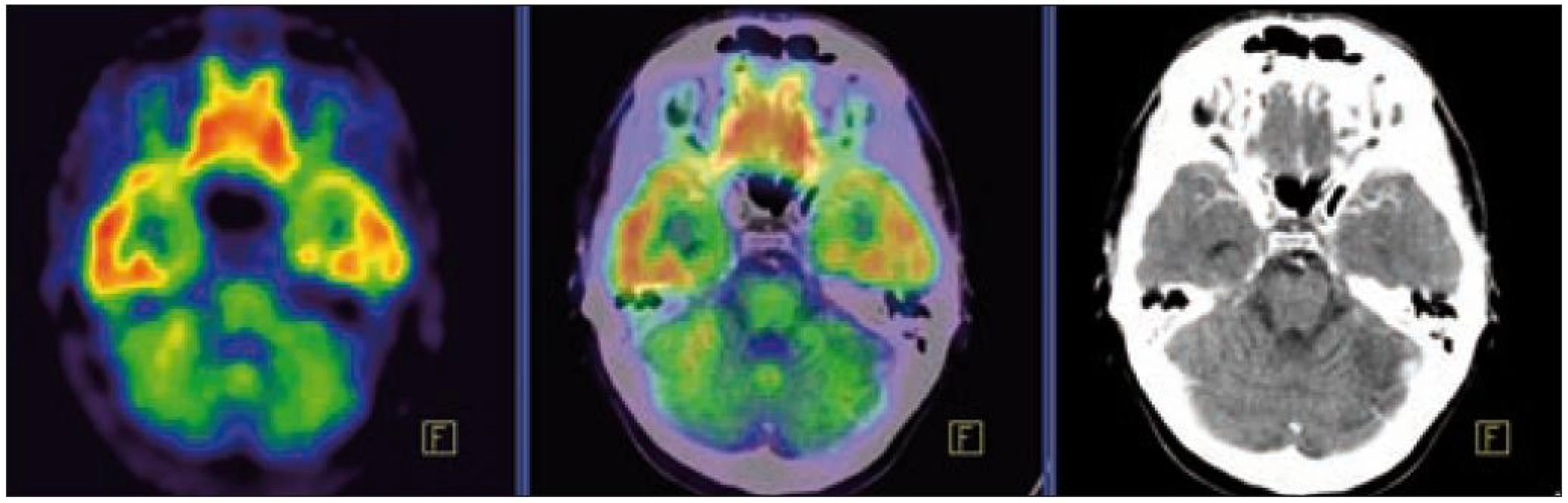
U 2. pacienta, narozeného 1974, byl diabetes insipidus prokázán v roce 2007, ve 33 letech věku. Současně byl zjištěn deficit dalších hormonů, dominantně androgenů. Při MR zobrazení byla zjištěna rozšířená stopka hypofýzy s patologickým infiltrátem. V roce 2009 bylo v rámci hledání příčiny diabetes insipidus provedeno PET-CT vyšetření. CT zobrazení prokázalo v plicním parenchymu četné drobné cystické útvary různého tvaru, o průměru 0,8–0,9 cm a ojedinělé drobné plicní nodularity a okrsky zesíleného intersticia. V PET obraze byla pouze hraniční akumulace fluorodeoxyglukózy v oblasti nazofaryngu.
V této fázi plicního postižení neměl mladý muž zatím žádné plicní příznaky a funkční vyšetření plic bylo normální. Následující bronchoalveolární laváž prokázala 10 % buněk exprimujících CD1a antigen a obsahujících protein S100. Tento nález odpovídal plicnímu postižení LCH.
V tomto případě jsme pomocí PET-CT zobrazení detekovali postižení plic LCH, cytologicky je ověřili a podobně jako v předchozím případě soudíme, že infiltrát stopky hypofýzy bude mít původ v LCH. Na kontrolním MR vyšetření po 4. z plánovaných 6 cyklů 2-chlorodeoxyadenosinem bylo popsáno zmenšení průměru infiltrátu hypofýzy z 5,5 na 3,0 mm.
Zde uvádíme pouze MR obraz a fúze PET a MR zobrazení (obr. 1 a 2).

Pacient s diabetes insipidius, následovaným neurologickým postižením při Erdheimově-Chesterově nemoci
3. pacient, narozený 1965, měl v roce 2004 (v 39 letech) prokázán diabetes insipidus. Po zavedení substituce adiuretinem byl bez dalších zdravotních potíží do roku 2008, kdy se postupně rozvinula dysartrie a následně porucha hybnosti ve formě frustní pravostranné hemiparézy. V roce 2006 byl zjištěn první patologický nález na MR mozku – zesílení stopky hypofýzy patologickou infiltrací o průměru 4–5 mm. V následujícím roce byly nalezeny další infiltráty v CNS, již mimo hypofýzu. Na opakovaných MR zobrazeních do roku 2008 narůstal počet infiltrátů CNS a zvětšovala se jejich velikost. Neurochirurgové provedli v roce 2008 pokus o biopsii stopky hypofýzy. Histologické vyšetření získaného malého vzorku tkáně neobjasnilo diagnózu, ale následky diagnostického výkonu způsobily panhypopituitarizmus.
V roce 2009 jsme provedli PET-CT vyšetření. CT zobrazila nepravidelnou strukturu skeletu s nápadnými sklerotickými ložisky v jinak prořídlé kostní struktuře, změny byly nejvíce zřetelné v dlouhých kostech dolních končetin, dále pak v kostech pánve, kalvy, v pažních kostech, zatímco z páteře byl postižen pouze jeden obratel. Dalším patologickým nálezem bylo zesílení stěny aorty až na 8 mm (coated aorta). V retroperitoneu byly zřetelné fibrotické změny. Aplikovaná fluorodeoxyglukóza se akumulovala v dlouhých kostech a v dalších kostních ložiscích, popsaných na CT zobrazení, a dále v zesílené stěně hrudní a břišní aorty, kde byla hodnota SUV 3,6. Scintigrafie skeletu 99mTc-pyrofosfátem znázornila stejná kostní ložiska jako zobrazení pomocí fluorodeoxyglukózy. Všechny tyto změny odpovídaly obrazu Erdheimovy-Chesterovy nemoci.
Diagnóza Erdheimovy-Chesterovynemoci byla ověřena biopsií ložiska v lopatě kosti pánevní. PET-CT vyšetření v tomto případě odhalilo kostní extrakraniální projevy Erdheimovy-Chesterovy nemoci, která způsobila diabetes insipidus jako první příznak nemoci.
Pacient s diabetes insipidus, horečkou nejasného původu a bolestí horních i dolních končetin při Erdheimově-Chesterově nemoci
4. pacient, narozený 1953, měl od roku 2005 diabetes insipidus a také horečku nejasného původu. Dále jej trápily difuzní bolesti kostí v oblasti dolních končetin. Na MR zobrazení mozku byl popsán patologický infiltrát stopky hypofýzy. Tkáň hypofýzy byla v dorzální části objemnější a přitom byla absence vysokého signálu neurohypofýzy.
V roce 2009 bylo provedeno první PET-CT vyšetření. V CT obraze bylo nalezeno zesílení stěny aorty, periaortální fibróza v hrudní a ještě více v břišní oblasti, diskrétní fibrózní změny perirenálně a hyperostotické a sklerotické změny kostí dolních a horních končetin. V kostech končetin, v oblasti epifýz, se zvýšeně akumulovala fluorodeoxyglukóza s hodnotami SUV až 10.
Biopsie lopaty kosti kyčelní byla provedena před PET-CT vyšetřením a byla bez patologického nálezu. V oblasti lopat pánevních kostí však nebyla zvýšená akumulace fluorodeoxyglukózy. Proto po PET-CT zobrazení byla ortopedy provedena cílená biopsie femuru. Histologicky byla prokázána Erdheimova-Chesterova choroba.
Shrnutí přínosu PET-CT vyšetření prostanovení příčiny diabetes insipidus, který se objevil v dospělosti, uvádí tab. 1.
Pacienti s LCH, kteří mají postiženy kosti kalvy a u nichž dochází k extraoseální intrakraniální expanzi LCH tkáně do CNS nebo mají s kostmi nesouvisející infiltrát v CNS
Pacient s expanzí hmot LCH z okcipitální kosti do nitra kalvy, vyvolávající poruchu zraku
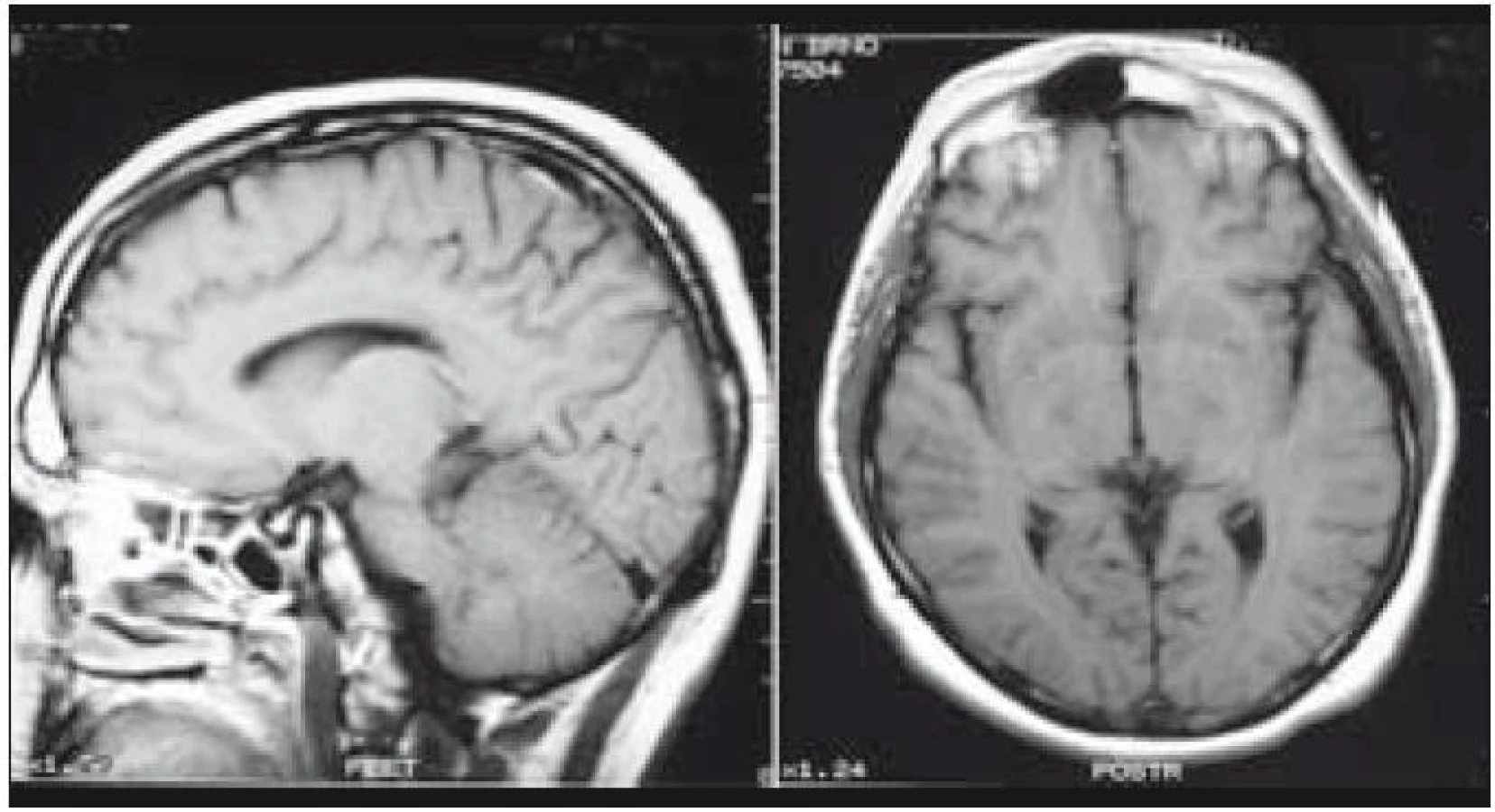
Muž, narozený 1978, měl v roce 2002 diagnostikované osteolytické ložisko ve femuru, z něhož byla histologicky ověřena LCH, původně považována za jednoložiskovou formu nemoci. V roce 2003 se mu v okcipitální lokalizaci objevilo měkké zduření o ploše 3 × 3 cm. Postupně, jak se ložisko zvětšovalo, si muž začal stěžovat na poruchy zraku s výpady zrakového pole. Z chirurgicky odstraněných měkkých hmot v okcipitální oblasti vyšla histologicky opět LCH. MR mozku znázornilo osteolýzu a infiltrát v okcipitální kosti, který se šířil oboustranně parasagitálně epidurálně až do oblasti zadní jámy lební, bez infiltrace dury. Prokázaná byla ale i další kostní ložiska, frontoparietálně vpravo a parietálně vlevo. U tohoto muže šlo tedy o multifokální postižení skeletu s expanzí do nitra kalvy.
Po léčbě kladribinem (2-chlorodeoxyadenosinem) intrakraniální expanze zcela vymizela a zcela vymizely i neurologické příznaky. Intrakraniální expanzi před léčbou a po léčbě znázorňují obr. 3 a 4. Metoda PET-CT byla použita až později, pro další sledování po léčbě, v roce 2010 měl tento muž negativní PET-CT vyšetření, které potvrdilo 7 let trvání remise.

Pacientka s bolestmi hlavy a s infiltrátem LCH v temporálním laloku
Žena, narozená 1953, měla diagnózu LCH zjištěnou ve 23 letech, takže za sebou má již 34letou historii opakovaných relapsů v oblasti lební baze, spánkové kosti a v poslední době i v krční páteři a také na kůži. V roce 2009 a 2010 se objevily bolesti hlavy.
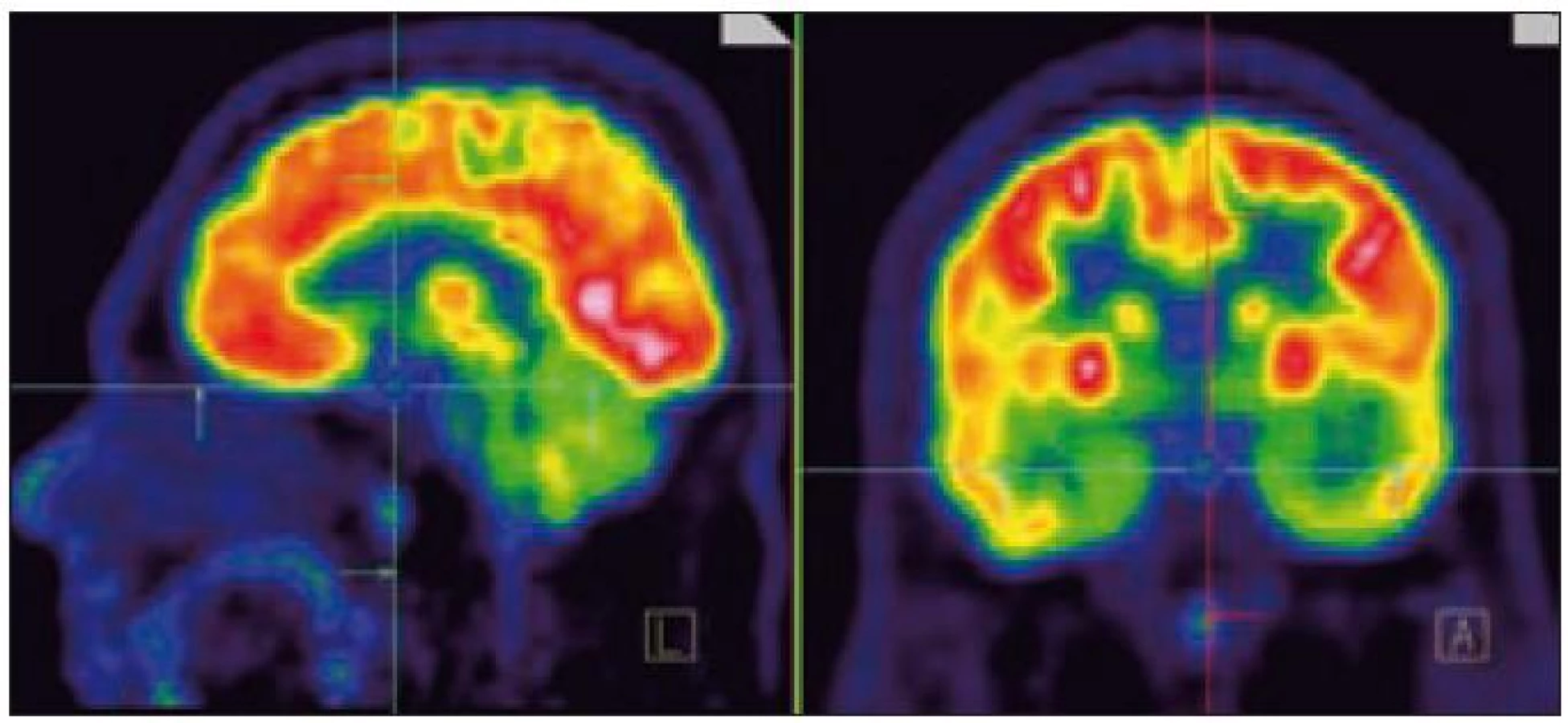
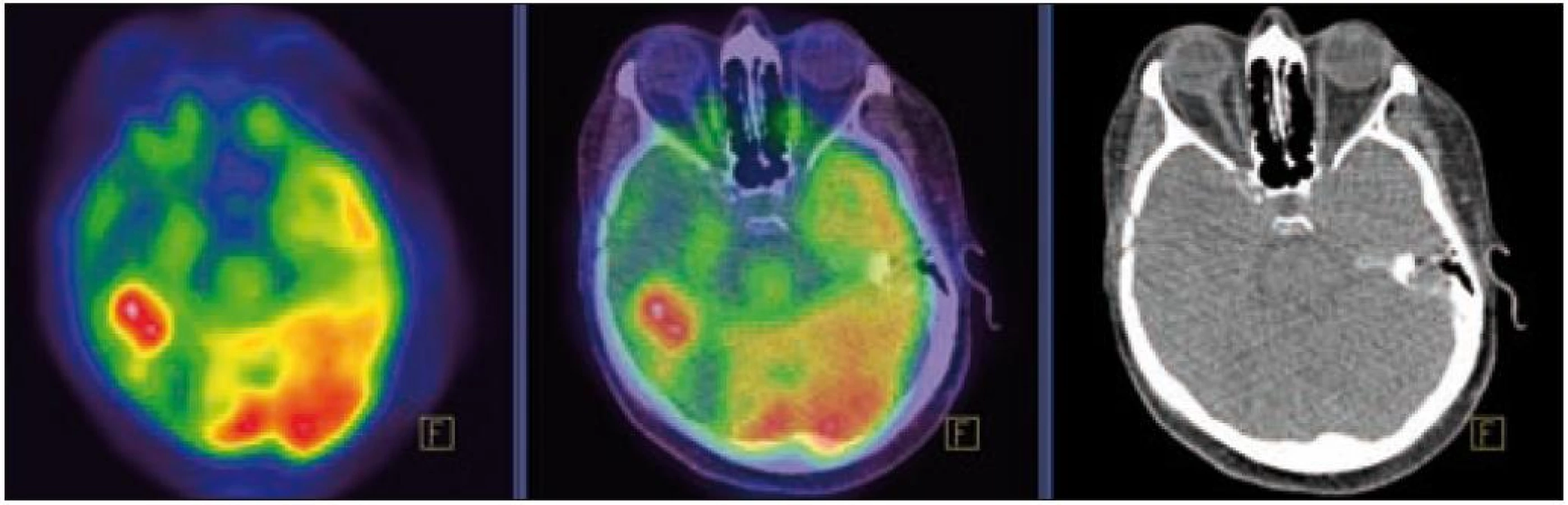
MR zobrazení hlavy potvrdilo postižení oblasti spánkové kosti, která byla již dříve opakovaně ozářena, kumulativní dávka činila 80 Gy. Zcela nově byly detekovány patologické změny v oblasti temporálního laloku, jak dokumentuje obr. 5 a 6. Interpretace změn, zřetelných na MR zobrazení, nebyla jednoznačná. Nebylo možné odlišit ložiskové poradiační změny v CNS od infiltrace mozkové tkáně v této oblasti. Vyšetření magnetickou rezonancí neumožnilo jednoznačný závěr o etiologii těchto změn. Proto bylo provedeno PET-CT vyšetření, které popsalo patologicky zvýšenou aktivitu v oblasti uvedeného ložiska. Na základě PET-CT vyšetření byla diagnóza upřesněna na LCH infiltrát temporálního laloku (obr. 7). Pokles akumulace fluorodeoxyglukózy při kontrolním vyšetření po 2 cyklech kladribinu (z SUV 12,53 na SUV 5,71) potvrdil senzitivitu této nemoci na aplikovanou léčbu.

Pacienti, u nichž se po mnohaletém průběhu LCH objevily neurologické příznaky typu ataxie a dysartrie
U 2 pacientů, u nichž od stanovení diagnózy uběhlo 20 a 34 let, se v průběhu sledování objevily neurologické problémy – dysartrie, poruchy chůze, závratě, porucha stability a koordinace pohybů. Stáli jsme tedy před otázkou, zda jde o relaps nemoci v CNS s infiltrací určitých struktur nebo o jiný typ poškození mozku. Odpovědi jsme se snažili získat pomocí PET-CT a MR vyšetření mozku.
Pacient s ataxií a dysartrií po 20 letech od stanovení diagnózy LCH
Muž, narozený v roce 1976, s LCH diagnostikovanou v dětském věku (diabetes insipidus a kožní infiltráty od 5 let věku), měl velmi časté recidivy nemoci a měl četná ložiska v kostech kalvy. V roce 2001 začal udávat diplopii a neurolog dále konstatoval ataxii a dysartrii. Provedené MR popsalo atrofické změny v oblasti bazálních ganglií a dále v bílé a šedé hmotě mozečku, odpovídající neurodegenerativním změnám, typickým pro LCH. Na kontrolním PET vyšetření byla zřetelně snížená akumulace fluorodeoxyglukózy v oblasti bazálních ganglií a v mozečku, bez jakéhokoliv dalšího ložiska v těle. Snížení akumulace fluorodeoxyglukózy v určité oblasti mozku signalizuje sníženou funkci, a tedy degenerativní změny. MR a PET-CT zobrazení umožnily uzavřít diagnózu jako pozdní neurodegenerativní postižení CNS bez prokazatelné aktivity LCH kdekoliv v těle. V průběhu dalších 7 let se neurologické poškození postupně zhoršovalo, pacient nebyl schopen ovládat svěrače, byl upoután na vozík a dysartrie ztěžovala komunikaci. V dubu 2008 pacient umřel, dle pitvy byla příčinou plicní embolie.
PET-CT a MR vyšetření v tomto případě potvrdilo hypometabolizmus v cerebellu, typický pro tento typ pozdních následků LCH.
Pacient s ataxií a dysartrií po 34 letech od stanovení diagnózy LCH
Muž, narozený v roce 1975, trpěl LCH od 1. roku života. První neurologické potíže se objevily v roce 1993 (dysatrie, nejistá chůze, závratě). Neurolog popsal centrální vestibulární syndrom, neúplný neocerebelární syndrom vpravo a naznačenou pravostrannou pyramidovou symptomatologii. Tyto problémy spontánně téměř ustoupily a mladý muž zvládal manuální zaměstnání. Při opakovaných kontrolách nebyla zjištěna recidiva nemoci. V září roku 2009 se však neurologický stav znovu náhle a výrazně zhoršil, objevily se opět závratě, nejistota při chůzi, zhoršená řeč, takže podobné potíže, jako měl v roce 1993, ale intenzivnější. Stáli jsme tedy před otázkou, zda jsou potíže způsobeny recidivou nemoci v CNS nebo jiným postižením CNS.
V září roku 2009 bylo provedeno PET-CT vyšetření, na němž byly v CT obraze zřetelné atrofické změny mozečku, rozšíření likvorových prostor. Při PET zobrazení byla nalezena souměrná a přiměřená aktivita v kůře mozkových hemisfér a v bazálních gangliích. Naproti tomu byla nalezena difuzně nízká akumulace fluorodeoxyglukózy v mozečkových hemisférách. PET-CT vyšetření trupu bylo bez patologie. Takže jediným patologickým nálezem při PET zobrazení byl hypometabolizmus (snížená akumulace fluorodeoxyglukózy) v oblasti cerebella a dle CT mozku zřetelná atrofie cerebella (obr. 8 a 9). MR zobrazení mozku došlo ke stejnému závěru, dominovala výrazná atrofie mozečku a rozšířená 4. komora (obr. 10–12). Informace o změnách v mozku získané pomocí PET-CT zobrazení a MR zobrazení odpovídají pozdním degenerativním změnám, které se výjimečně objeví po mnoha letech trvání této nemoci. Uvedená vyšetření zcela vyloučila možnost vzniku nového aktivního ložiska LCH v mozku nebo jinde v těle.
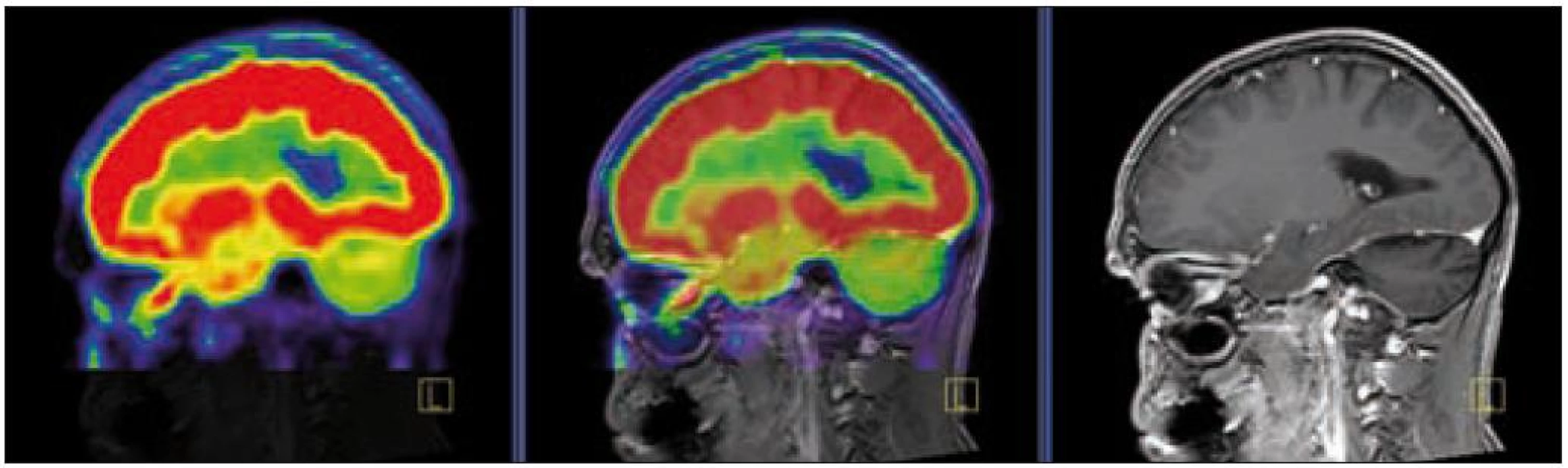


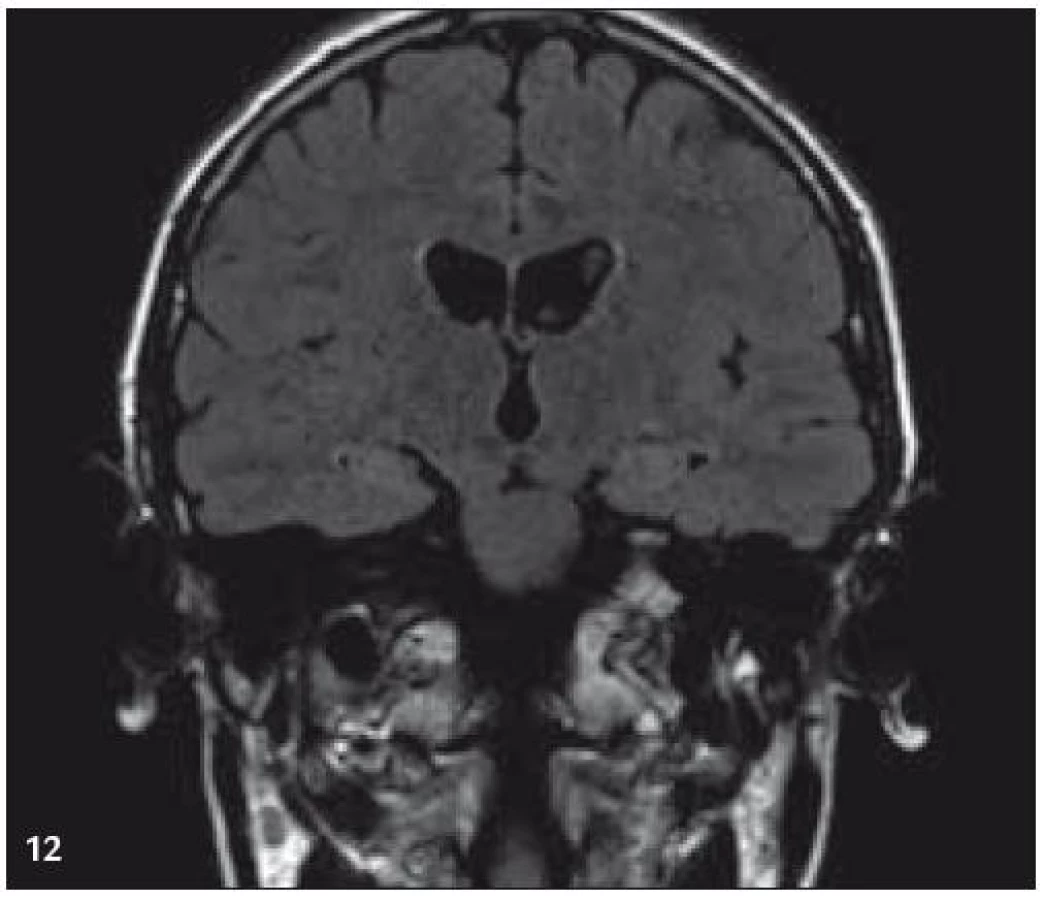
PET-CT vyšetření v tomto případě opět jednoznačně potvrdilo degenerativní změny v oblasti cerebella a zřetelně je odlišilo od možné nové infiltrace CNS v rámci recidivy nemoci.
Diskuze
LCH je vzácnou nemocí, obvykle v dospělosti postihuje kosti, plíce a kůži. V podstatě však může i v dospělosti postihnout kterýkoliv orgán. Z hlediska vlivu nemoci na kvalitu života je nejzávažnější poškození centrálního nervového systému. U dětí je postižení CNS častější než u dospělých, a proto odborná literatura popisuje častěji mozkovou formu LCH v dětském věku než v dospělosti [3–7]. V dospělosti, podobně jako je tomu u dětí, bývá nejčastější postižení hypotalamu a hypofýzy.
Na rozdíl od ostatních tkání, v nichž jsou popisovány spontánní recidivy, je poškození CNS opravdu jen zcela výjimečně přechodné [8,9], obvykle bývá trvalé [10].
Vzhledem k nemožnosti zkoumat průběh těchto komplikací opakovanými biopsiemi, musí poznání vycházet ze zobrazovacích metod, nemnohých výsledků biopsií a autoptických studií a případně analýz cerebrospinálního moku [11].
Postižení hypotalamu a hypofýzy
Buňky LCH mají nevysvětlenou afinitu právě k hypotalamu a k hypofýze. Klinicky se to nejčastěji projeví deficitem antidiuretického hormonu, čili diabetes insipidus. Bylo by však chybou domnívat se, že infiltrace stopky hypofýzy či hypotalamu způsobuje pouze diabetes insipidus. Obvykle bývá deficit i dalších hypofyzárních hormonů, který však nemusí způsobovat jasné klinické příznaky.
Protože LCH a diabetes insipidus patří k sobě, je vhodné si u každého pacienta s nově vzniklým diabetes insipidus položit otázku, zda právě LCH nezpůsobila diabetes insipidus. A dále, pokud prokážeme diabetes insipidus, je nutné cíleně pátrat po případném deficitu dalších hypofyzárních hormonů [10,15–17]. U dvou našich pacientů s infiltrátem stopky hypofýzy LCH je současně snížena hladina gonadotropinu a u jednoho našeho pacienta s Erdheimovou-Chesterovou nemocí je přítomen kompletní iatrogenní panhypopituitarizmus.
Diabetes insipidus v dospělosti mohou vyvolat i jiné choroby, PET-CT má však potenciál zachytit extrakraniální projevy i jiných nemocí, které mohou způsobovat diabetes insipidus [11,18–21,23–26].
Pro průkaz poškození hypotalamu a stopky hypofýzy či hypofýzy samotné se používá MR zobrazení. Nejčastěji je popisován infiltrát v oblasti stopky infundibula, méně často pak patologický infiltrát v hypofýze či obraz prázdného tureckého sedla [27–30].
Při patologické infiltraci jiných orgánů, než je CNS, se obvykle provádí biopsie a diagnóza se stanovuje na podkladě histologického hodnocení odebraného materiálu. Provedení biopsie infundibula hypofýzy však není bez rizika, jak nakonec můžeme ilustrovat na našem pacientovi, u něhož se po biopsii stopky hypofýzy rozvinul kompletní panhypopituitarizmus, aniž by tento zákrok vedl ke stanovení diagnózy. Komplikace při tomto zákroku jsou popisovány i jinými autory [31–35].
V případě našich 4 nemocných s diabetes insipidus přineslo PET-CT vyšetření informaci o extrakraniálních patologických ložiscích. Jednou to bylo ložisko s vysokou akumulací fluo-rodeoxyglukózy na kůži perianálně, z něhož byla histologicky prokázána LCH. V druhém případě CT komponenta celotělového PET-CT vyšetření odhalila strukturální změny plicního parenchymu typické pro plicní formu LCH u nemocného bez respiračních příznaků. Ve třetím a čtvrtém případě PET-CT vyšetření odhalilo vysokou akumulaci fluorodeoxyglukózy v dlouhých kostech, typickou pro Erdheimovu-Chesterovu chorobu.
Domníváme se proto, že u dospělého pacienta s nově vzniklým diabetes insipidus je vhodné provést PET-CT vyšetření s cílem detekovat extrakraniální projevy té nemoci, která způsobila diabetes insipidus, a jejich charakter pak ověřit biopsií.
Infiltráty CNS mimo hypotalamo-hypofyzární osu
Tyto infiltráty mohou vznikat jak propagací z primárního kostního ložiska do CNS, tak přímo v mozkových plenách či v CNS bez souvislosti s kostí. Popisují se případy nově vzniklé epilepsie, jejíž etiologií bylo ložisko LCH [36] či jiné neurologické příznaky dle lokalizace ložiska LCH v mozku [37–40]. Z literárních popisů je jasné, že tento typ postižení je méně častý než hypotalamicko-hypofyzární infiltrace. Tato ložiska jsou obvykle detekována pomocí CT a MR zobrazení.
V našem souboru máme jednoho pacienta s intrakraniální expanzí LCH v oblasti okcipitální, která způsobovala poruchu zraku, výpady zorného pole a jednu pacientku, u níž byla prokázána infiltrace v oblasti spánkového laloku bez MR prokazatelné spojitosti s kostním postižením. U této nemocné nebylo možné MR zobrazení jednoznačně interpretovat jako ložisko LCH v mozkové tkáni, protože podobnou změnu signálu na MR mohly způsobit změny v mozkové tkáni po radioterapii. PET-CT vyšetření v tomto případě prokázalo vysokou akumulaci fluorodeoxyglukózy, odpovídající maligní tkáni. Pokles akumulace fluorodeoxyglukózy v průběhu chemoterapie potvrdil maligní infiltraci a senzitivitu tohoto infiltrátu v CNS na podanou léčbu.
Léčba infiltrace CNS histiocytózou z Langerhansových buněk
Klasické léčebné postupy pro multifokální či multisystémovou formu nemoci, založené na merkaptopurinu a vinblastinu [41], nelze použít, protože tyto léky nepronikají v dostatečně účinné koncentraci do CNS. V současnosti se za lék první volby pro pacienty s infiltrací mozku LCH považuje kladribin, který v dostatečně účinné dávce proniká hematoencefalickou bariérou [42–46].
Tento lék jsme podali také našim pacientům s infiltrátem LCH ve stopce hypofýzy. V prvním případě jsme dosáhli vymizení infiltrátu infundibula a remise trvá již 5 let od ukončení léčby. V druhém případě je k dispozici zatím jenom kontrolní vyšetření po 4. cyklu chemoterapie, které prokázalo zmenšení průměru infiltrátu z 5,5 na 3,0 mm.
Z dvou pacientů s Erdheimovou-Chesterovou nemocí došlo k výraznému zlepšení pouze u jednoho z nich.
V případě infiltrátu LCH, který postihoval temporální lalok, jsme pomocí PET-CT zobrazení prokázali léčebnou odpověď již po 2. cyklu 2-chlorodeoxyadenosinu. V ložisku v temporálním laloku poklesla akumulace fluorodeoxyglukózy o více než o 50 % (SUV pokleslo z 12,53 na 5,71), takže dosaženou léčebnou odpověď hodnotíme jako parciální remisi.
Pozdní neurodegenerativní změny, obvykle počínající v mozečku a v bazálních gangliích, způsobující ataxii a poruchu řeči
U pacientů s diagnózou LCH stanovenou v dětství se po mnoha letech (obvykle až v dospělosti) velmi vzácně vyskytují takzvané pozdní neurodegenerativní změny. Tento typ postižení mozku postupně progreduje a je nevratný. Etiologie tohoto procesu není zcela známa a nemáme zatím ani účinnou léčbu, kterou by bylo možné proces pozastavit.
Pozdní neurodegenerativní změny postihují nejčastěji cerebellum, nucleusdentatus, cerebellární bílou hmotu a mozkový kmen. Projevují se hypore-flexií, ataxií, závratěmi, dysartrií, nystagmem, tremorem, diplopií, psychomotorickou retardací a neuropsychologickými defekty.
Současné patofyziologické informace o tomto procesu vycházejí z nemnohých autoptických a bioptických studií, které neprokázaly buňky, exprimující znak CD1+, v ložiscích postižených neurodegenerativními změnami. V ložiscích dominovala výrazná zánětlivá infiltrace, obsahující CD8 lymfocyty, což odpovídá T buněčnému zánětlivému procesu, který je provázen destrukcí neuronů a axonů se sekundární demyelinizací, připomínající paraneoplastickou encefalitidu [47–59].
První metodou, která zobrazila neurodegenerativní změny, bylo CT a posléze s rozvojem odpovídající technologie pak MR [60–64]. PET-CT vyšetření bylo pro detekci těchto změn použito až v poslední době a dle 3 publikací, které hodnotí přínos PET-CT pro tento typ poškození, umožňuje právě PET-CT zobrazení sledování dynamiky tohoto procesu [65–67].
V našich dvou případech odpovídala lokalizace snížení akumulace fluorodeoxyglukózy rozsahu pozdních denegerativních změn dle MR vyšetření. Nepřítomnost jiného patologického ložiska v těle nemocných odpovídala trvající remisi LCH.
Závěr pro praxi
- PET-CT vyšetření považujeme za velmi přínosné pro nemocné s diabetes insipidus vzniklým v dospělosti, může odhalit extrakraniální projevy té nemoci, která vedla ke vzniku diabetes insipidus, a nasměrovat cílený histologický odběr na tyto extrakraniální projevy.
- PET-CT vyšetření se osvědčilo při neurologických symptomech pacientů s LCH. Umí odlišit LCH infiltraci CNS s vysokou akumulací fluorodeoxyglukózy od pozdních neurodegenerativních změn.
Tato publikace byla připravena v rámci projeku MUNI/A/1012/2009 s názvem „Optimalizace diagnostiky a terapie maligních chorob a komplikací, které tyto maligní nemoci provázejí, s využitím nových molekulárně biologických metod“, a také je součástí aktivit v rámci grantů IGA MZ: NR9225, NS10387 a NS10406.
prof. MUDr. Zdeněk Adam, CSc.
www.fnbrno.cz
e-mail: z.adam@fnbrno.cz
Doručeno do redakce: 9. 9. 2010
Sources
1. Adam Z, Szturz P, Pour L et al. Histiocytóza z Langerhansových buněk u dospělých osob. Postgrad Med 2010; 12 : 704–711.
2. Adam Z, Krejčí M, Pour L et al. Odlišné průběhy recidivující anebo multisystémové formy histiocytózy z Langerhansových buněk. Popis 22 případů z jednoho pracoviště. Vnitř Lék 2010; 56 : 542–557.
3. Grois N, Prayer D, Prosch H et al. CNS LCH Co-operative Group. Neuropathology of CNS disease in Langerhans cell histiocytosis. Brain 2005; 128 : 829–838.
4. Report of the Histiocyte Society workshop on “Central nervous system (CNS) disease in Langerhans cell histiocytosis (LCH)”. Med Pediatr Oncol 2009; 29 : 73–78.
5. Grois N, Fahrner B, Arceci RJ et al. Histiocyte Society CNS LCH Study Group. Central nervous system disease in Langerhans cell histiocytosis. J Pediatr 2010; 156 : 873–881.
6. D’Ambrosio N, Soohoo S, Warshall C et al. Craniofacial and intracranial manifestations of Langerhans cell histiocytosis: report of findings in 100 patients. AJR Am J Roentgenol 2008; 191 : 589–597.
7. Davidson L, McComb JG, Bowen I et al. Craniospinal Langerhans cell histiocytosis in children: 30 years’ experience at a single institution. J Neurosurg Pediatr 2008; 1 : 187–195.
8. Ryan P, Walterfang M, Scholes A et al. Recovery of cognitive function in neuropsychiatric Langerhan’s cell histiocytosis. Psychiatry Clin Neurosci 2006; 60 : 629–632.
9. Gunny R, Clifton A, Al-Memar A. Spontaneous regression of supratentorial intracerebral Langerhans’ cell histiocytosis. Br J Radiol 2004; 77 : 685–687.
10. Nanduri VR, Bareille P, Pritchard J et al. Growth and endocrine disorders in multisystem Langerhans’ cell histiocytosis. Clin Endocrinol (Oxf) 2000; 53 : 509–515.
11. Stockschlaeder M, Sucker C. Adult Langerhans cell histiocytosis. Eur J Haematol 2006; 76 : 363–368.
12. Grois N, Prosch H, Waldhauser F et al. Pineal gland abnormalities in Langerhans cell histiocytosis. Pediatr Blood Cancer 2004; 43 : 261–266.
13. Horn EM, Coons SW, Spetzler RF et al. Isolated Langerhans cell histiocytosis of the infundibulum presenting with fulminant diabetes insipidus. Childs Nerv Syst 2006; 22 : 542–544.
14. Prosch H, Grois N, Prayer D et al. Central diabetes insipidus as presenting symptom of Langerhans cell histiocytosis. Pediatr Blood Cancer 2004; 43 : 594–599.
15. Prosch H, Grois N, Bökkerink J et al. Central diabetes insipidus: Is it Langerhans cell histiocytosis of the pituitary stalk? A diagnostic pitfall. Pediatr Blood Cancer 2006; 46 : 363–366.
16. Amato MC, Elias LL, Elias J et al. Endocrine disorders in pediatric – onset Langerhans Cell Histiocytosis. Horm Metab Res 2006; 38 : 746–751.
17. De Buyst J, Massa G, Christophe C et al. Clinical, hormonal and imaging findings in 27 children with central diabetes insipidus. Eur J Pediatr 2007; 166 : 43–49.
18. Carpinteri R, Patelli I, Casanueva FF et al. Pituitary tumours: inflammatory and granulomatous expansive lesions of the pituitary. Best Pract Res Clin Endocrinol Metab 2009; 23 : 639–650.
19. Müssig K, Beschorner R. Rare differential diagnosis of diabetes insipidus. Dtsch Med Wochenschr 2008; 133 : 2159–2160.
20. Rupp D, Molitch M. Pituitary stalk lesions. Curr Opin Endocrinol Diabetes Obes 2008; 15 : 339–345.
21. Makras P, Alexandraki KI, Chrousos GP et al. Endocrine manifestations in Langerhans cell histiocytosis. Trends Endocrinol Metab 2007; 18 : 252–257.
22. Ghirardello S, Garrè ML, Rossi A et al. The diagnosis of children with central diabetes insipidus. J Pediatr Endocrinol Metab 2007; 20 : 359–375.
23. Ghirardello S, Malattia C, Scagnelli P et al. Current perspective on the pathogenesis of central diabetes insipidus. J Pediatr Endocrinol Metab 2005; 18 : 631–645.
24. Maghnie M, Cosi G, Genovese E et al. Central diabetes insipidus in children and young adults. N Engl J Med 2000; 343 : 998–1007.
25. Maghnie M. Diabetes insipidus. Horm Res 2003; 59 (Suppl 1): 42–54.
26. Maghnie M, Ghirardello S, De Bellis A et al. Idiopathic central diabetes insipidus in children and young adults is commonly associated with vasopressin-cell antibodies and markers of autoimmunity. Clin Endocrinol (Oxf)2006; 65 : 470–478.
27. Varan A, Cila A, Akyüz C et al. Radiological evaluation of patients with pituitary langerhans cell histiocytosis at diagnosis and at follow-up. Pediatr Hematol Oncol 2008; 25 : 567–574.
28. Demaerel P, Van Gool S. Paediatric neuroradiological aspects of Langerhans cell histiocytosis. Neuroradiology 2008; 50 : 85–92.
29. Prayer D, Grois N, Prosch H et al. MR imaging presentation of intracranial disease associated with Langerhans cell histiocytosis. AJNR Am J Neuroradiol 2004; 25 : 880–891.
30. Halefoglu AM. Magnetic resonance imaging of thickened pituitary stalk proceeding to Langerhans cell histiocytosis in a child. Australas Radiol 2006; 50 : 175–178.
31. Killory BD, Ponce FA, Wait SD et al. Endoscopic intraventricular biopsy of infundibular Langerhans cell histiocytosis: case report. Neurosurgery 2009; 65: E214–E215.
32. Charalampaki P, Reisch R, Ayad A et al. Endoscopic endonasal pituitary surgery: surgical and outcome analysis of 50 cases. J Clin Neurosci 2007; 14 : 410–415.
33. Sudhakar N, Ray A, Vafidis JA. Complications after trans-sphenoidal surgery: our experience and a review of the literature. Br J Neurosurg 2004; 18 : 507–512.
34. Grois N, Pötschger U, Prosch H et al. Risk factors for diabetes insipidus in langerhans cell histiocytosis. Pediatr Blood Cancer 2006; 46 : 228–230.
35. Tashiro T, Sano T, Xu B et al. Spectrum of different types of hypophysitis: a clinicopathologic study hypophysitis in 31 cases. Endocr Pathol 2002; 13 : 183–185.
36. Jain RS. Langerhans cell histiocytosis presenting as adult onset epilepsy. Int J Clin Pract 2003; 57 : 739–741.
37. Patton N, Lai T, Robbins P et al. Presumed choroidal langerhans cell histiocytosis following a previously resected solitary central nervous system lesion in an adult. Arch Ophthalmol 2006; 124 : 1193–1195.
38. Manning L, Sellal F. Hypothalamic amnesia and frontal lobe function disorders after Langerhans cell histiocytosis. J Neurol Neurosurg Psychiatry 2003; 74 : 1348.
39. Ghosal N, Kapila K, Kakkar S et al. Langerhans cell histiocytosis infiltration in cerebrospinal fluid: a case report. Diagn Cytopathol 2001; 24 : 123–125.
40. Cagli S, Oktar N, Demirtas E. Langerhans’ cell histiocytosis of the temporal lobe and pons. Br J Neurosurg 2004; 18 : 174–180.
41. von Stebut E, Schadmand-Fischer S, Bräuninger W et al. Successful treatment of adult multisystemic Langerhans cell histiocytosis with psoralen-UV-A, prednisolone, mercaptopurine and vinblastine. Arch Dermatol 2008; 144 : 649–653.
42. Binning MJ, Brockmeyer DL. Novel multidisciplinary approach for treatment of langerhans cell histiocytosis of the skull base. Skull Base 2008; 18 : 53–58.
43. Montella L, Insabato L, Palmieri G. Imatinib mesylate for cerebral Langerhans’-cell histiocytosis. N Engl J Med 2004; 351 : 1034–1035.
44. Allen CE, McClain KL. Langerhans cell histiocytosis: a review of past, current and future therapies. Drugs Today (Barc) 2007; 43 : 627–643.
45. Watts J, Files B. Langerhans cell histiocytosis: central nervous system involvement treated successfully with 2-chlorodeoxyadenosine. Pediatr Hematol Oncol 2001; 18 : 199–204.
46. Dhall G, Finlay JL, Dunkel IJ et al. Analysis of outcome for patients with mass lesions of the central nervous system due to Langerhans cell histiocytosis treated with 2-chlorodeoxyadenosine. Pediatr Blood Cancer 2008; 50 : 72–79.
47. van der Knaap MS, Arts WF, Garbern JY et al. Cerebellar leukoencephalopathy: most likely histiocytosis-related. Neurology 2008; 71 : 1361–1367.
48. Wnorowski M, Prosch H, Prayer D et al. Pattern and course of neurodegeneration in Langerhans cell histiocytosis. J Pediatr 2008; 153 : 127–132.
49. Van’t Hooft I, Gavhed D, Laurencikas Eet al. Neuropsychological sequelae in patients with neurodegenerative Langerhans cell histiocytosis. Pediatr Blood Cancer 2008; 51 : 669–674.
50. Gavhed D, Akefeldt SO, Osterlundh G et al. Biomarkers in the cerebrospinal fluid and neurodegeneration in Langerhans cell histiocytosis. Pediatr Blood Cancer 2009; 53 : 1264–1270.
51. Shuper A, Stark B, Yaniv Y et al. Cerebellar involvement in Langerhans’ cell histiocytosis: a progressive neuropsychiatric disease. J Child Neurol 2000; 15 : 824–826.
52. Weiss SE, O’Connor L, Welsh JS. Refinement of radiation therapy based on PET data in an adult with Langerhans cell histiocytosis of soft tissues. Clin Adv Hematol Oncol 2006; 4 : 290–292.
53. Mittheisz E, Seidl R, Prayer D et al. Central nervous system-related permanent consequences in patients with Langerhans cell histiocytosis. Pediatr Blood Cancer 2007; 48 : 50–56.
54. Imashuku S, Ishida S, Koike K et al. Japan LCH Study Group. Cerebellar ataxia in pediatric patients with Langerhans cell histiocytosis. J Pediatr Hematol Oncol 2004; 26 : 735–739.
55. Grois N, Prayer D, Prosch H et al. Neuropathology of CNS disease in Langerhans cell histiocytosis. Brain 2005; 128 : 829–838.
56. Martin-Duverneuil N, Idbaih A, Hoang-Xuan K et al. French Langerhans Cell Histiocytosis Study Group. MRI features of neurodegenerative Langerhans cell histiocytosis. Eur Radiol 2006; 16 : 2074–2082.
57. Mittheisz E, Seidl R, Prayer D et al. Central nervous system-related permanent consequences in patients with Langerhans cell histiocytosis. Pediatr Blood Cancer 2007; 48 : 50–56.
58. Grois N, Fahrner B, Arceci RJ et al. Histiocyte Society CNS LCH Study Group. Central nervous system disease in Langerhans cell histiocytosis. J Pediatr 2010; 156 : 873–881.
59. Bös M, Grothe C, Urbach H et al. Cerebellar syndromes in Langerhans’ cell histiocytosis. Nervenarzt 2007; 78 : 437–440.
60. Prosch H, Grois N, Wnorowski M et al. Long-term MR imaging course of neurodegenerative Langerhans cell histiocytosis. AJNR Am J Neuroradiol 2007; 28 : 1022–1028.
61. Ertan G, Huisman TA. Susceptibility-weighted imaging in neurodegeneration in Langerhans cell histiocytosis. J Pediatr 2010; 156 : 1032.
62. Imashuku S, Okazaki NA, Nakayama M et al. Japan LCH Study Group. Treatment of neurodegenerative CNS disease in Langerhans cell histiocytosis with a combination of intravenous immunoglobulin and chemotherapy. Pediatr Blood Cancer 2008; 50 : 308–311.
63. Steiner M, Prayer D, Asenbaum S et al. Modern imaging methods for the assessment of Langerhans’ cell histiocytosis-associated neurodegenerative syndrome: case report. J Child Neurol 2005; 20 : 253–257.
64. Ribeiro MJ, Idbaih A, Thomas C et al. 18F-FDG PET in neurodegenerative Langerhans cell histiocytosis: results and potential interest for an early diagnosis of the disease. J Neurol 2008; 255 : 575–580.
65. Calming U, Bemstrand C, Mosskin M et al. Brain 18FDG PET scan in central nervous system langerhans cell histiocytosis. J Pediatr 2002; 141 : 435–440.
66. Büchler T, Cervinek L, Belohlavek O et al. Langerhans cell histiocytosis with central nervous system involvement: follow-up by FDG-PET during treatment with cladribine. Pediatr Blood Cancer 2005; 44 : 286–288.
67. Mahnel R, Tan KH, Fahlbusch R et al. Problems in differential diagnosis of non Langerhans cell histiocytosis with pituitary involvement: case report and review of literature. Endocr Pathol 2002; 13 : 361–368.
68. Salsano E, Savoiardo M, Nappini S et al. Late-onset sporadic ataxia, pontine lesion, and retroperitoneal fibrosis: a case of Erdheim-Chester disease. Neurol Sci 2008; 29 : 263–267.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2010 Issue Supplementum 2
Most read in this issue
- Hemofagocytující lymfohistiocytóza
- Erdheimova-Chesterova nemoc v obrazech
- Systémová mastocytóza
- Histiocytóza z Langerhansových buněk u dětí a dospívajících