
Hemofagocytující lymfohistiocytóza
Hemophagocytic lymphohistiocytosis syndrome
Hemophagocytic lymphohistiocytosis (HLH) represents a heterogenous group of specific immune-systeme deficiencies, characterized by uncontrolled proliferation of T lymphocytes and macrophages, resulting in overproduction of cytokines and inadequate hemophagocytic activity in lymphoreticular systeme and CNS. Cytokines overproduction plays a major role in a tissue damage and brings out typical clinical and laboratory features – persistant fever, hepatosplenomegaly, peripheral blood cytopenia, hypertriglyceridemia, hypofibrinogenemia and bone marrow hemophagocytosis. Full-blown HLH is a life-threatening disease rapidly progressing to multiorgan failure and death. Diagnostics of HLH is often difficult under the condition of absent specific diagnostic marker. Diagnostic guidelines proposed by The Histiocyte Society are based on combination of clinical, biochemical and immunological signs supported by morfological evidence of hemophagocyting macrophages in bone marrow. HLH comprises two different conditions that may be difficult to distinguish one from another: primary HLH – a heterogenous group of rare genetic disorders and a secondary form – HLH developing as a consequence of inadequate immune systeme activation initiated by infections, malignancies or systemic autoimmune diseases. According to HLH-2004 treatment proposal remission could be reached in both conditions by immunosuppression (dexamethasone, etoposide and steroids), but in primary HLH stem cell transplantation offers the only currative option. A series of 17 primary HLH patients diagnosed in the Czech Republic between 1999 and 2009, representing our experience with this rare disease, has outcome comparable to data published by European HLH group (4yrs OS 74%, EFS 56%). Diagnostic dilemma in HLH is demonstrated through several case reports of both primary and secondary HLH patients, revealing significance of clinical experience, awareness of differential diagnosis and application of modern immunological analyses in diagnostic procedure in HLH. All these parametres contribute to early onset of treatment determinating prognosis of HLH patients.
Key words:
hemophagocytic lymphohistiocytosis – immunodeficiency – lymphoproliferation – macrophage activation syndrome – immunosuppressive therapy – stem-cell transplantation
Authors:
M. Suková 1; E. Mejstříková 2; E. Vodičková 3; R. Špíšek 4; R. Formánková 1; D. Sumerauer 1; T. Freiberger 5; P. Sedláček 1; J. Starý 1
Authors‘ workplace:
Klinika dětské hematologie a onkologie 2. lékařské fakulty UK a FN Motol Praha, přednosta prof. MUDr. Jan Starý, DrSc.
1; CLIP cytometrie Kliniky dětské hematologie a onkologie 2. lékařské fakulty UK a FN Motol Praha, vedoucí laboratorního centra prof. MUDr. Jan Trka, Ph. D.
2; Oddělení klinické hematologie FN Motol Praha, přednostka prim. MUDr. Ivana Hochová
3; Ústav imunologie 2. lékařské fakulty UK a FN Motol Praha, přednostka prof. MUDr. Jiřina Bartůňková, DrSc.
4; Centrum kardiovaskulární a transplantační chirurgie Brno, ředitel doc. MUDr. Petr Němec, CSc.
5
Published in:
Vnitř Lék 2010; 56(Supplementum 2): 157-169
Category:
Langerhans cell histiocytosis and some other Hematology rare diseases
Overview
Hemofagocytující lymfohistiocytóza (HLH) je heterogenní skupina onemocnění vznikajících na podkladě specifické vrozené nebo získané poruchy imunitního systému. Podkladem pro vznik syndromu HLH, typických klinických a laboratorních příznaků, je aktivace a nekontrolovaná proliferace T-lymfocytů a makrofágů provázená masivní produkcí cytokinů a hemofagocytární aktivitou makrofágů v lymforetikulárním systému a CNS. Typickými projevy HLH jsou horečka, hepatosplenomegalie a cytopenie s laboratorním obrazem: hypertriglyceridemie, hypofibrinogenemie a hemofagocytózy v kostní dřeni. Primární HLH představuje skupinu vzácných geneticky podmíněných primárních imunodeficiencí. Sekundární HLH jsou sporadická onemocnění, rozvíjející se v souvislosti s infekcí, malignitou nebo systémovým autoimunitním onemocněním. HLH je život ohrožující onemocnění s akutním průběhem, bez léčby vede rychle k rozvoji multiorgánového selhání a smrti. Diagnostika je vzhledem k absenci specifického markeru ve většině případů obtížná, přesto musí být rychlá. Diagnostický algoritmus HLH, doporučovaný evropskou pracovní skupinou, je založen na kombinaci klinicko-biochemicko-imunologických příznaků a morfologickém průkazu hemofagocytózy v kostní dřeni. Recentně se rozvíjející molekulární diagnostika, stejně jako specializované imunologické testy jsou podkladem pro potvrzení geneticky podmíněné primární HLH. Rozlišení mezi primární a sekundární HLH je zásadní pro volbu léčebné strategie. U obou typů onemocnění lze navodit remisi imunosupresivní léčbou, primární HLH lze vyléčit pouze transplantací kostní dřeně. Naše zkušenosti s diagnostikou a léčbou HLH představujeme na souboru 17 pacientů s primární HLH, diagnostikovaných a léčených v ČR v letech 1999–2009, jejichž osud (4letý OS 74 %, EFS 56 %) je srovnatelný s daty publikovanými evropskou HLH skupinou. Diagnostická dilemata dokladujeme podrobněji na několika kazuistikách primární i sekundární HLH, demonstrujících zásadní význam klinických zkušeností, znalosti diferenciální diagnózy a využití moderních imunologických metod. Nejen naše zkušenosti potvrzují, že včasné zahájení účinné léčby, tedy rychlá diagnostika, je zásadním prognostickým faktorem u tohoto závažného onemocnění.
Klíčová slova:
hemofagocytující lymfohistiocytóza – imunodeficience – lymfoproliferace – syndrom aktivovaných makrofágů – imunosupresivní léčba – transplantace krvetvorných buněk
Úvod
Hemofagocytující lymfohistiocytóza(HLH) je syndrom charakterizovaný nekontrolovanou proliferací aktivovaných T-lymfocytů a makrofágů. Nadměrná sekrece cytokinů a hemofagocytární aktivita makrofágů v lymforetikulárním systému a CNS má za následek rozvoj typických klinických a laboratorních příznaků. HLH je heterogenní onemocnění, vzniklé na podkladě specifické vrozené (primární HLH) nebo získané (sekundární HLH) poruchy imunitního systému (tab. 1). Primární HLH jsou onemocnění vzácná, vyskytující se zejména u malých dětí a mladých dospělých, sekundární HLH může vzniknout u imunokompromitovaného jedince v kterémkoli věku. Defektní imunitní systém pacientů reaguje nepřiměřeně na infekci (většinou virovou), aniž by byl schopen vytvořit efektivní imunitní odpověď, nekontrolovaná proliferace aktivovaných T-lymfocytů a makrofágů vede postupně k selhání jater, kostní dřeně a lymforetikulárního systému. Bez léčby je průběh onemocnění fatální, včasná imunosupresivní léčba dokáže navodit remisi u obou typů HLH. Primární – geneticky determinovaná onemocnění – lze vyléčit pouze transplantací kmenových buněk.

Klinické a laboratorní příznaky
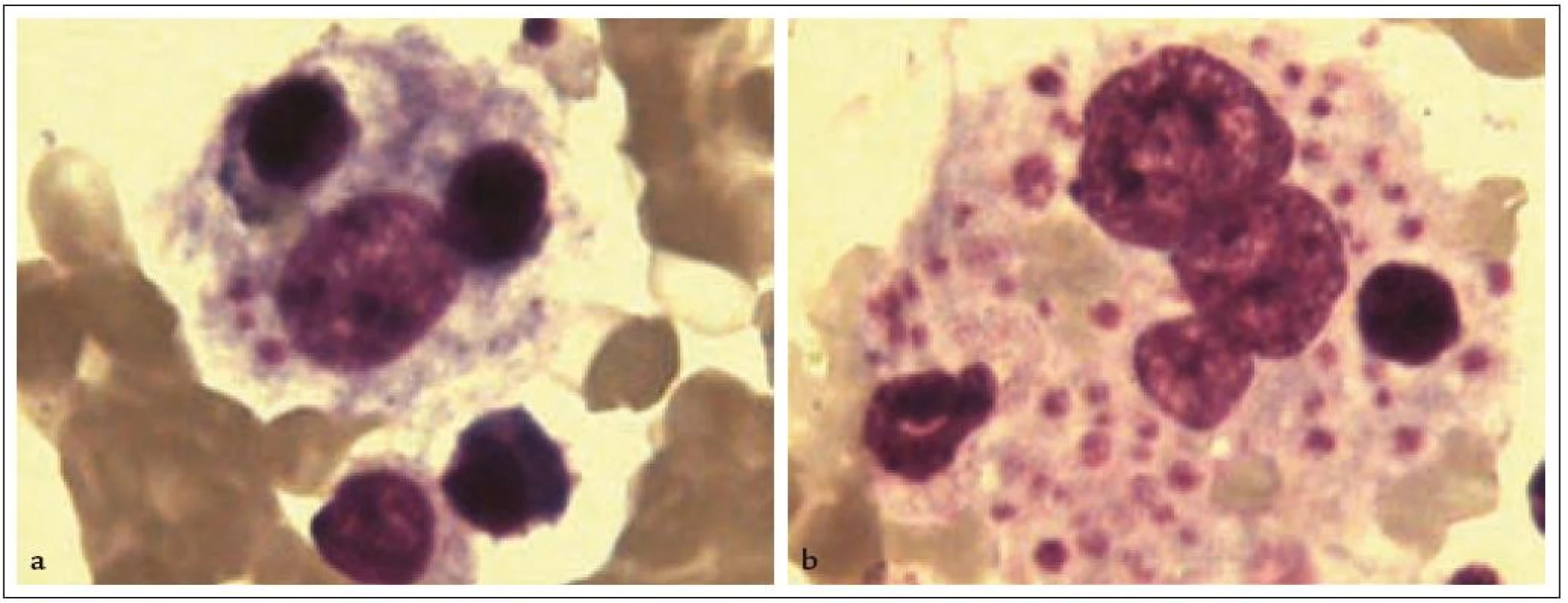
Syndrom HLH je charakterizovaný souborem klinických a laboratorních příznaků, které shrnuje tab. 2. Klinické projevy – horečka, hepatosplenomegalie a symptomatická periferní cytopenie – stejně jako patognomonický nález zvýšeného množství makrofágů, které nadměrně fagocytující krevní elementy v kostní dřeni (obr. 1), jsou sice příznaky pro HLH typické, ale ne specifické. Obdobný klinický obraz a zvýšené množství hemofagocytujících makrofágů v kostní dřeni nacházíme i u pacientů se sepsí, multiorgánovým selháním a polytransfundovaných. Podobně nespecifické jsou i charakteristické biochemické markery HLH (zvýšení triacylglycerolů, snížení fibrinogenu a zvýšení feritinu v séru), odrážející aktivaci buněk monocytomakrofágového systému. Markery aktivace T-lymfocytů a NK buněk, nadprodukce cytokinů a defektní cytotoxické funkce T-lymfocytů jsou specifičtější znaky, prokazatelné ale pouze pomocí speciálních imunologických metodik. V podstatě, s výjimkou chybění exprese perforinu v NK buňkách a T-lymfocytech u části pacientů s FHL, neexistuje žádný pouze pro HLH specifický klinický ani laboratorní příznak. Definitivní průkaz primární HLH je postaven na nálezu specifické mutace, molekulárně genetické vyšetření je ale časově náročné a dostupné v případě některých genů pouze v zahraničí [42]. Přesto je rychlá diagnostika nezbytná, neboť přirozený průběh nemoci je akutní s tendencí k rychlému rozvoji multiorgánového selhání, komplikujícím superinfekcím, krvácení při trombocytopenii a koagulopatii a bez léčby vede neodvratně k úmrtí. Proto jsou v diagnostice využívána kritéria postavená na komplexu klinicko-biochemicko-imunologicko-morfologických znaků charakterizující HLH při splnění 5 z 8 uvedených [22].

Patogeneze [3]
Syndrom HLH vzniká v imunodeficitním terénu, je spuštěn infekcí, většinou virovou, jako induktor je nejčastěji obviňován EB virus, u řady pacientů se nepodaří spouštějící infekci identifikovat. Infekce vyvolá nepřiměřenou aktivaci a proliferaci T-lymfocytů (zejména cytotoxických CD8+), které produkují nadměrné množství cytokinů – interferonu γ (IFN γ) a GM-CSF. Důsledkem je aktivace makrofágů, které proliferují v orgánech retikuloendoteliálního systému a secernují další cytokiny – faktor nekrotizující nádory (tumor necrosis factor – TNF), interleukin-1 (IL-1), interleukin-6 (IL-6) a další. Akumulace proliferujících lymfocytů a makrofágů vede k zvětšení sleziny, jater, lymfatických uzlin. Aktivace makrofágů má za následek nadměrnou fagocytózu krevních elementů, jejímž důsledkem je periferní cytopenie, na které se rovněž podílí tlumivý efekt TNF a IFN γ. Aktivované lymfocyty a makrofágy infiltrují i mozkomíšní mok a překonávají hematoencefalickou bariéru, což vysvětluje časté CNS postižení s „meningitidou“ provázenou pleiocytózou a multifokálními nekrózami v parenchymu [18]. Charakteristické laboratorní markery nemoci vznikají jako produkty aktivace makrofágů a nadprodukce cytokinů: hypertriglyceridemie jako důsledek inhibice lipoproteinové lipázy prostřednictvím TNF, hypofibrinogenemie v důsledku excesivního uvolňování aktivátoru plazminogenu stimulovanými makrofágy. Zvýšená hladina feritinu (v řádu tisíců, desetitisíců i statisíců µg/l) je následkem jeho zvýšené akumulace v makrofázích při hemofagocytóze a uvolňování při jejich stimulaci. Aktivované a proliferující CD8+ T-lymfocyty a NK buňky nejsou schopny likvidovat infikované buňky, následkem je progrese infekce, další replikace viru a pokračující aktivace antigen prezentujících buněk. Nekontrolovaná cytokinová bouře vede k poškození orgánů a multiorgánovému selhání [3].
HLH je heterogenní onemocnění. Klinické a laboratorní příznaky mohou být u jednotlivých forem nemoci různě vyjádřeny, patogeneza se může v detailech lišit. Následující text je věnován konkrétním chorobám, které se pod obrazem HLH manifestují.
Primární HLH
Familiární hemofagocytující lymfohistiocytóza (FHL)
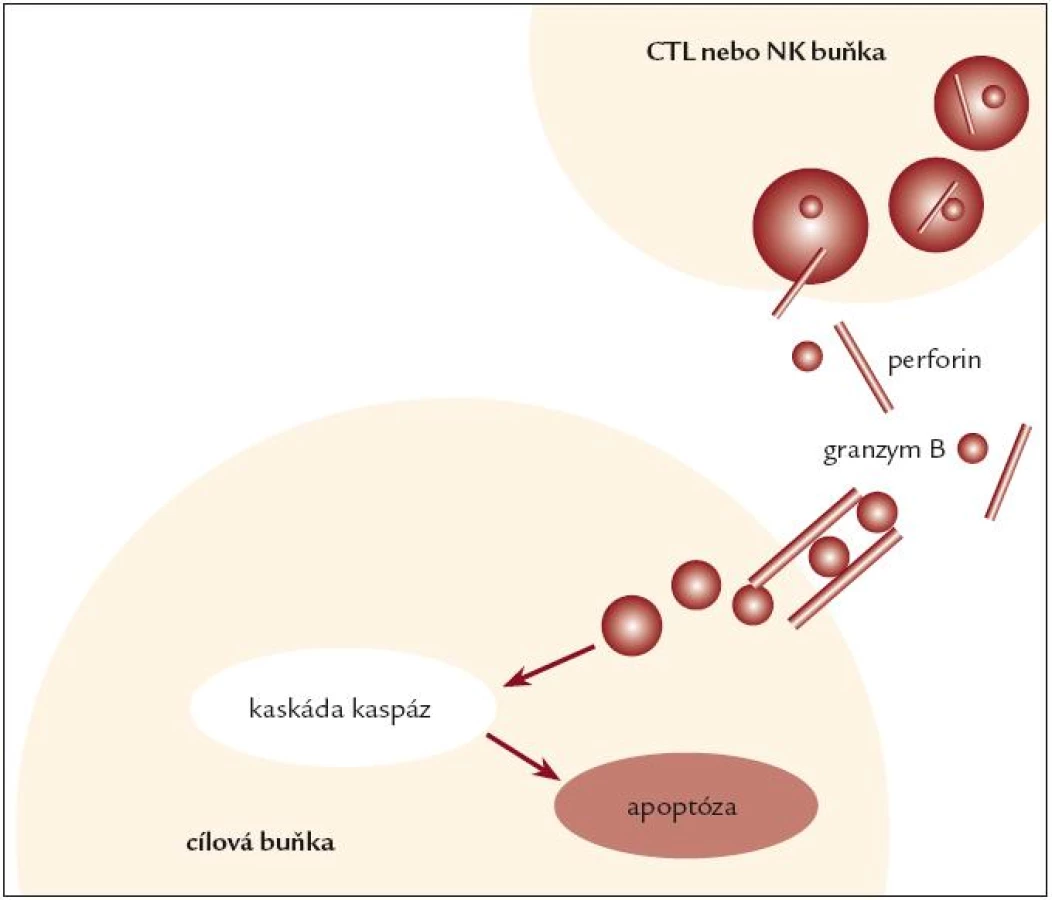
FHL je autozomálně recesivně dědičné onemocnění, vyskytující se v kavkazské rase s incidencí přibližně 1 případ na 50 000 porodů. První popis onemocnění pochází od Farquhara z roku 1952, přehledový článek systematicky popisující tuto jednotku byl publikován v roce 1983 [24]. Pojem familiární může být zavádějící, neboť HLH může postihnout 1. dítě v rodině či je v rodině zdravý starší sourozenec. FHL se typicky manifestuje v raném dětství perzistující intermitentní horečkou a následně se rozvíjejícími klinickými a laboratorními příznaky lymfoproliferace a aktivace makrofágů. Průběh je typicky perakutní s rozvojem plně vyjádřené nemoci během dnů, vzácněji FHL probíhá indolentně s rozvojem plného obrazu v řádu týdnů nebo formou remitující. CNS postižení je časté na začátku a i v průběhu nemoci a postihuje až 100 % dětí [18], klinický obraz kolísá od asymptomatické aseptické meningitidy po mnohočetnou nekrotickou meningoencefalitidu s ireverzibilním neurologickým postižením, které nevyléčí ani transplantace kostní dřeně. Medián manifestace onemocnění je podle mezinárodního registru HLH ve věku 3 měsíců, multicentrická studie HLH 94 uvádí 13 měsíců [2,21], byly popsány i případy intrauterinního počátku FHL. Výskyt nemoci po 3. roce života je výjimečný, i když možný [1]. Medián přežití dětí s FHL je 6 měsíců, čím je mladší dítě při manifestaci nemoci, tím je jeho prognóza horší. FHL je geneticky heterogenní onemocnění, v současnosti jsou známy 4 kauzální genetické defekty prokazované u pacientů s FHL s různou četností v závislosti na etnické příslušnosti. Nejčastější (20–40 %) příčinou nemoci v kavkazské populaci je mutace genu pro perforin (PRF1) na chromozomu 10q21–22 [10,13,35]. Mutace Munc 13-4 (UNC13D na chromozomu 17q25) kódující protein zodpovědný za exocytolýzu granul je podkladem nemoci u asi 20 % pacientů [14]. Vzácnějším genetickým podkladem FHL jsou mutace v genu STX na chromozomu 6q24 kódující translační protein syntaxin [41] a konečně recentně popsaná mutace Munc 18-2, kódující syntaxin binding protein [8]. Funkčním důsledkem těchto genetických poruch je defekt cytotoxické funkce CD8+ lymfocytů a NK buněk. Cytotoxická aktivita T-lymfocytů je u virových infekcí přednostně zajišťována systémem perforin/granzym, který vyvolá apoptózu cílových infikovaných buněk (obr. 2). U HLH způsobené mutací v genu kódujícím perforin tato imunologická reakce selhává, a je tak vysvětlením chybějící cytotoxické funkce T-lymfocytů a NK buněk in vitro a neschopnosti zvládnout infekci in vivo. Pro patogenezi onemocnění je důležitá skutečnost, že přes defekt v efektorové funkci mají T-lymfocyty a NK buňky zachovanou schopnost aktivace a produkce cytokinů. Děti s mutací perforinového genu mají deficit perforinu v T-lymfocytech a NK buňkách prokazatelný nejsnáze průtokovou cytometrií pomocí monoklonální protilátky proti perforinu. Ostatní pacienti s HLH mají expresi perforinu v lymfocytech normální [5]. Laboratorní průkaz porušené cytotoxické funkce CD8+ lymfocytů a NK buněk je jedním ze základních testů potvrzujících diagnózu FHL [20]. Je prokazatelný u postižených jedinců již před rozvojem nemoci, cytotoxický test lze tedy použít jako vyhledávací test u asymptomatických sourozenců [39].
V léčbě FHL se dlouhodobě úspěšně používají imunosupresivní léky s cytotoxickým efektem na aktivované proliferující T-lymfocyty (kortikoidy a antitymocytární globulin), léky indukující apoptózu makrofágů (etoposid) a cyklosporin A, účinně tlumící časné fáze aktivace T-lymfocytů a ovlivňující produkci IL-1, IL-6 a TNF makrofágy [20]. Intratekálně aplikovaný metotrexát spolu s vysokou dávkou kortikoidů se používá jako cytotoxické agens indukující apoptózu aktivovaných lymfocytů při infiltraci CNS. Imunosupresivní léčba má potenciál navodit remisi, nicméně s rizikem relapsu při další infekci. Jedinou kurativní léčbou FHL je transplantace krvetvorných buněk (SCT), která navodí normalizaci defektní cytotoxické funkce T-lymfocytů a NK buněk. Od roku 1994 byla zahájena mezinárodní studie léčby dětí s FHL kombinací dexametazonu, etoposidu a cyklosporinu A s následnou transplantací krvetvorných buněk od příbuzných i nepříbuzných dárců provedenou časně v průběhu nemoci, jejíž výsledky byly publikovány v roce 2002 [21]. 113 dětí absolvovalo léčbu protokolem HLH 94, imunosupresivní léčbou bylo dosaženo remise u 53 % pacientů, 11 % zemřelo na progresi před provedením transplantace. Transplantováno bylo 65 dětí (většinou od nepříbuzného dárce), z nichž žije 62 %, většina úmrtí byla v souvislosti s potransplantačními komplikacemi. Při mediánu sledování 3,1 roku je 3leté přežití pro celý soubor pacientů 55 %. Nejvýznamnějším prognostickým faktorem v analyzované skupině byl věk při manifestaci nemoci, s významně vyšší pravděpodobností přežití u dětí starších jednoho roku (72 % vs 42 %). Výstupy ze studie HLH-1994 a pokrok v molekulární diagnostice HLH byly zohledněny v revizi diagnostických kritérií [22] a inovaci léčebného protokolu – recentně používaná studie HLH-2004 (obr. 4).

Chediak-Higashiho syndrom
Chediak-Higashiho syndrom (CHS) je autozomálně recesivně dědičné onemocnění, charakterizované parciálním okulokutánním albinizmem, vyšším výskytem pyogenních infekcí a nálezem abnormálně velikých granul v různých krevních buňkách (neutrofily, monocyty, eozinofily, lymfocyty) v periferní krvi i kostní dřeni (obr. 3a, 3b). Jde o primární imunodeficienci s porušenou chemotaxí a baktericidní kapacitou granulocytů a monocytů a poruchou cytotoxické funkce T-lymfocytů a NK buněk v důsledku neschopnosti lysozomálních granul secernovat cytolytické proteiny (perforin/granzym). Příčinou nemoci jsou mutace v genu LYST na chromozomu 1q42-q43, který reguluje funkci lysozomálních granul [7]. Příčinou albinizmu je porucha uvolňování obsahu granul z melanocytů, nález abnormálně velkých melanozomů při mikroskopickém vyšetření vlasů je patognomonický (obr. 3c). Většina pacientů umírá v dětství v akcelerované fázi nemoci, která probíhá pod obrazem HLH spuštěné virovou infekcí, u těch, kteří přežijí do dospělosti, se často vyvíjí neurologické postižení projevující se mentální retardací. Transplantace krvetvorných buněk onemocnění vyléčí, není ale vždy schopna předejít rozvoji neurologického postižení.
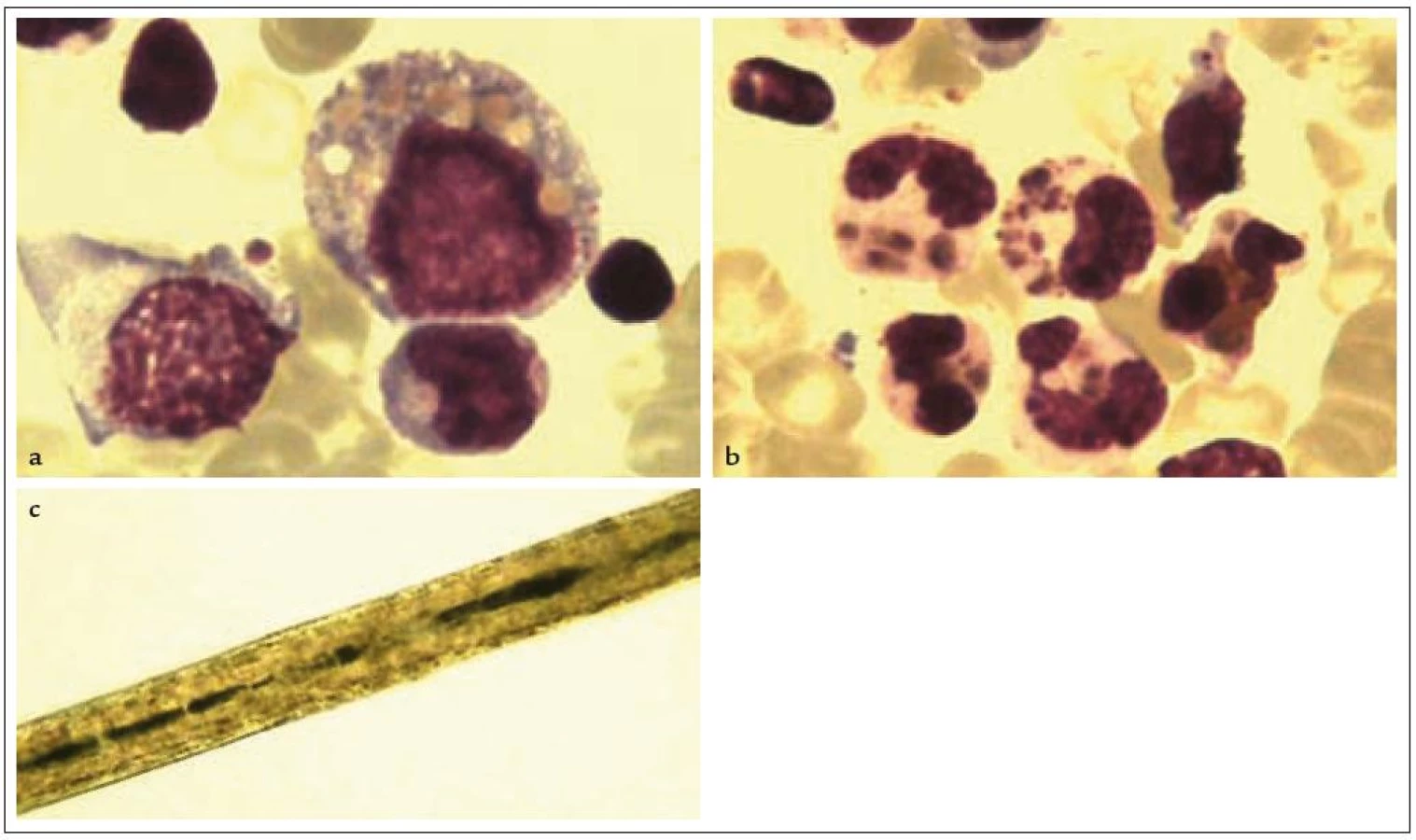
Griscelliho syndrom
Griscelliho syndrom je onemocnění příbuzné s Chediak-Higashiho syndromem, od kterého se liší nepřítomností velkých granul v krevních elementech a vzhledem melanozomů v melanocytech kůže. Jde o autozomálně recesivně dědičnou poruchu pigmentu, projevující se nálezem „stříbrných“ vlasů spojenou s defektem cytotoxické funkce T-lymfocytů a NK buněk. Důsledkem je stejně jako u Chediak-Higashiho syndromu vznik akcelerované fáze nemoci pod obrazem HLH v dětství, u přeživších hrozí v dospělosti neurologické postižení z infiltrace CNS lymfocyty a histiocyty. Diagnózu lze potvrdit nálezem typické distribuce pigmentu při mikroskopickém vyšetření vlasů, velikost melanozomů v melanocytech je na rozdíl od CHS normální [26]. Příčinou nemoci je mutace v genu Rab27a, jehož proteinový produkt je potřebný pro exocytózu cytotoxických granul. Deficit Rab27a způsobuje selhání cytotoxické funkce lymfocytu stejně jako defekt v transportu pigmentu z melanocytů do keratinocytů [27]. Onemocnění je vyléčitelné transplantací krvetvorných buněk.
Lymfoproliferativní syndrom vázaný na X-chromozom (XLP)
V roce 1975 Purtillo popsal fatální lymfoproliferativní syndrom, kterému podlehlo 6 z 18 mužských příbuzných skotské rodiny Duncanových [29]. Odtud také historický název nemoci. Podstatou nemoci je specifický primární imunodeficit, charakterizovaný neschopností organizmu účinně kontrolovat EBV infekci. Klinický obraz XLP je heterogenní. Primoinfekce EBV se nejčastěji (asi v 50 %) projeví jako fatální infekční mononukleóza, probíhající pod obrazem HLH, s mortalitou přesahující 90 %. U přibližně 1/3 postižených primoinfekce EBV způsobí vznik dysgamaglobulinemie (resp. obraz CVID) s vysokým rizikem vzniku nehodgkinského lymfomu, který se většinou vyvíjí před 30. rokem života a může být prvním projevem onemocnění (u 30 % pacientů). Vzácněji se XLP může manifestovat jako aplastická anémie nebo lymfoidní vaskulitida. Jde o geneticky determinované onemocnění, příčinou je v 60–80 % případů mutace genu SAP (SH2D1A) na chromozomu Xq25 [4], v 17 % případů mutace genu XIAP (BIRC4) [30]. Protein SAP se účastní přenosu signálu z receptorů CD2 rodiny SLAM (signalling lymphocytic activation molecule) na T-lymfocytech a 2B4 na NK buňkách, X-linked inhibitor of apoptosis (XIAP) má klíčovou roli v diferenciaci NK buněk. Vzhledem k faktu, že B-lymfocyty infikované EB virem silně exprimují molekulu CD48, která je ligandou receptoru 2B4, pak porucha obou uvedených mechanizmů vede k selhání cytotoxické funkce NK buněk právě proti EBV+ B-lymfocytům, které nekontrolovaně proliferují [17]. Pacienti s XLP vytváří IgM protilátky proti EBV, je pro ně ale charakteristická neschopnost vytvořit EBNA IgG protilátky. K izotypovému přesmyku nedojde pro defektní aktivaci pomocných CD4+ T-lymfocytů. Defekty humorální imunity jsou nacházeny i u matek-přenašeček. Syndrom fatální infekční mononukleózy lze dostat pod dočasnou kontrolu eliminací proliferujících EBV infikovaných B-lymfocytů podáním monoklonální protilátky anti-CD20 (rituximab) a imunosupresivní léčbou (etoposid + kortikoidy + cyklosporin A). Vyléčení lze dosáhnout pouze transplantací kostní dřeně, která napraví genetický podklad onemocnění.
Sekundární HLH
HLH související s infekcí
Rozvoj HLH po virových infekcích popsal Risdall v roce 1979 a nazval tuto jednotku VAHS (virus-associated hemophagocytic syndrome) [31]. V jeho souboru 19 pacientů bylo v době manifestace HLH 14 léčeno imunosupresivní léčbou pro stav po transplantaci ledviny nebo lupus erytematodes a pouze 5 nemělo prokázané jiné základní onemocnění. U většiny pacientů byla dokumentována virová infekce, nejčastěji EBV. Vysazení imunosupresivní léčby mělo u 13 pacientů příznivý efekt na HLH. Obecné teze vyplývající z Risdallova pozorování jsou platné i dnes. Nejčastějším spouštěčem VAHS jsou herpetické viry, zejména EBV, popsána byla ale i při infekci adenovirem nebo parvovirem B19. Bakterie či mnohobuněční parazité jako vyvolavatelé HLH jsou vzácnější, popis existuje v souvislosti s infekcí Mycoplasma pneumonie, tuberkulózou, malárií, syfilis [25,31]. Infekce může být příčinou syndromu HLH i u lidí bez prokázané primární či sekundární imunodeficience, imunokompromitovaný organizmus je ale predispozicí. Názory na léčbu HLH asociované s infekcí nejsou jednotné. V případě vzniku v souvislosti s imunosupresivní léčbou se doporučuje její vysazení. Trvá-li HLH i po vysazení imunosupresivní léčby, je vhodné zahájit terapii kortikoidy, etoposidem a/nebo cyklosporinem A. Výjimkou z těchto doporučení je leishmanióza, kde předpokladem úspěchu je cílená antiinfekční léčba.
Viscerální leishmanióza
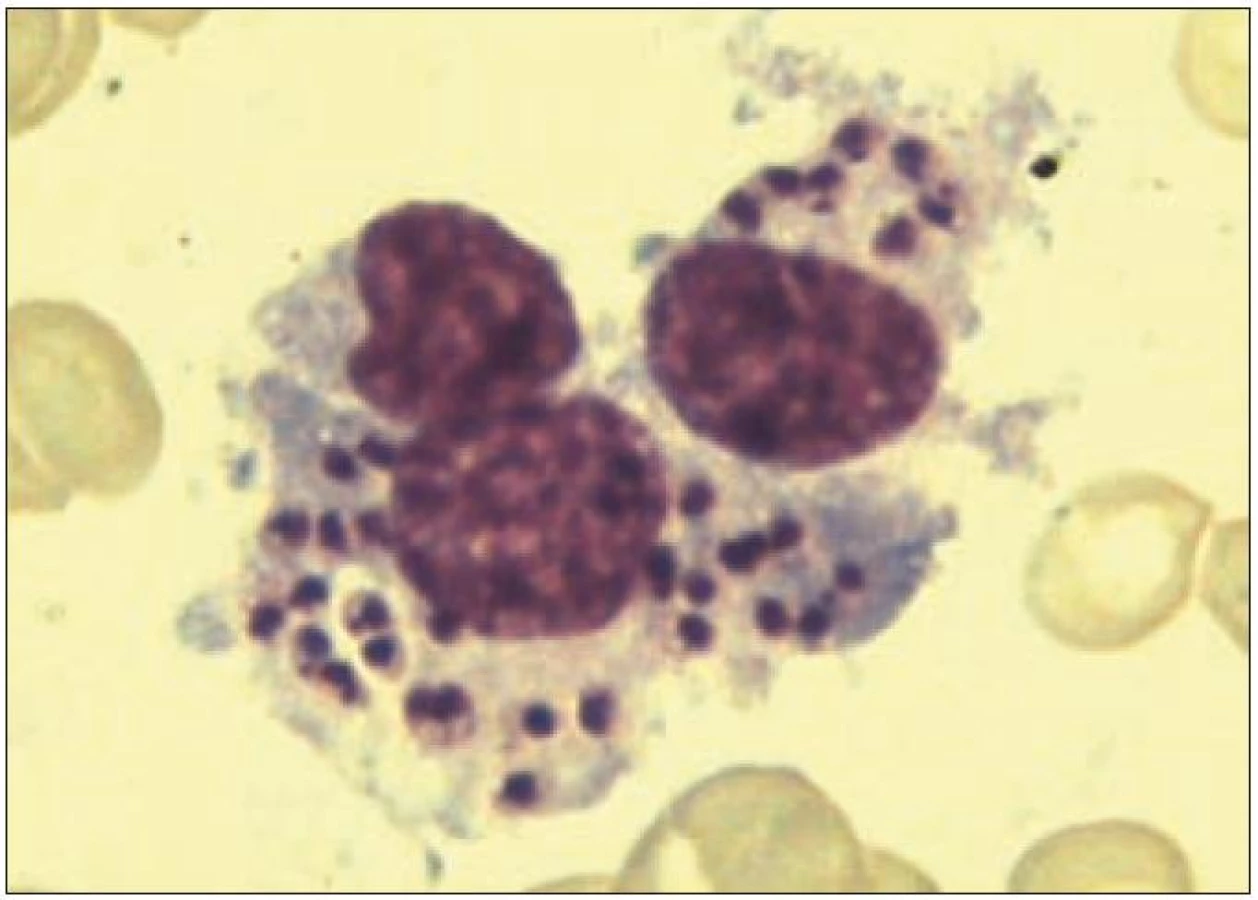
Viscerální leishmanióza (kala-azar) je infekce s ohniskovým výskytem v tropické a subtropické Africe, Asii a Jižní Americe, v Evropě se nemoc endemicky vyskytuje v oblastech Středomoří, zejména v Dalmácii, jižní Itálii, na Maltě a dále v severní Africe (Tunisko). Jde o onemocnění, jehož incidence v ČR stoupá, a je nutné na něj myslet při cestovatelské anamnéze 2 týdny až několik měsíců před manifestací HLH. Původcem nemoci jsou jednobuněční parazité rodu Leishmania, přenášení na člověka krev sajícími samičkami komárů rodu Phlebotomus, rezervoárem infekce jsou psi. Ve Středomoří je původcem viscerální leishmaniózy nejčastěji druh Leishmania infantum [16]. Amastigotní stadia leishmanií, která jsou zodpovědná za rozvoj nákazy, se infikují a množí uvnitř makrofágů, způsobují chronickou intracelulární infekci vedoucí k aktivaci makrofágů a následné hemofagocytóze. Jsou detekovány vysoké hladiny TNF a rozvíjí se klinický a laboratorní obraz HLH. Klasickými projevy leishmaniózy jsou horečka, hepatosplenomagalie a pancytopenie. Infekce je průkazná nálezem amastigotních stadií parazita v makrofázích kostní dřeně (obr. 5), potvrzen sérologicky průkazem protilátek. V iniciálních stadiích nemoci, v období epidemického okna, se amastigoti leishmanií v kostní dřeni vyskytují jen v malém množství nebo mohou úplně chybět [36]. Pro přímý průkaz parazita se alternativně doporučuje punkce sleziny, to je ale výkon, který při pancytopenii představuje větší riziko než aspirace kostní dřeně. Přínosem pro rychlou diagnostiku je zavedení detekce parazita v krvi metodou PCR. Účinnou léčbou infekce je lipozomální forma amfotericinu B.
HLH spojená s maligním nádorovým onemocněním
HLH může být prvním projevem maligního nádorového onemocnění nebo se vyvinout jako komplikace v průběhu protinádorové léčby. HLH je nejčastěji asociována s nehodgkinským lymfomem, velkobuněčným anaplastickým lymfomem nebo periferním T-lymfomem, jejichž diagnostiku může zásadně komplikovat [6,23]. HLH byla popsána i jako komplikace akutní lymfoblastické leukemie, mediastinálního nádoru z germinálních buněk a, jistě ne náhodně, multisystémové formy histiocytózy z Langerhansových buněk [12]. Obdobně jako u ostatních sekundárních HLH je za spouštěč považována virová infekce (EBV), v terénu neoplastické proliferace (zejména buněk T typu) snadno dochází k nepřiměřené produkci cytokinů a aktivaci makrofágů. Vznikne-li HLH v průběhu chemoterapie základního onemocnění, je prvním krokem zvážení přerušení léčby.
HLH spojená se systémovým autoimunitním onemocněním
Vzácně může HLH komplikovat manifestaci nebo průběh systémové formy juvenilní idiopatické artritidy (sJIA) nebo systémového lupusu erythematodu (SLE), v této souvislosti je často používán termín „syndrom aktivovaných makrofágů“ (MAS). Za spouštěč HLH jsou označovány viry nebo změna léčby základního onemocnění [32]. Na vzniku HLH u sJIA se nepochybně podílí defektní exprese perforinu v lymfocytech, v jejímž důsledku je lymfoproliferace a aktivace makrofágů konstantním projevem aktivní nemoci [40,42]. MAS syndrom v průběhu systémového autoimunitního onemocnění je závažnou život ohrožující komplikací vyžadující intenzivní léčbu. Časté je postižení CNS, projevující se podrážděností, zmateností, bolestí hlavy, někdy i bezvědomím a křečemi. Lékem první volby jsou bolusy kortikoidů, není-li rychlá klinická odpověď, je nutné neodkladně zahájit léčbu cyklosporinem A, který navodí u většiny pacientů remisi [34]. Obávanou komplikací sJIA je vznik fatální HLH časně po autologní transplantaci krvetvorných buněk.
Vlastní pozorování
V letech 1999–2010 bylo v ČR diagnostikováno 17 dětí s primární HLH, 11 chlapců, 6 dívek (tab. 3). Jako primární HLH byli zařazeni na základě specifického imunologického obrazu buď s molekulárním průkazem geneticky definované jednotky, nebo při recidivujícím průběhu: FHL: 11, Chédiak-Higashi syndrom: 2, XLP 2, nezařazená FHL s fatálním průběhem EBV infekce: 2. FHLbyla v souladu s povahou onemocnění diagnostikována převážně v kojeneckém věku, medián manifestace byl 2,5 měsíce (0–27 měsíců). U ostatních primárních HLH byl medián manifestace 16,3 měsíců (23–205 měsíců). Familiární výskyt byl pozorován u 3 sourozeneckých dvojic – v prvním případě byla diagnóza HLH stanovena při souběžném fatálním průběhu infekční mononukleózy u bratra a sestry. Ve druhém případě molekulárně potvrzená FHL u mladšího bratra ozřejmila diagnózu u staršího sourozence, který zemřel o 10 let dříve pod obrazem makrofágového syndromu při rezistentní systémové JIA. Ve třetím případě až relaps HLH u staršího sourozence vedl k průkazu nové mutace asociované s FHL, která byla potvrzena prenatálně i u mladšího bratra, který byl následně jako jediný ze souboru transplantován preemptivně, před manifestací HLH. Ze skupiny primárních HLH je aktuálně molekulárně zařazeno 6 pacientů, což představuje 50 % z alespoň limitovaně vyšetřených a zohledňuje fakt, že pokrok v poznání molekulárního podkladu HLH je zcela recentní. Vyšetření perforinu bylo provedeno u 8 z 11 pacientů s FHL, pouze v jednom případě jsme prokázali kompletní deficit perforinu [38]. Molekulární podklad byl potvrzen u 4/8 vyšetřených FHL (mutace PRF1 1krát, MUNc 13-4 1krát, MUNc 18-2 2krát) a u 2 ze 4 klinicky suspektních XLP (SH2D1A 1krát, BIRC4 1krát). Imunosupresivní léčbu (kombinací dexamethazon + etoposid + CsA) k navození remise před SCT podstoupilo 15 ze 17 pacientů. Kromě dítěte diagnostikovaného prenatálně byla frontline transplantace indikována u pacienta s XLP, který se manifestoval jako aplastická anémie po chemoterapii pro B-NHL. Remisi HLH se podařilo navodit u 10/15 léčených pacientů (67 %), 5 zemřelo v akcelerované fázi před transplantací. Transplantováno bylo 12 pacientů (celkem 13 transplantací) ve věku s mediánem 20 měsíců (2–207 měsíců). Dárcem byl 3krát sourozenec, 7krát nepříbuzný a 3krát haploidentický rodič, jako štěp byla použita 5krát kostní dřeň, 3krát pupečníková krev, 5krát periferní kmenové buňky (3krát po T depleci). V přípravě k transplantaci byl pro pacienty s FHL volen myeloablativní režim BuCy(VP)ATG, pro pacienty s XLP s vědomím rizika vyšší toxicity režim s redukovanou intenzitou (FluMelCampath). Štěp přihojilo 10/12 pacientů (83 %), 2 děti zemřely časně po transplantaci – pacient s FHL den 15 na sepsi s multiorgánovým selháním, pacient s XLP den 13 pod obrazem multiorgánového selhání při toxicitě conditioningu, TRM v našem souboru představuje 17 %. Remise HLH bylo po transplantaci dosaženo u 9/12 pacientů (75 %). Jedna pacientka zemřela 6 měsíců po SCT na CNS relaps HLH, jeden pacient byl retransplantován pro rejekci štěpu a při komplikující HHV6 infekci vyvinul chronické CNS postižení s rezistentní epilepsií a psychomotorickým regresem. Třetí pacient vyvinul 6 měsíců po SCT sekundární EBV indukovaný DLBCL úspěšně léčený chemoterapií. Při mediánu sledování 36 měsíců je v našem souboru primární HLH celkové přežití 74 %, přežití bez události 54 %, úspěšnost léčby je v kontextu s evropskými zkušenostmi srovnatelná [21].
K ilustraci klinického průběhu, dia-gnostického algoritmu a různých léčebných přístupů u primární a sekundární HLH uvádíme vybrané kazuistiky pacientů s obrazem HLH na různém podkladě.
Kazuistika 1
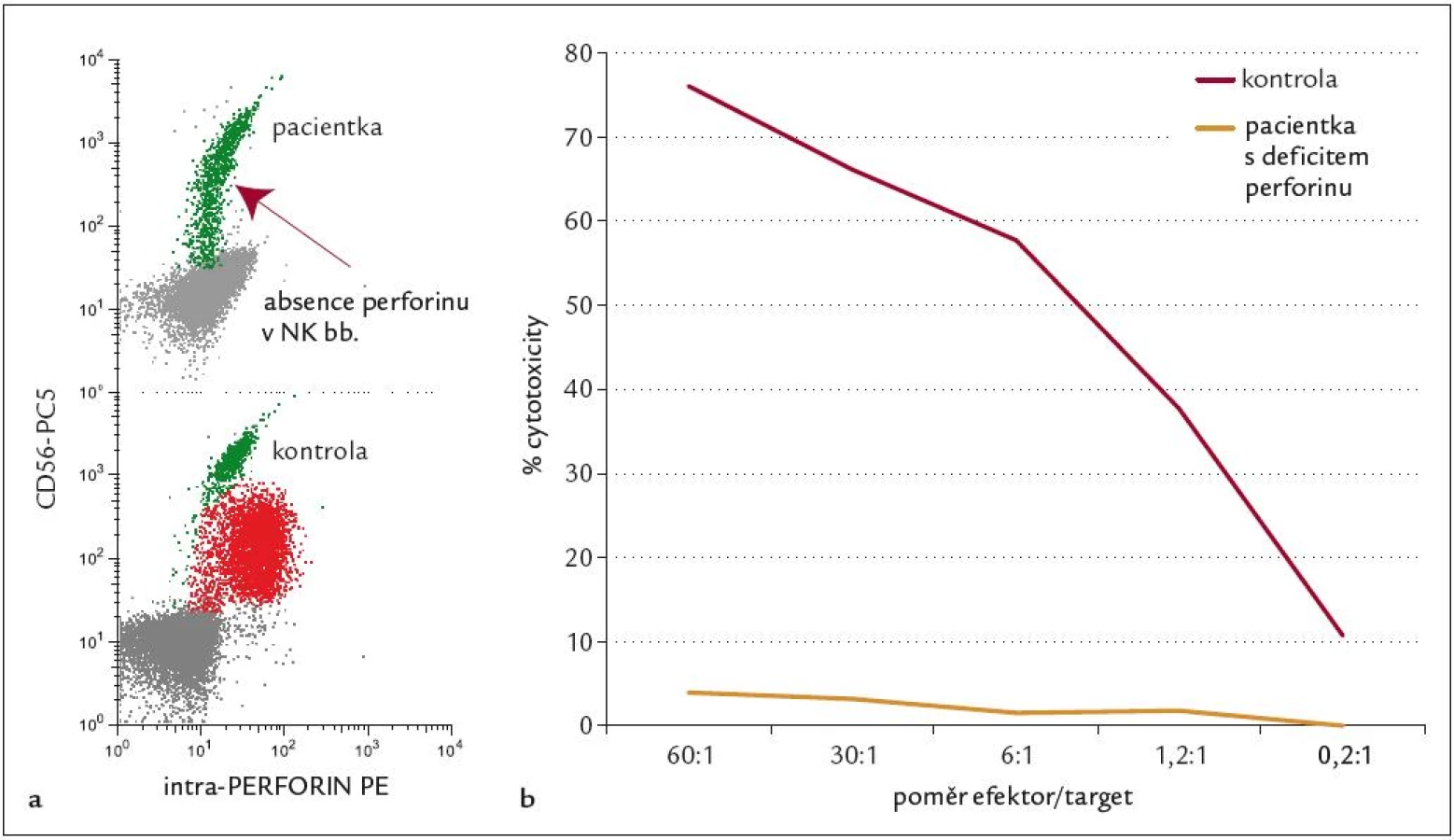
Dívka, věk 1 měsíc. Diagnóza: FHL. RA: sestra 4 roky, zdráva. NO: fyziologický novorozenec, porodní váha 2 570 g, poporodní adaptace normální. 29. denživota náhle horečka 39 °C, pro zchvá-cenost přijata na spádovém dětském oddělení, zjištěna hepatosplenomegalie (játra +5 cm, slezina +5 cm), trom-bocytopenie (55 × 109/l). Pro podezření na sepsi zahájena léčba antibio-tiky a kortikoidy, při zhoršování celkového stavu a anemizaci vyšetřena kostní dřeň, kde zachyceny suspektní atypické buňky, proto s podezřením na hemoblastózu přeložena na II. dětskou kliniku. Během 24 hod se rozvinul kompletní obraz HLH – s horečkami, krvácivou diatézou, progresí hepatosplenomegalie. Laboratorně dominovala těžká pancytopenie (leukocyty 9,4 × 109/l, Hb 6,5 g/l, trombocyty 16 × 109/l), hypofibrinogenemie (0,58 g/l) s koagulopatií charakteru DIC (APTT 72,0, Quick 25 %, D-dimer > 1 000 ng/ml, AT III 32 %), hypertriglyceridemie (3,29 mmol/l ) a extrémní hodnoty feritinu v séru (40 640 µg/l). V kostní dřeni je nápadné zmnožení makrofágů s vyjádřenou hemofagocytózou. Vyšetření periferní krve průtokovou cytometrií prokazuje výraznou aktivaci T-lymfocytů (CD3+DR+ 57% z lymfocytů, 92% CD3+CD8+ T lymfocytů exprimovalo aktivační marker HLA DR) a chybění perforinu na NK buňkách (obr. 6). Posléze doplněné testy cytotoxické aktivity T-lymfoblastů vykazovaly téměř nulové hodnoty (pod 5 %), geneticky determinovaný deficit perforinu byl prokázán i na molekulární úrovni (mutace genu PRF1). Nálezy jsou v plném souladu s diagnózou FHL, urgentně byla proto zahájena léčba dexametazonem a etoposidem, komplikovaná již první den sepsí Acinetobacter s respiračním selháním, které vyžadovalo 6 dnů umělé plicní ventilace. Přes nepříznivý průběh se remise HLH (dle parametrů krevního obrazu a výrazného snížení exprese HLA DR na CD3+8+ T lymfocytech) podařilo dosáhnout 13. den léčby a přes další komplikující pseudomonádovou sepsi byla na imunosupresivní léčbě remise udržována až do transplantace kostní dřeně 2,5 měsíce od diagnózy. Dítě bylo transplantováno ve věku 3,5 měsíce, PBSC od nepříbuzného dárce se shodou 9/10, po myeloablativním režimu BuCyVP16 + ATG, s časným přihojením štěpu a bez GVHD. Pro nárůst autologní krvetvorby byla den 125 podána transfuze dárcovských lymfocytů, která stabilizovala smíšený chimerizmus. V současné době je pacientka 5 let po transplantaci v trvající remisi FHL, ode dne +180 je evidována normalizace i cytotoxických testů. Případ dokumentuje klasický agresivní průběh HLH u nejmenších dětí a dokladuje typický laboratorní obraz FHL, který je ale spíš výjimečný.
Kazuistika 2
Chlapec, 17 let. Diagnóza: X-vázaný lymfoproliferativní syndrom. Rodinná anamnéza byla v době onemocnění negativní, bratr 19 let, zdráv. Zpětně je zjištěno úmrtí bratra matky na fatální infekční mononukleózu. V 16 letech, rok před diagnózou se objevila krční lymfadenopatie provázená subfebriliemi a váhovým úbytkem. Vyšetřením byla potvrzena generalizovaná lymfadenopatie a oboustranný fluidotorax, histologickým vyšetření axilární uzliny a pleurálního výpotku potvrzen zralý B-NHL (CD 20+), s kauzální translokací t(8,14), vzhledem k rozsahu klinické stadium III. Onkologem indikována léčba podle protokolu B-NHL BFM 2004, R4, tj. 6 bloků chemoterapie (obsahující dexametazon, vincristine, cyklophosfamid, metotrexát, etoposid a antracykliny) v kombinaci s rituximabem. Indukční fáze léčby byla komplikována recidivujícími hrudními výpotky s nutností dlouhodobé drenáže a kachektizací, v souvislosti s další chemoterapií opakovaně zaznamenána myelotoxicita těžkého stupně. Kompletní remise bylo dosaženo po 1. bloku chemoterapie, její trvání potvrzeno včetně PET-CT po ukončení posledního bloku. Tři měsíce po ukončení chemoterapie v rámci dispenzarizace zjištěna progresivní pancytopenie (leukocyty 1,8 × 109/l, Hb 4,6 g/dl, trombocyty 55 × 109/l, ANC 334 × 109/l). Vyšetření kostní dřeně ukázalo významnou hypocelularitu s absencí prekurzorů hematopoézy, histologicky byla dřeň hodnocena jako aplastická, s buněčností pod 10 %, bez dokumentovaného zmnožení makrofágů či hemofagocytózy. Stav byl uzavřen jako těžká aplastická anémie při trvající remisi NHL. Vzhledem ke koincidenci uvedených jednotek a imunofenotypickému obrazu (aktivace T-lymfocytů – CD3+DR+ 20 %, 57% CD3+8+ lymfocytů exprimovalo aktivační marker HLA DR) bylo vysloveno podezření na XLP, který byl posléze potvrzen průkazem mutace SH2D1A genu (delece exon 1). Pacient byl indikován k alogenní transplantaci kmenových buněk od HLA identického bratra, u kterého byla předem vyloučena přítomnost kauzální mutace XLP. Pacient byl transplantován PBSC 3 měsíce od diagnózy aplastické anémie, bez předchozí imunosuprese, po přípravném režimu s redukovanou intenzitou (fludarabine + melfalan + Campath). Potransplantační průběh byl bez komplikací, s časným přihojením, bez GVHD a s trvající remisí při kompletním chimérizmu 12 měsíců po transplantaci.
Kazuistika 3
Chlapec, 3 roky. Diagnóza: Viscerální leishmanióza. Přijat na kliniku pro týden trvající horečky až 40 °C, které se objevily 1 měsíc po návratu z letního pobytu v Chorvatsku. Byla patrná nápadná hepatosplenomegalie, játra +5 cm a slezina +8 cm pod oblouk. V laboratorním vyšetření panyctopenie – KO: Hb 88 g/l, erytrocyty 3,6 × 1012/l, MCV 74, leukocyty 3,9 × 109/l, S 5 %, T 5 %, Mmy 1 %, Mono 19 %, Lymfo 70 %, trombocyty 68 × 109/l, normální hladina sérového fibrinogenu 2 g/l (norma 1,5–4 g/l), vysoký feritin 4 039 mg/l (norma 7–142 µg/l), hypertriglyceridemie 3,17 mmol/l (norma 1,2–1,6 mmol/l), LD 19,3 µkat/l (norma 3–8,4 µkat/l). V aspirátu kostní dřeně byl normocelulární nátěr s normálním poměrem řad, bylo zastiženo 0,80 % hemofagocytujících makrofágů, při cíleném detailním prohlížení ojediněle i makrofág obsahující amastigoty leishmanie. Vzhledem k faktu, že se jednalo o druhý případ v krátkém časovém odstupu, cílená sérologická diagnostika byla provedena časně – potvrdila diagnózu viscerální leishmaniosy. Sérologie Leishmania infantum v krvi byla pozitivní se vzestupným trendem: 1. vyšetření titr 1 : 32, za 2 týdny 1 : 256 (E. Nohýnková, III. klinika infekčních a tropických nemocí 1. LF UK Praha). Pacient byl léčen standardně, lipozomálním amphotericinem B, celkem 2 týdny s následnými reindukcemi v intervalu 1 týden. Ústup klinických projevů i úprava pancytopenie trvaly několik týdnů. 6 měsíců po ukončení 4. reindukce se objevila recidiva hepatosplenomegalie, horeček, provázená mírnou pancytopenií. Aspirace kostní dřeně potvrdila znovu amastigotní stadia v kostní dřeni, sérologie zůstala negativní. Desetidenní terapie lipozomálním amphotericinem B s reindukcemi vedla k definitivní remisi, která trvá 5 let.
Kazuistika 4
Chlapec 11 roků, 5 měsíců. Diagnóza: HLH jako manifestace velkobuněčného anaplastického nonhodgkinského lymfomu. Přijat pro 1 měsíc trvající horečky 39–40 °C s rozvojem pancytopenie a hepatomegalie. Ve spádovém dětském oddělení provedena aspirace kostní dřeně s nálezem četných hemofagocytujících makrofágů, vysloveno podezření na maligní histiocytózu, zahájena terapie kortikoidy a s odstupem 10 dnů chlapec přeložen na II. dětskou kliniku. Při přijetí dominovala hepatomegalie (játra +4 cm) bez splenomegalie, nápadně suchá kůže a rty, rezistence v levém třísle – sonograficky suspektní zvětšená lymfatická uzlina. V laboratorním vyšetření dominovala panyctopenie (Hb 89 g/l, erytrocyty 3,47 × 1012/l, leukocyty 1,9 × 109/l, S 55 %, T 18 %, Eo 1 %, Mo 2 %, Lymfo 24 %, trombocyty 5 × 109/l), hypofibrinogenemie (1,48 g/l) (norma 1,5–4 g/l), hypoproteinemie (celková bílkovina 47,7 g/l) (norma 58–77 g/l), hypertriglyceridemie (3,47mmol/l) (norma 1,2–1,6 mmol/l), zvýšení sérového feritinu 2 891 µg/l (norma 7–142 µg/l) a LD (13,2 µkat/l) (norma 3–8,4 µkat/l). Imunologicky nebyla patrná aktivace T-lymfocytů ani klonalita (CD3+HLADR+ 2 %), v aspirátu kostní dřeně bylo zachyceno zmnožení makrofágů (3,6 %), z nichž část fagocytovala lymfocyty, erytrocyty, normoblasty i neutrofily. Plánovaná exstirpace uzliny v třísle odložena pro akutní rozvoj ARDS. Z téhož důvodu při klinickém a laboratorním obrazu HLH byla zahájena imunosupresivní terapie podle protokolu HLH 94, která vedla k úpravě klinického stavu i laboratorních nálezů. S vědomím v.s. sekundárního HLH byla léčba po 2 měsících vysazena. 3 měsíce poté, 5 měsíců od diagnózy HLH, byl pacient přijat pro bolesti v levé kyčli a stoupající zánětlivé parametry. CT vyšetření prokázalo prosáknutí měkkých tkání v levé jámě kyčelní s lokálním zvětšením lymfatických uzlin, následné histologické vyšetření extirpované uzliny potvrdilo diagnózu velkobuněčného anaplastického nehodgkinského lymfomu. Nádorové buňky reagovaly pozitivně s monoklonálními protilátkami anti-CD30 a ALK, molekulárně genetické vyšetření prokázalo pro tento typ lymfomu typický fuzní gen ALK-NPM při t(2;5). Terapie podle protokolu pro nehodgkinské lymfomy vedla pouze ke krátké remisi, následoval relaps, pro který byla indikována megachemoterapie s autologní transplantací kostní dřeně.
Kazuistika 5
Chlapec, 10 let. Diagnóza: Syndrom aktivovaných makrofágů asociovaný s juvenilní idiopatickou artritidou. Pacient byl 4 roky léčen v revmatologické ordinaci pro systémovou formu juvenilní idiopatické artritidy (sJIA, HLA DRB4 pozitivní) kombinací kortikoidy + metotrexát (MTX), který byl 4 měsíce před atakou HLH při dosažené remisi sJIA vysazen. Zhoršení celkového stavu se projevilo únavou a subfebriliemi. Byl hospitalizován pro „bronchopneumonii“ (proužkovitá kondenzace plicního parenchymu) s výpotkem – oboustranným hydrotoraxem, stav byl hodnocen jako relaps základního onemocnění. V průběhu 14 dnů se stav výrazně horšil, objevila se cefalea, následně tonicko-klonické křeče, jen s částečnou odpovědí na antikonvulziva, s přetrvávajícím opistotonem, žmouláním, třesy končetin a záškuby mimického svalstva. Vyšetřen liquor, který byl bez pleiocytózy, Pandy negativní (bílkovina 0,90 g/l, cukr 3,29 mmol/l, buňky: 7,6 × 106/l: Lymfo 13 %, Mono 5 %, neutrofily 37 %). Doplněno vyšetření magnetickou rezonancí, na jehož základě (vpravo frontálně a oboustranně parietálně drobná skvrnitá hypodenzní ložiska – susp. ischemické změny při základním onemocnění) bylo vysloveno podezření na vaskulitidu. Kloubní nález zůstával přesto normální, bez známek synovitidy. Souběžně s CNS projevy se vyvinula hepatosplenomegalie (játra + 4 cm, slezina dle USG 120 × 44 mm s nehomogenní strukturou) a v průběhu 14 dnů plný obraz syndromu aktivovaných makrofágů: horečky se septickým charakterem, pancytopenie (Hb 8,0 g/l, erytrocyty 2,80 × 1012/l, leukocyty 2,7 × 109/l, neutrofily 2,1 × 109/l, trombocyty 110 × 109/l), ikterus s hepatopatií (bilirubin 89 µmol/l, AST 14,45 µkat/l, ALT 7,09 µkat/l, GGT 9,43 µkat/l), vysoký feritin v séru > 30 000 ng/l (norma 20–300 ng/l), hypertriglyceridemie (3,76 mmol/l) (norma 0,45–2,2 mmol/l), extrémně zvýšené LDH 234,7 µkat/l (norma 3,8–7,8 µkat/l). Fibrinogen zůstával na dolní hranici normy 1,96 g/l (norma 1,8–4,5 g/l). Vyšetření periferní krve průtokovou cytometrií potvrdilo aktivaci T-lymfocytů a zvýšení IL-2 receptoru (309,2 pmol/l) (norma 25–115 pmol/l), aspirát kostní dřeně byl normocelulární s relativně přiměřeným zastoupením všech hemopoetických řad, byly přítomny atypické buňky RES s výraznou fagocytární aktivitou, zejména erytrofagocytózou. Stav uzavřen jako MAS komplikující léčbu sJIA, jako spouštěč HLH se zdála suspektní kombinace CMV infekce (PCR pozitivita: plazma, leukocyty, buňky kostní dřeně) a infekce parvovirem B19 (pozitivita „nested PCR“ v kostní dřeni). Pacient byl léčen podle protokolu HLH 94 (kortikoidy, etoposid, CsA), kterou byla postupně zvládnuta ataka HLH včetně CNS projevů, po ukončení etoposidu pokračoval v posílené imunosupresivní léčbě kombinací metylprednisolon + CsA + MTX, která dlouhodobě udržovala remisi JIA, k recidivě HLH v dalším průběhu nedošlo [37].
Diagnostický a léčebný algoritmus
Rozlišení mezi primární a sekundární formou HLH má zásadní význam pro volbu léčebné strategie (indikace k transplantaci kostní dřeně u primární HLH vs léčba základního onemocnění u sekundární HLH). Na počátku onemocnění je ale často velmi obtížné, ne-li nemožné. Uvedený algoritmus shrnuje postupné diagnostické kroky, které lze použít v diferenciální diagnostice. Manifestuje-li se HLH u kojence, je na 1. místě nutné uvažovat o geneticky podmíněných příčinách HLH. Nejčastější FHL je asociována s chybějící expresí perforinu v NK buňkách. Diagnózu lze potvrdit molekulárně genetickým vyšetřením v zahraničí. Je-li exprese perforinu normální, je nutné provést vyšetření cytotoxické aktivity T-lymfocytů. Ta je porušená u FHL, u Chediakova-Higashiho a Griscelliho syndromu, zatímco u XLP bývá normální. Diagnózu Chediak-Higashiho syndromu potvrdíme průkazem atypické granulace leukocytů v periferní krvi. Jedná-li se o chlapce a HLH indukovanou EBV infekcí, je XLP vysoce suspektní. Pak je třeba provést funkční vyšetření 2B4 receptoru NK buněk (FACS) a molekulárně genetické vyšetření SAP genu. Přítomnost mutace potvrdí diagnózu XLP, není-li mutace přítomna, může se jednat o deficit XIAP, u něhož je molekulární diagnostika dostupná v zahraničí. V nejběžnější situaci, kdy cytotoxická aktivita lymfocytů je defektní a není potvrzen molekulární podklad, je diagnózou první volby FHL [5]. Všechna tato onemocnění patří do skupiny primární HLH a jsou, po navození remise, indikací k transplantaci kostní dřeně. I takto postavený diagnostický algoritmus má bohužel své limity. Funkční testy cytotoxické aktivity lymfocytů nejsou dostatečně senzitivní ani specifické, normální nález zcela nevylučuje FHL, naopak snížení cytotoxické aktivity můžeme najít i u sekundární HLH asociované s infekcí nebo během imunosupresivní léčby. Důležitým rozlišovacím markerem zůstává často věk pacienta. U starších dětí a dospělých jsou genetická onemocnění méně pravděpodobná, přesto by, zejména s ohledem na XLP, měla zůstávat v diferenciální diagnóze. Výskyt FHL po 3. roce věku je výjimečný, pravděpodobnější je sekundární HLH indukovaná infekcí, malignitou (zejména NHL) nebo systémovým autoimunitním onemocněním. Nutno ale připomenout, že infekce je spouštěčem i geneticky determinované HLH a že obraz HLH vyvine jen specificky imunokompromitovaný jedinec. Panel mikrobiologických a virologických testů je dle cestovatelské anamnézy vhodné rozšířit i na viscerální leishmaniózu. Komplikuje-li HLH průběh maligního onemocnění, je na individuálním rozhodnutí přerušení chemoterapie nádoru a cílená léčba HLH. Komplikuje-li HLH autoimunitní onemocnění, jsou lékem první volby kortikoidy a cyklosporin A. Pokud není zřejmé primární onemocnění, je vždy vhodné zahájit cytotoxickou léčbu HLH s cílem navození remise, ve které je při pochybách léčba přerušena a pacient monitorován. Vznikne-li relaps onemocnění, jde velmi pravděpodobně o primární HLH na podkladě defektu imunity, který nelze současnými metodami diagnostikovat a pacient je indikován k transplantaci.
Závěr
Hemofagocytující lymfohistiocytóza je diagnosticky i léčebně velmi svízelným onemocněním. Klinické projevy nejsou specifické a kromě deficitu perforinu u části dětí s FHL neexistuje ani specifický laboratorní marker HLH. Na druhou stranu, zejména u nejmenších dětí, je průběh HLH často foudroyantní s tendencí s časnému multiorgánovému selhání, což nutí k potřebě rychlé diagnostiky a rozhodnutí o zahájení cytotoxické léčby, která přináší nepochybná rizika. Právě ve snaze rychle definovat stav indikovaný k léčbě byla vypracována diagnostická kritéria definující HLH jako komplex klinických, biochemických, imunologických a morfologických parametrů. Pro rozhodnutí o dalším postupu je zásadní rozlišit mezi primární (geneticky determinovanou) HLH a sekundární, jiným onemocněním indukovanou HLH. Stanovení správné diagnózy vyžaduje možnost vyšetření exprese perforinu, cytotoxické funkce T-lymfocytů a NK buněk, funkční vyšetření 2B4 receptoru, stanovení aktivovaných CD8+ lymfocytů a molekulárně genetické vyšetření k průkazu mutace genů. Nicméně v počátečním stadiu onemocnění je rozlišení mezi primární a sekundární formou HLH často velmi obtížné a přes veškerou snahu není vždy možné zařadit nemoc do jedné z uvedených jednotek. Léčebnou alternativou první volby je v současnosti terapie podle protokolu HLH 2004 – kombinace steroidů, etoposidu a cyklosporinu A, jejímž cílem je navození remise HLH. U primární, geneticky determinované HLH je jedinou kurativní možností následná alogenní transplantace kmenových buněk, která je alternativou i pro relabující pacienty bez prokazatelného molekulárního podkladu HLH. Je zjevné, že i přes velké pokroky v imunologické a molekulární diagnostice HLH má i v současné době zkušenost s klinikou a diferenciální diagnózou zásadní význam pro prognózu tohoto závažného onemocnění.
Práce byla podpořena granty MSM00216 20812, NS10480-3, MZO 00064203 VZ FNM.
MUDr. Martina Suková
www.fnmotol.cz
e-mail: martina.sukova@fnmotol.cz
Doručeno do redakce: 9. 9. 2010
Sources
1. Allen M, De Fusco C, Legrand F et al. Familial hemophagocytic lymphohistiocytosis: how late can the onset be? Haematologica 2001; 86 : 499–503.
2. Aricó M, Janka G, Fischer A et al. Hemophagocytic lymphohistiocytosis. Report of 122 children from the International Registry. FHL Study Group of the Histiocyte Society. Leukemia 1996; 10 : 197–203.
3. Aricó M, Danesino C, Pende D et al. Pathogenesis of haemophagocytic lymphohistiocytosis. Br J Haematol 2001; 114 : 761–769.
4. Aricó M, Imashuku S, Clementi R et al. Hemophagocytic lymphohistiocytosis due to germline mutations in SH2D1A, the X-linked lymphoproliferative disease gene. Blood 2001; 97 : 1131–1133.
5. Aricò M, Allen M, Brusa S et al. Haemophagocytic lymphohistiocytosis: proposal of a diagnostic algorithm based on perforin expression. Br J Haematol 2002; 119 : 180–188.
6. Blatt J, Weston B, Belhorn T et al. Childhood non-Hodgkin lymphoma presenting as hemophagocytic syndrome. Pediatr Hematol Oncol 2002; 19 : 45–49.
7. Certain S, Barrat F, Pastural E et al. Protein truncation test of LYST reveals heterogenous mutations in patients with Chediak-Higashi syndrome. Blood 2000; 95 : 979–983.
8. Côte M, Ménager M, Burgess A et al. Munc 18-2 deficiency causes familial hemophagocytic lymphohistiocytosis type 5 and impairs cytotoxin granule exocytosis in patient NK cells. J Clin Invest 2009; 119 : 3765–3773.
9. Dufourcq-Lagelouse R, Pastural E, Barrat FJ et al. Genetic basis of hemophagocytic lymphohistiocytosis syndrome (Review). Int J Mol Med 1999; 4 : 127–133.
10. Göransdotter Ericson K, Fadeel B, Nilsson-Ardnor S et al. Spectrum of perforin gene mutations in familial hemophagocytic lymphohistiocytosis. Am J Hum Genet 2001; 68 : 590–597.
11. Farquhar JW, Claireaux AE. Familial haemophagocytic reticulosis. Arch Dis Child 1952; 27 : 519–525.
12. Favara BE, Jaffe R, Egeler RM. Macrophage activation and hemophagocytic syndrome in Langerhans cell histiocytosis: report of 30 cases. Pediatr Dev Pathol 2002; 5 : 130–140.
13. Feldmann J, Le Deist F, Ouachée-Chardin M et al. Functional consequences of perforin gene mutations in 22 patients with familial haemophagocytic lymphohistiocytosis. Br J Haematol 2002; 117 : 965–972.
14. Feldmann J, Callebaut I, Raposo G et al. Munc 13-4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3). Cell 2003; 115 : 461–473.
15. Fisman DN. Hemophagocytic syndromes and infection. Emerg Infect Dis 2000; 6 : 601–608.
16. Gagnaire MH, Galambrun C, Stéphan JL. Hemophagocytic syndrome: a misleading complication of visceral leishmaniasis in children-a series of 12 cases. Pediatrics 2000; 106: E58.
17. Gaspar HB. X-linked lymphoproliferative disease: clinical, diagnostic and molecular perspective. Br J Haematol 2002; 119 : 585–595.
18. Haddad E, Sulis ML, Jabado N et al. Frequency and severity of central nervous system lesions in hemophagocytic lymphohistiocytosis. Blood 1997; 89 : 794–800.
19. Henter JI, Elinder G, Ost A et al. Diagnostic guidelines for hemophagocytic lymphohistiocytosis. The FHL Study Group of the Histiocyte Society. Semin Oncol 1991; 18 : 29–33.
20. Henter JI. Biology and treatment of familial hemophagocytic lymphohistiocytosis: importance of perforin in lymphocyte-mediated cytotoxicity and triggering of apoptosis. Med Pediatr Oncol 2002; 38 : 305–309.
21. Henter JI, Samuelsson-Horne AC, Aricò M et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood 2002; 100 : 2367–2373.
22. Henter JI, Horne AC, Aricó M et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2007; 48 : 124–131.
23. Jaffe ES, Costa J, Fauci AS et al. Malignant lymphoma and erythrophagocytosis simulating malignant histiocytosis. Am J Med 1983; 75 : 741–749.
24. Janka GE. Familial hemophagocytic lymphohistiocytosis. Eur J Pediatr 1983; 140 : 221–230.
25. Janka G, Imashuku S, Elinder G et al. Infection – and malignancy-associated hemophagocytic syndromes. Secondary hemophagocytic lymphohistiocytosis. Hematol Oncol Clin North Am 1998; 12 : 435–444.
26. Kumar M, Sackey K, Schmalstieg F et al. Griscelli syndrome: rare neonatal syndrome of recurrent hemophagocytosis. J Pediatr Hematol Oncol 2001; 23 : 464–468.
27. Ménasché G, Feldmann J, Houdusse A et al. Biochemical and functional characterization of Rab27a mutations occuring in Griscelli syndrome patients. Blood 2003; 101 : 2736–2742.
28. Pileri SA, Grogan TM, Harris NL et al. Tumours of histiocytes and accessory dendritic cells: an immunohistochemical approach to classification from the International Lymphoma Study Group based on 61 cases. Histopathology 2002; 41 : 1–29.
29. Purtillo DT, Cassel CK, Yang JP et al. X-linked recessive progressive combined variable immunodeficiency (Duncan’s disease). Lancet 1975; 1 : 935–941.
30. Rigaud S, Fondanèche MC, Lambert N et al. XIAP deficiency in humans causes an X-linked lymphoproliferative syndrome. Nature 2006; 444 : 110–114.
31. Risdall RJ, McKenna RW, Nesbit ME et al. Virus-associated hemophagocytic syndrome. Cancer 1979; 44 : 993–1002.
32. Sawhney S, Woo P, Murray KJ. Macrophage activation syndrome: a potentially fatal complication of rheumatic disorders. Arch Dis Child 2001; 85 : 421–426.
33. Stejskal J, Hrodek O, Eleder M. Familiární hemofagocytující lymfohistiocytóza. Česk Patol 1990; 26 : 14–24.
34. Stéphan JL, Koné-Paut I, Galambrun Cet al. Reactive haemophagocytic syndrome in children with inflammatory disorders. A retrospective study of 24 patients. Rheumatology 2001; 40 : 1285–1292.
35. Stepp SE, Dufourcq-Lagelouse R, Le Deist F et al. Perforin gene defects in familial hemophagocytic lymhohistiocytosis. Science 1999; 286 : 1957–1959.
36. Suková M, Starý J, Housková J et al. Hemofagocytující lymfohistiocytóza jako manifestace viscerální leishmaniózy. Čas Lék Čes 2002; 141 : 581–584.
37. Starý J, Housková J, Špíšek R et al. Hemofagocytující lymfohistiocytóza – diagnostické a léčebné dilema. Čes Slov Pediat 2004; 59 : 70–82.
38. Špíšek R, Mejstříková E, Formánková R et al. Familiární hemofagocytující lymfohistiocytóza na podkladě deficitu perforinu úspěšně léčená transplantací hematopoetických kmenových buněk – první diagnostikovaný případ v České republice. Čas Lék čes 2006; 145 : 50-54.
39. Sullivan KE, Delaat CA, Douglas SD et al. Defective natural killer cell function in patients with hemophagocytic lymphohistiocytosis and in first degree relatives. Pediatr Res 1998; 44 : 465–468.
40. Wulffraat NM, Rijkers GT, Elst E et al. Reduced perforin expression in systemic juvenile idiopathic arthritis is restored by autologous stem-cell transplantation. Rheumatology 2003; 42 : 375–379.
41. zur Stadt U, Schmidt S, Kasper B et al. Linkage of familial hemophagocytic lymphohistiocytosis (FHL) type-4 to chromosome 6q24 and identification of mutations in syntaxin 11. Hum Mol Genet 2005; 14 : 827–834.
42. zur Stadt U, Beutel K, Kolberg S et al. Mutation spectrum in children with primary hemophagocytic lymphohistiocytosis: molecular and functional analyses of PRF1, UNC13D, STX11 and RAB27A. Hum Mutat 2006; 27 : 62–68.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2010 Issue Supplementum 2
Most read in this issue
- Hemofagocytující lymfohistiocytóza
- Erdheimova-Chesterova nemoc v obrazech
- Systémová mastocytóza
- Histiocytóza z Langerhansových buněk u dětí a dospívajících