
Systémová mastocytóza
Systemic mastocytosis
Systemic mastocytosis is a heterogenous disorder characterized by abnormal growth and accumulation of abnormal mast cells in one or more organs. Mast cells are derived from CD34+/KIT+ pluripotent hematopoietic cells in the bone marrow. Most adult mastocytosis patients carry gain-of-function c-kit receptor mutations, most commonly D816V in the tyrosine kinase domain. The clinical phenotype of systemic mastocytosis is variable. Many of patients are suffering from urticaria pigmentosa, followed by the flushing, cramping, abdominal pain, diarrhea, bone pain and hepatosplenomegaly. Most adult patients have systemic disease (a condition generally confirmed by bone marrow biopsy). The natural history of systemic mastocytosis ranges from indolent forms to very aggressive subtypes.
Key words:
diagnostics – KIT – management – masitinib – systemic mastocytosis
Authors:
M. Doubek 1; T. Kozák 2; V. Vašků 3; P. Szturz 1; M; Tichá 1; L. Křen 4
Authors‘ workplace:
Interní hematoonkologická klinika Lékařské fakulty MU a FN Brno, pracoviště Bohunice, přednosta prof. MUDr. Jiří Mayer, CSc.
1; Oddělení klinické hematologie FN Královské Vinohrady Praha, přednosta prim. doc. MUDr. Tomáš Kozák, CSc., MBA
2; 1. dermatovenerologická klinika Lékařské fakulty MU a FN u sv. Anny Brno, přednosta doc. MUDr. Vladimír Vašků, CSc.
3; Ústav patologie Lékařské fakulty MU a FN Brno, pracoviště Bohunice, přednosta doc. MUDr. Josef Feit, CSc.
4
Published in:
Vnitř Lék 2010; 56(Supplementum 2): 188-194
Category:
Langerhans cell histiocytosis and some other Hematology rare diseases
Overview
Systémová mastocytóza je heterogenní onemocnění charakterizované množením a akumulací patologických mastocytů v jednom nebo více orgánech. Mastocyty vychází z CD34+/KIT+ pluripotentních hematopoetických buněk z kostní dřeně. Mnoho dospělých nemocných s mastocytózou má v mastocytech aktivační mutaci genu pro receptor c-kit, nejčastěji D816V v tyrozinkinázové doméně. Fenotyp systémové mastocytózy je různorodý. Mnoho nemocných má kožní změny typu urticaria pigmentosa, které provází červenání kůže, křeče, bolesti břicha, průjem, bolesti kostí a hepatosplenomegalie. Zejména dospělí nemocní mají velmi často systémové postižení, které je potvrzeno biopsií kostní dřeně. Průběh nemoci je u některých typů indolentní, u jiných může být velmi agresivní.
Klíčová slova:
diagnostika – KIT – léčba – masitinib – systémová mastocytóza
Úvod
Systémová mastocytóza je myeloproliferativní choroba, která vychází z mastocytů, tedy tkáňových makrofágů [23]. Onemocnění vzniká patologickou kumulací těchto buněk v různých orgánech, velmi často v kůži.
I když v českých zemích a na Slovensku se této nemoci několik pracovišť dlouhodobě věnuje [11,17,21], naše zkušenost naznačuje, že často o ní nemá širší lékařská veřejnost plné povědomí, a mnozí nemocní tak mohou mít i roky potíže, aniž by měli stanovenu přesnou diagnózu, nebo jsou léčeni a sledováni jen pro kožní projevy nemoci, aniž by bylo vyšetřeno, zda nemají také systémové postižení.
Patogeneze
Mastocyty mají původ v CD34+/KIT+ pluripotentních hematopoetických buňkách kostní dřeně [9,10,26]. Progenitory mastocytů exprimují znaky CD13 a CD34 [9] a lze je zjistit nejen v kostní dřeni, ale i periferní krvi [26]. Nejdůležitějším faktorem, který řídí růst a diferenciaci mastocytů, je faktor kmenových buněk (stem cell factor – SCF – c-kit ligand). Tento cytokin je ligandem receptoru c-kit (CD117), jenž se objevuje na myeloidních progenitorech, progenitorech mastocytů a zralých mastocytech [26,28].
Mastocyty se od jiných myeloidních buněk liší skladbou svých povrchových antigenů (CD antigeny, receptor pro IgE s vysokou afinitou k této protilátce), mediátory, které produkují (viz níže) i dlouhou dobou, po kterou v těle přežívají – několik měsíců až let [20,26]. Expresi různých CD antigenů na normálních a maligních mastocytech a na dalších krevních buňkách ukazuje tab. 1.
![Exprese antigenů na normálních a patologických mastocytech a dalších krevních buňkách [26].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/90395d81a88e4b5eaba0494a4ec3fa67.jpeg)
Systémová mastocytóza byla oddělena od dalších myeloidních neoplazií Lennertem a Parwareschem v roce 1979 [12]. Klonální podstata nemoci byla potvrzena roku 1995, kdy byla zjištěna asociace mutace Asp-816-Val (D816V) genu označovaného KIT pro receptor c-kit (substituce aspartát/valin v kodonu 816) [16]. Tato mutace se vyskytuje téměř výhradně jen u systémové mastocytózy a ne u jiných myeloidních malignit. U pacientů s indolentní systémovou mastocytózou lze tuto mutaci najít pouze v mastocytech, zatímco u agresivních mastocytóz i v dalších hematopoetických liniích, např. v monocytární řadě [26]. Mutaci KIT má většina dospělých nemocných se systémovou mastocytózou [8,16]. V některých případech systémové mastocytózy bývá také zjišťována translokace genu PDGFRβ [t(4;5)] či fuzní gen FIP1L1/ PDGFRα [18,20]. Jde především o systémové mastocytózy spojené s lymfadenopatií a eozinofilií. Sporadicky lze najít i cytogenetické změny: del 20(q12), +9, t(8;21) [23,26].
Jak už bylo zmíněno, patologické mastocyty u nemocných se systémovou mastocytózou uvolňují řadu látek: histamin, heparin, tumor nekrotizující faktor α (TNFα), vaskulární endoteliální růstový faktor (VEGF), leukotrieny (LTC4), transformující růstový faktor (TGFβ), tkáňový aktivátor plazminogenu (tPA), prostaglandin D2, interleukiny (IL-1, - 2, - 3, - 5, - 6, - 9, - 10, - 13), chemokiny (IL-8, MCP-1 – monocyte chemoattractant protein, MIP-1α – macrophage inflammatory protein) a proteázy [26]. Tyto biologicky aktivní látky jsou zodpovědné za některé klinické příznaky nemoci (viz níže).
Epidemiologie
Systémová mastocytóza je onemocnění vzácné, jeho přesná incidence není známa. Je pravděpodobné, že onemocnění zůstává u mnoha nemocných nediagnostikováno. Nemoc se může vyskytnout v každém věku, častěji ale po 20. roku života. Poměr mužů a žen s touto nemocí se pohybuje od 1 : 1 do 1 : 3 [23].
Klasifikace systémové mastocytózy
Podle klasifikace Světové zdravotnické organizace se mastocytóza dělí především na kožní mastocytózy a systémové mastocytózy [23].
Kožní mastocytózy jako všechny typy mastocytóz vykazují klinické rysy nebo jejich podstatnou část vyvolanou nadměrnou produkcí mediátorů uvolňovaných mastocyty. Jde především o urtikarielní otok, podklad tzv. erektilních eflorescencí [2].
Kožní mastocytózy mají několik forem:
- urticaria pigmentosa infantum s hemoragickou a bulózní variantou,
- urticaria pigmentosa adultorum,
- telangiectasia macularis eruptiva perstans,
- difuzní kožní mastocytóza,
- mastocytom,
- jiné manifestace – např. erytrodermická [2,6,23].
Mastocytomy se manifestují jako hnědavé uzly obvykle do 6. měsíce věku. Při traumatizaci mohou vyvolat systémové symptomy – „flushing“ a hypotenzi. Představují 15–20 % všech mastocytóz v dětském věku. Souvislost se systémovými mastocytózami nebývá.
U urticaria pigmentosa v dětském věku se hustý výsev žlutohnědých, hnědočervených či šedohnědých makul nebo lehce prominujících papul objevuje především na trupu. Makuly jsou obvykle neostře ohraničené. Oblasti exponované slunečnímu záření jsou postiženy méně často. Mírné podráždění v oblasti makuly či papuly vede ke vzniku urtikarielního pupenu a erytému – tzv. Darierovo znamení. Část dětí má nevelké potíže s příznaky jen v místě tření kůže ve stále stejných oblastech, např. jako důsledek tření plenami. Výjimečně se objevují v místech skvrn či papul hemoragie. Vystupňovaná reakce může vést až k tvorbě puchýřů, což vidíme spíše jen v kojeneckém věku. Spouštěcími faktory pro tento vývoj může být infekce či imunizace. Tendence k tvorbě puchýřů obvykle vymizí do puberty.
Projevy urticaria pigmentosa u dospělých mají hnědavé zbarvení. Dermografizmus s výrazným erytémem nebo kopřivkovými pupeny je silně vyjádřen. S puchýři se u této formy nesetkáváme. U rozsáhlého postižení se může objevit komplex systémových příznaků z uvolňovaných mediátorů (především histaminu) až po ztrátu vědomí.
Projevy typu urticaria pigmentosa se vyskytují u více než 90 % nemocných s indolentní systémovou mastocytózou a u méně než 50 % nemocných se systémovou mastocytózou spojenou s dalším klonálním hematologickým onemocněním.
Telangiectasia macularis eruptiva perstans je vzácným onemocněním, představuje jen 1 % mastocytóz a manifestuje se převážně jen u dospělých. Projevuje se červenohnědými telangiektatickými makulami s nepravidelnými okraji. Darierovo znamení často nebývá pozitivní.
Difuzní kožní mastocytóza je velmi vzácnou formou. Vyskytuje se téměř výlučně u dětí, může přetrvávat do dospělosti. Kůže má difuzně vzhled pomerančové kůry s červenohnědavými barevnými okrsky. Je provázena živým dermografizmem s možností tvorby puchýřů [6,24].
Pacienti s kožní mastocytózou by měli být důkladně vyšetřeni, neboť řada nemocných může mít zatím asymptomatické postižení dalších orgánů.
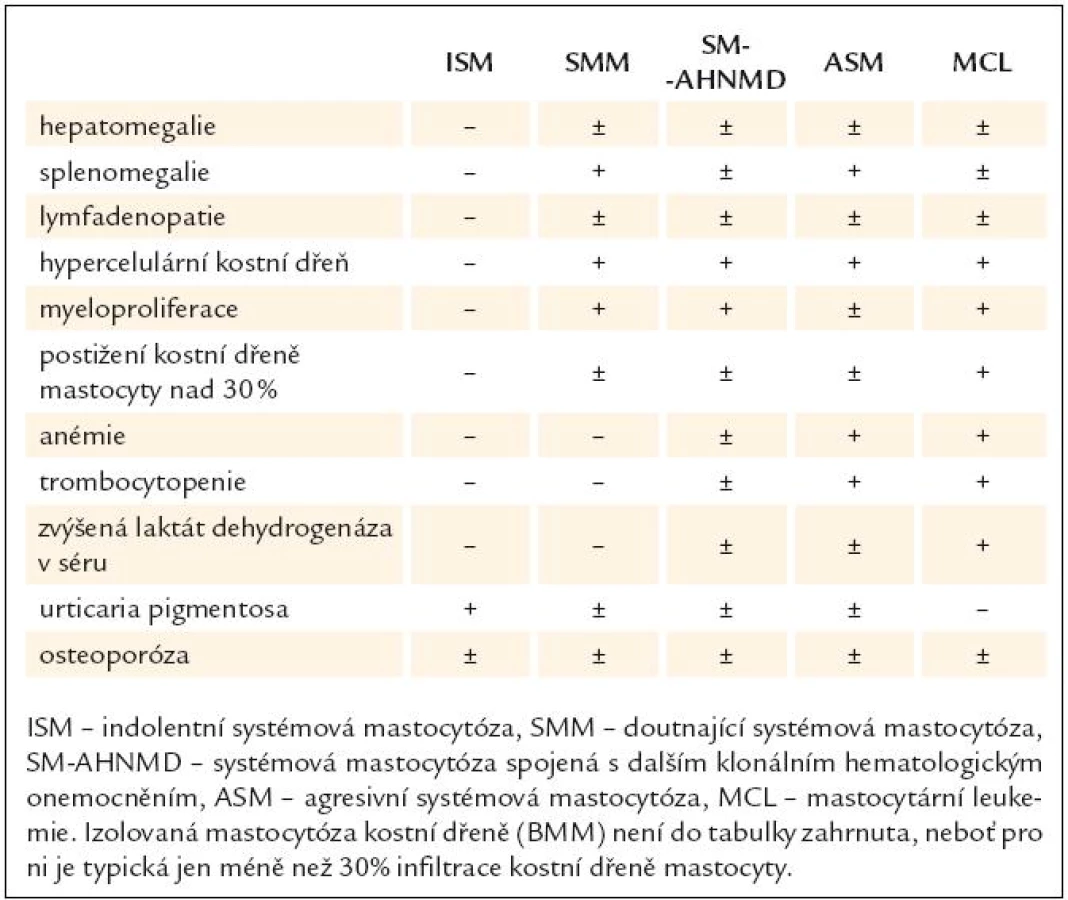
Systémová mastocytóza se dělí na indolentní systémovou mastocytózu (ISM), která má 2 podskupiny, tzv. izolovanou mastocytózu kostní dřeně (BMM) a doutnající systémovou mastocytózu (SMM), systémovou mastocytózu spojenou s dalším klonálním hematologickým onemocněním (akutní myeloidní leukemií, myelodysplastickým syndromem, myeloproliferativním onemocněním, chronickou myeloidní leukemií nebo neHodgkinovými lymfomy) (SMAHNMD), agresivní systémovou mastocytózu (ASM) a mastocytární leukemii (MCL) [23].
Velmi vzácně se mohou vyskytnout i lokalizované mastocytární tumory – mastocytární sarkom a extrakutánní mastocytom [23].
Klinické a laboratorní nálezy u jednotlivých typů mastocytózy představuje tab. 2.
Příznaky
Příznaky systémové mastocytózy jsou velmi různorodé. Bývají způsobeny jednak látkami, které se z patologických mastocytů uvolňují, a jednak postižením orgánů, které je způsobeno jejich infiltrací maligními buňkami.
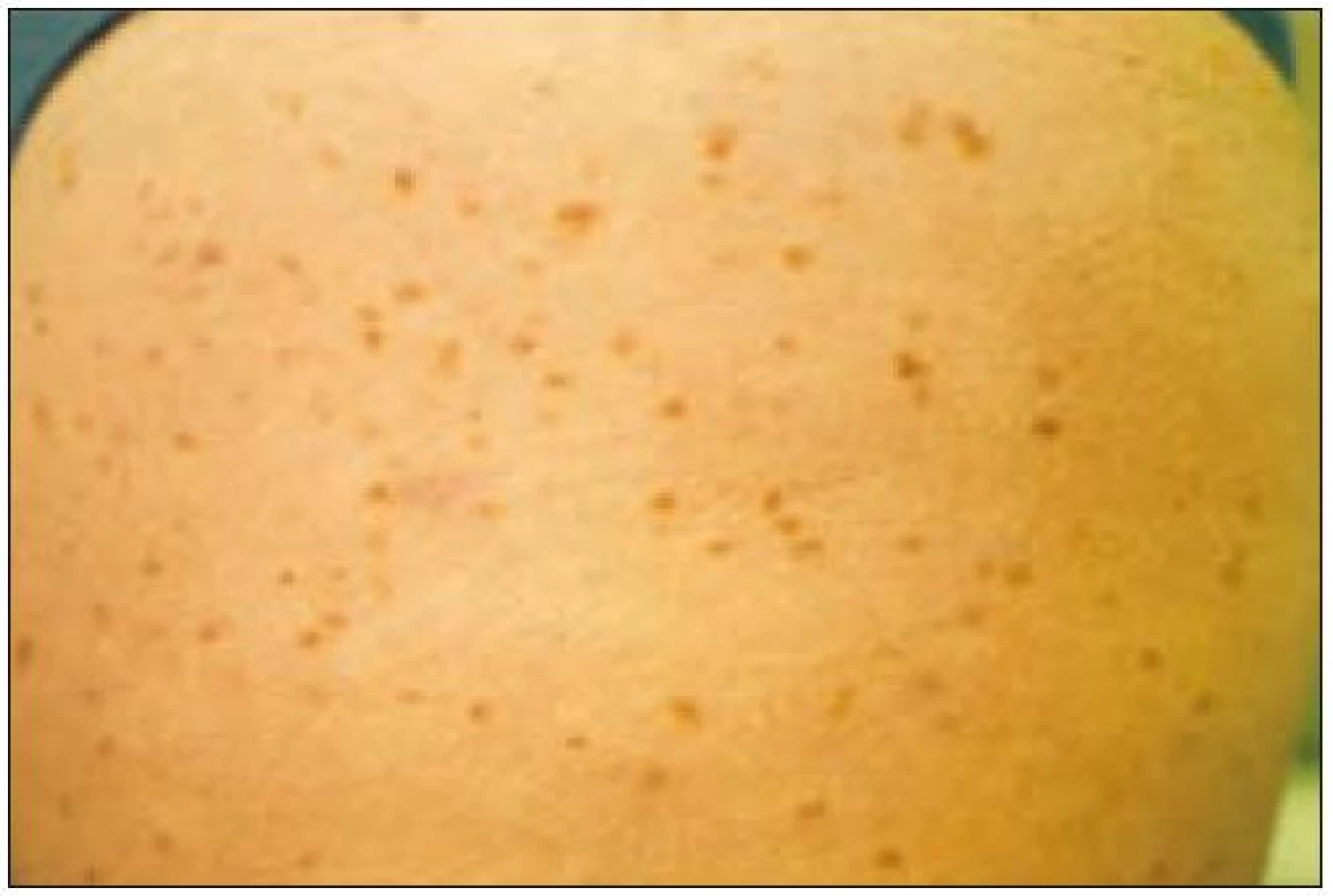
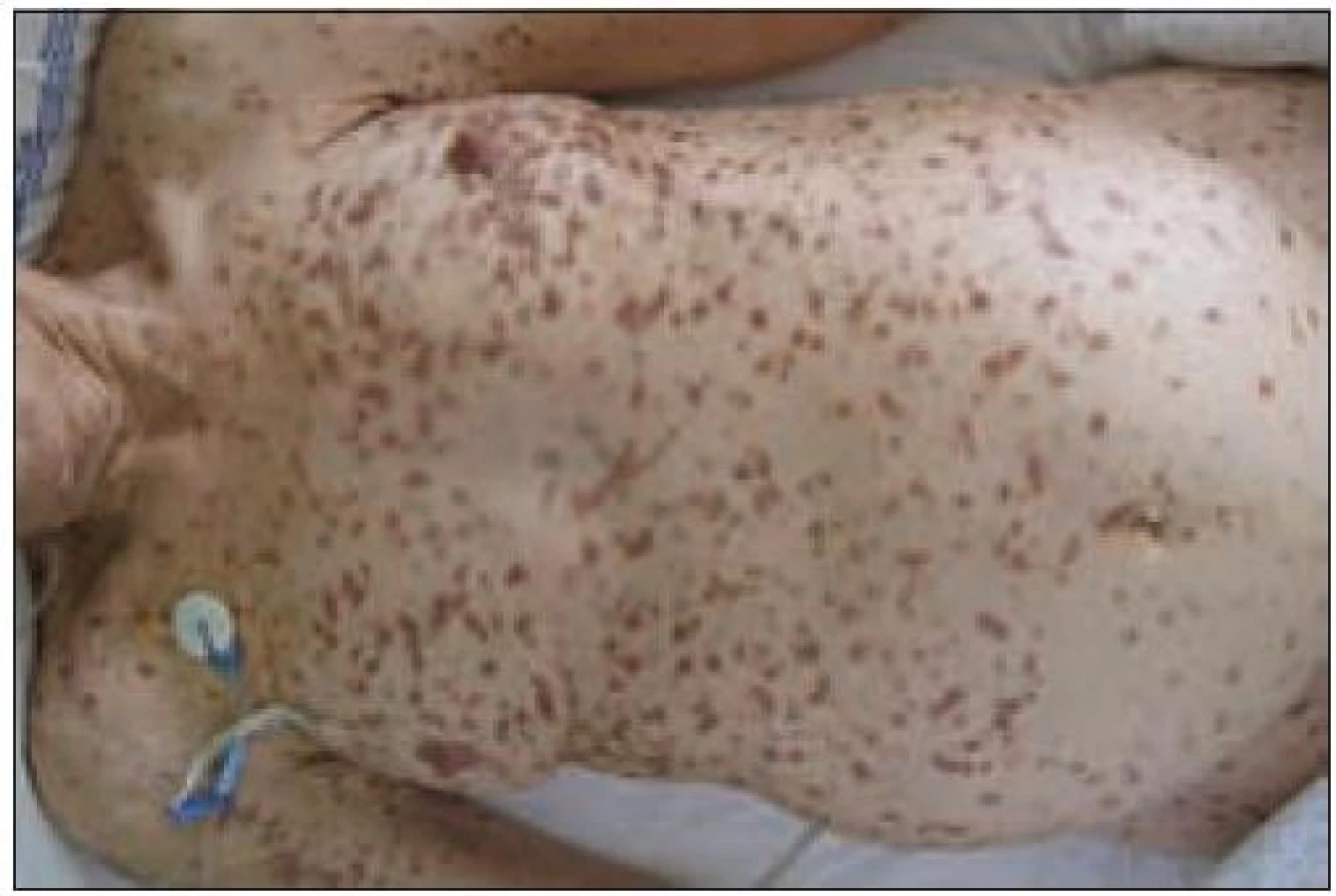
Pacienti trpí velmi často kožními příznaky typu urticaria pigmentosa (obr. 1 a 2), osteoporózou a postižením jaterních funkcí. Kožní příznaky mohou trvat roky, aniž by byly správně diagnostikovány jako projev systémové mastocytózy! Je také zajímavé, že tento příznak provází většinou indolentní formy mastocytózy a je méně častý u agresivních forem nemoci. Kožní postižení má asi 80 % nemocných se systémovou mastocytózou [23]. U agresivních forem systémové mastocytózy může být přítomno postižení kterékoliv tkáně a orgánu. Častá je hmatná splenomegalie, hepatomegalie, lymfadenopatie, ascites, cytopenie v periferní krvi způsobená infiltrací kostní dřeně nebo hypersplenizmem, osteoporóza, osteolýza nebo malabsorpce [15,26] (tab. 2).
Kromě těchto příznaků mají nemocní i řadu příznaků systémových, které jsou způsobeny biologicky aktivními látkami, jež se z mastocytů uvolňují: bolesti hlavy, otoky, průjmy, časté močení, svědění kůže, ataky zarudnutí kůže, náhlé poklesy nebo zvyšování krevního tlaku, horečka, nevolnost, hubnutí, deprese [26].
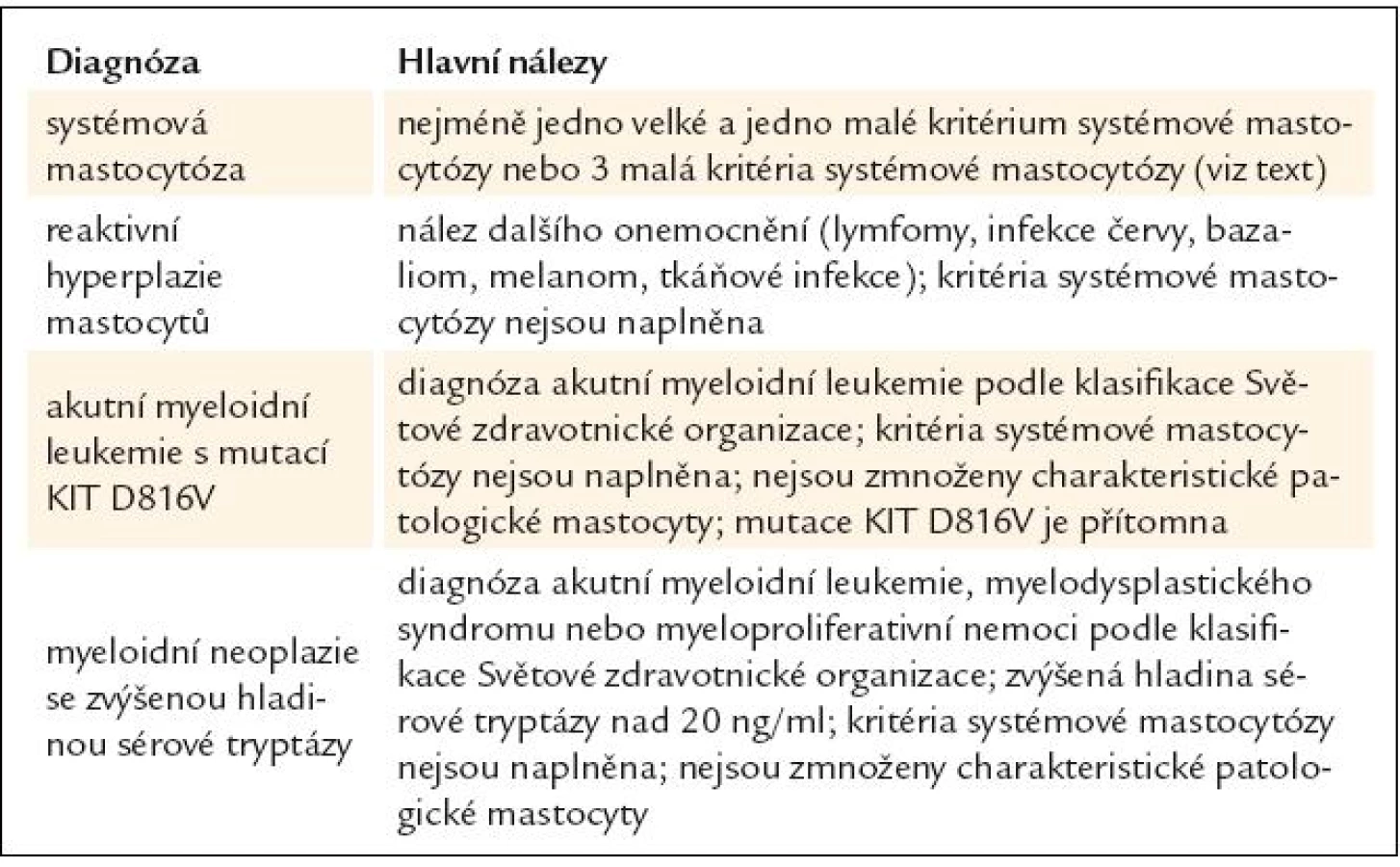
Diferenciální diagnostiku systémové mastocytózy, reaktivní hyperplazie mastocytů, akutní myeloidní leukemie s mutací KIT D816V a myeloidních neoplazií se zvýšenou hladinou tryptázy uvádí tab. 3.
Diagnostika
Ke stanovení diagnózy systémové mastocytózy je zapotřebí provedení histologického, hematologického a biochemického vyšetření, případně i další vyšetření zobrazovacími metodami.
K specializovanému vyšetření patří histologické vyšetření kožních lézí, denzitometické vyšetření kostí, vyšetření kostní dřeně trepanobiopsií a vyšetření periferní krve. Histolog v kostní dřeni vidívá shluky mastocytů. Molekulárně biologické vyšetření často v buňkách kostní dřeně (nebo jiné postižené tkáně) prokazuje bodovou mutaci KIT genu (D816V). Molekulárně genetické vyšetření periferní krve nebývá zejména u pacientů s ISM přínosné. To dokazují i česká data, podle nichž má pozitivitu mutace KIT jen 46 % nemocných, i když podle literatury je to naprostá většina systémových mastocytóz. Řada pacientů z české analýzy měla na přítomnost mutace vyšetřenu jen periferní krev [8,11,16] (viz níže).
Velkým kritériem pro stanovení diagnózy systémové mastocytózy je nález více než 3 denzních infiltrátů mastocytů v biopsii kostní dřeně (viz níže).
Malými kritérii jsou:
- nález více než 25 % mastocytů v kostní dřeni,
- průkaz mutace KIT (D816V),
- exprese znaků CD2 a/nebo CD25 na mastocytech a
- zvýšená hladina sérové tryptázy.
Diagnóza systémové mastocytózy je splněna, když u nemocného zjišťujeme jedno velké a jedno malé kritérium, nebo 3 malá kritéria [23].
Hladina sérové tryptázy je většinou normální (pod 20 ng/ml) u pacientů s izolovanou kožní mastocytózou, zatímco u pacientů se systémovou mastocytózou bývá až na výjimky zvýšená.
V séru pacientů se systémovou mastocytózou může být zvýšená hladina kalcitoninu nebo prostaglandinu F2a. V moči lze detekoval histamin nebo N-metylhistamin [26].
Histopatologické nálezy u systémové mastocytózy
Velkým kritériem pro stanovení diagnózy systémové mastocytózy je nález více než 3 denzních infiltrátů mastocytů v biopsii kostní dřeně. Takovýto infiltrát je agregátem více než 15 atypických mastocytů a lze ho najít v kostní dřeni nebo v jiné tkáni či orgánu (obr. 3) [23].
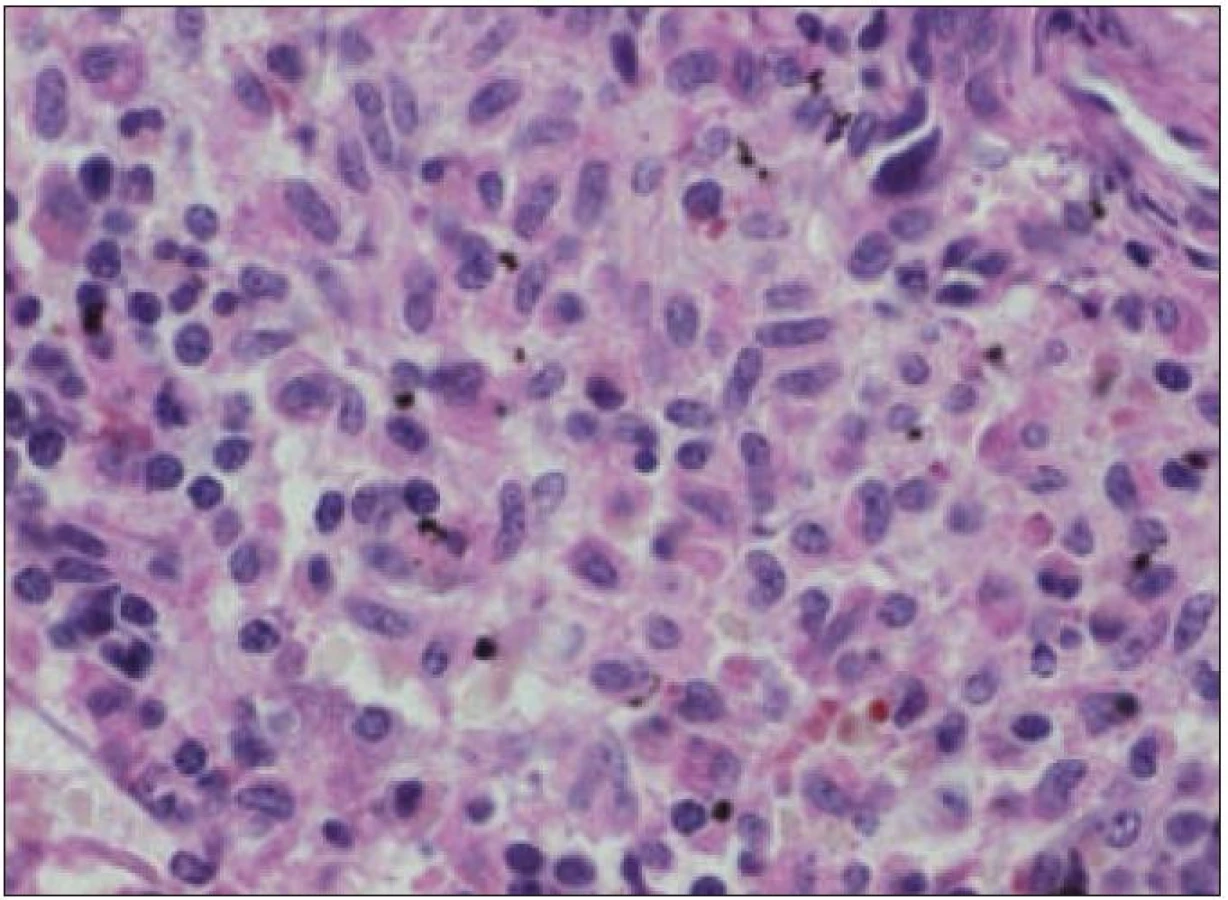
Morfologické charakteristiky typických a atypických mastocytů shrnuje tab. 4. Typické cytomorfologické nálezy v kostní dřeni a periferní krvi nemocných se systémovou mastocytózou pak uvádí tab. 5.

![Typické cytomorfologické nálezy v kostní dřeni a periferní krvi u jednotlivých typů systémové mastocytózy [26].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/ea87ec6ec46ee97808783500a942e625.jpeg)
K průkazu mastocytů používají histopatologové různá speciální barvení. Nejčastěji používané je imunohistochemické značení protilátkou proti tryptáze mastocytů. Z histochemických metod lze užít k identifikaci cytoplazmatických granulí metachromatická barvení (toluidinová modř nebo barvení podle Giemsy) či názorné barvení kresylovou violetí (obr. 4).
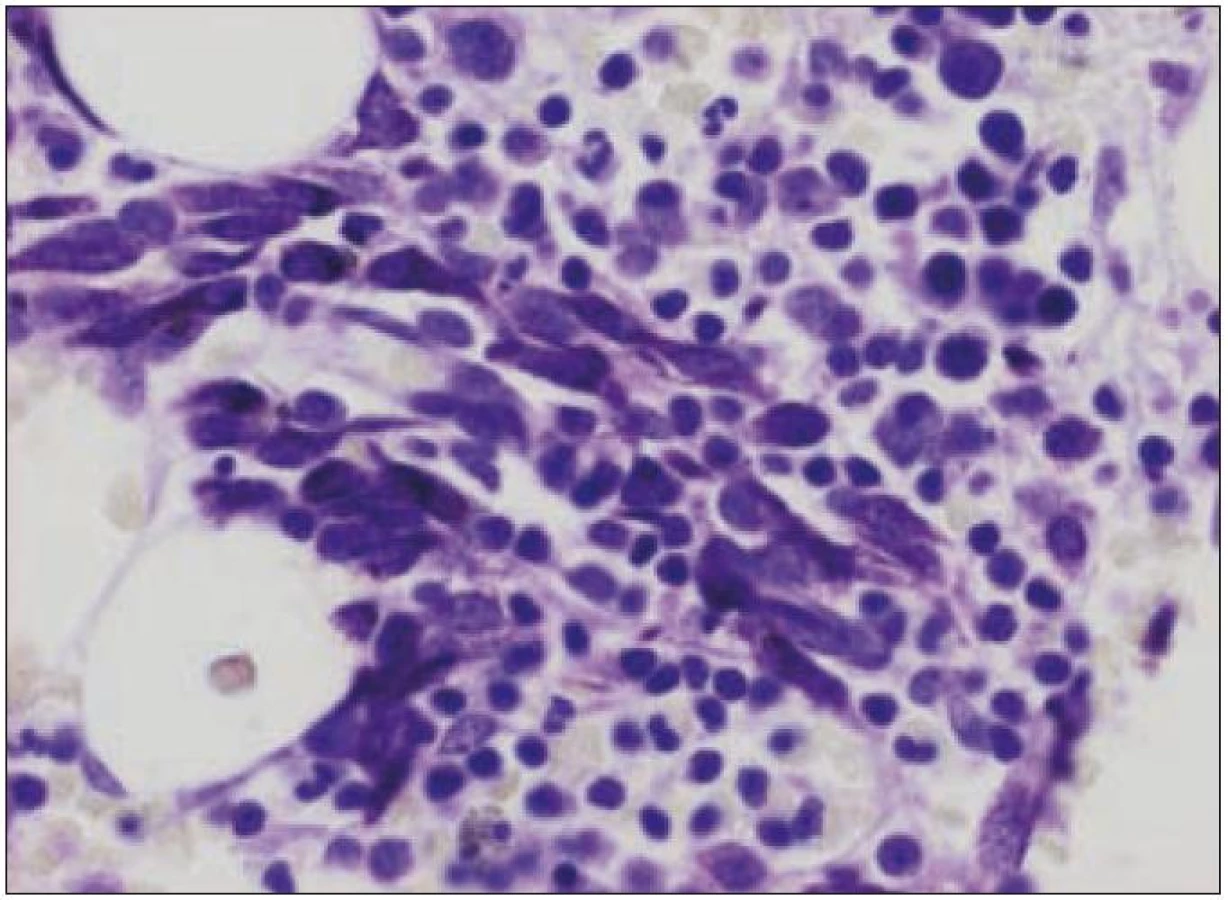
Terapie
Léčbu pacientů se systémovou mastocytózou lze rozdělit na léčbu systémových příznaků, lokální léčbu kožních příznaků a cílenou terapii proti proliferaci maligních mastocytů.
Terapie systémových příznaků
V léčbě systémových (mediátory uvolněnými z patologických buněk způsobených) příznaků se uplatňují antihistaminika (antagonisté H1 i H2 receptorů). Antihistaminika je nutné podávat u pacientů se symptomy dlouhodobě. V případě, že antihistaminika efekt nemají, je zapotřebí nasadit kortikoidy. V léčbě zažívacích potíží provázejících systémovou mastocytózu se osvědčil omeprazol či další inhibitory protonové pumpy a také kromoglykát sodný. U pacientů, u nichž došlo k rozvoji anafylaktické reakce, je zapotřebí intenzivní léčba tohoto stavu vysokými dávkami kortikoidů a katecholaminů. Analgetika jsou indikována v léčbě bolestivých, zejména kostních lézí. V terapii osteopenie a osteoporózy jsou indikovány bisfosfonáty, kalcium a vitamin D [26,27].
Lokální terapie kožních příznaků
U dětí lze pozorovat velmi často spontánní regresi kožních lézí. U dospělých je to ale jev zřídkavý (méně než 10 % dospělých) [3].
Symptomaticky se široce používají steroidní externa a emoliencia, dále lokálně aplikované inhibitory kalcienurinu. Osvědčuje se také fototerapie, resp. PUVA (fotochemoterapie) na principu kombinace UVA záření se syntetickými psoraleny, především 8metoxypsoralenem celkově podávaným. Dobré výsledky přináší také metoda UVA I se zářením v oblasti 340–400 nm o vysoké energii. U teleangiectasia macularis perstans lze použít lasery [6].
Cílená terapie proti proliferaci maligních mastocytů
Tato terapie je indikována u nemocných u všech pacientů s agresivními formami systémové mastocytózy a u pacientů s indolentní systémovou mastocytózou, kteří mají systémové symptomy onemocnění.
Interferon α je lékem, který u nemocných potlačuje systémové příznaky choroby. Aplikuje se většinou v dávce 3 miliony jednotek podkožní injekcí 3krát týdně. Dávky však mohou být i mnohem vyšší. Není vhodný pro nemocné s agresivní systémovou mastocytózou a mastocytární leukemií, u nichž má maximálně jen přechodný efekt [5,26,27].
Imatinib, tyrozinkinázový inhibitor, je účinný především u pacientů se systémovou mastocytózou, kteří nemají mutaci KIT D816V. U těchto nemocných je po podání léku pozorováno až 80 % příznivých léčebných odpovědí (zejména ústup systémových příznaků nemoci). Mutace KIT D816V způsobuje rezistenci na imatinib [14,19]. Imatinib je dále 100% účinný u nemocných s pozitivním nálezem fuzního genu FIP1L1/PDGFRα [27].
Nilotinib, tyrozinkinázový inhibitor, jenž je odvozen od imatinibu, prokázal v pokusech na myším modelu efekt na buňky s mutací KIT D814V, které odpovídají lidské mutaci D816V [4].
Dasatinib je další z tyrozinkinázových inhibitorů, který má širší spektrum inhibice tyrozinkináz než imatinib. Jeho efekty v monoterapii jsou ale u systémové mastocytózy omezené. Větší úspěšnost léčby tímto lékem lze možná očekávat v kombinaci s chemoterapií (např. kladribin) [1,22,25].
Masitinib je nový tyrozinkinázový inhibitor, který má selektivně inhibiční efekt na KIT, způsobuje zástavu buněčného cyklu a apoptózu mastocytů. Masitinib na rozdíl od imatinibu inhibuje mastocyty s mutovaným i wild type KIT. Kromě KIT inhibuje i PDGFR, FGF-R3 a FAK (focal adhesion kinase). Studie fáze II s tímto lékem u systémové mastocytózy prokázaly významnou redukci systémových příznaků nemoci u pacientů (zatím nepublikovaná data firmy AB Science, Paříž, Francie).
Chemoterapie a další léčebné možnosti – v léčbě pomalu progredujících systémových mastocytóz může mít efekt aplikace kladribinu (2-chlordeoxyadenosin; po jeho podání je dosahováno 32–65 % parciálních remisí), nebo cyklosporinu A. Paliativně lze podávat hydroxyureu. U mladších nemocných s mastocytární leukemií lze užít chemoterapeutické režimy jako u pacientů s akutní myeloidní leukemií a lze u nich uvažovat i o alogenní transplantaci kostní dřeně. Prognóza mastocytární leukemie je však i přes tuto razantní terapii velmi špatná [26,27].
Nemocní se SM-AHNMD samozřejmě profitují z léčby základního onemocnění.
Průběh a prognóza
Prognóza onemocnění se liší podle typu systémové mastocytózy. Pacienti s ISM mají medián přežití 198 měsíců, pacienti s ASM 41, pacienti se SMAHNMD 24 a nemocní s MCL pouhé 2 měsíce [13].
K nezávislým rizikovým faktorům systémové mastocytózy se řadí: věk ≥ 65 let, váhový úbytek, hladina hemoglobinu ≤ 100 g/l, trombocyty v periferní krvi ≤ 100 × 109/l, hypalbuminemie a > 5 % blastů v kostní dřeni [13].
Mastocytóza v České republice
V roce 2010 byla zveřejněna data získaná sledováním 30 nemocných se systémovou mastocytózou léčených na Oddělení klinické hematologie FN Královské Vinohrady Praha a na Interní hematoonkologické klinice FN Brno [11]. V tomto souboru bylo 19 žen a 11 mužů; medián věku byl 55,5 roku (25–79). 19 nemocných mělo ISM, 5 SM-AHNMD (při myelodysplastickém syndromu, akutní myeloidní leukemii, esenciální trombocytemii a 2krát při non-Hodgkinově lymfomu), 4 ASM a 2 MCL. Urticaria pigmentosa mělo 9 nemocných, anafylaktické reakce 7 a osteolýzu 4. U 12 nemocných z 26 vyšetřených (46 %) byla zjištěna mutace KIT D816V. 11 nemocných bylo léčeno interferonem α s efektem stabilizace nemoci u 9 a efektem snížení aktivity nemoci u 2. Kladribin byl podán 4 nemocným, u 3 z nich navodil parciální remisi. Imatinib podaný ve 3 případech vždy stabilizoval onemocnění, cytosin arabinosid podaný 1 nemocnému průběh nemoci neovlivnil. Za dobu sledování zemřelo 5 nemocných (3 nemocné se SM-AHNMD, 1 nemocný s MCL a 1 nemocný s ASM [11].
Nemocní s mastocytózami se začínají sdružovat do pacientských organizací. Je to patrné i v České republice. O jejich aktivitách se lze dozvědět více na www.mastocytosis.eu [7] a v brzké době i na www.mastocytosa.cz.
Závěr
Systémová mastocytóza je raritní myeloproliferativní onemocnění s různým průběhem, na které je ale třeba myslet především v diferenciální diagnostice anafylaktických reakcí, organomegalie, uzlinového syndromu, osteolytických ložisek a samozřejmě urticaria pigmentosa.
prof. MUDr. Michael Doubek, Ph.D.
www.fnbrno.cz
e-mail: mdoubek@fnbrno.cz
Doručeno do redakce: 9. 9. 2010
Sources
1. Aichberger KJ, Sperr WR, Gleixner KV et al. Treatment response to cladribine and dasatinib in rapidly progressing aggressive mastocytosis. Eur J Clin Invest 2008; 38 : 869–873.
2. Amon U. Mastocytózy. In: Braun-Falco O,Plewig G, Wolff HH (eds). Dermatológia a venerológia. Martin: Osveta 2001 : 1313–1319.
3. Brockow K, Scott LM, Worobec AS et al. Regression of urticaria pigmentosa in adult patients with systemic mastocytosis: correlation with clinical patterns of disease. Arch Dermatol 2002; 138 : 785–790.
4. von Bubnoff N, Gorantla SH, Kancha RK et al. The systemic mastocytosis-specific activating cKit mutation D816V can be inhibited by tyrosine kinase inhibitor AMN107. Leukemia 2005; 19 : 1670–1671.
5. Butterfield JH, Tefferi A, Kozuch GF. Successful treatment of systemic mastocytosis with high-dose interferon-alfa: long-term follow-up of a case. Leuk Res 2005; 29 : 131–134.
6. Carter MC, Metcalfe DD. Biology of Mast Cells and the Mastocytosis Syndromes. In: Wolff K, Goldsmith LA, Katz SI et al (eds). Fitzpatrick’s Dermatology in General Medicine. New York: McGraw-Hill 2008 : 1434–1443.
7. European Mastocytosis Support Network. 20. 7. 2010. Available from www.masocytosis.eu.
8. Garcia-Montero AC, Jara-Acevedo M, Teodosio C et al. KIT mutation in mast cells and other bone marrow hematopoietic cell lineages in systemic mast cell disorders: a prospective study of the Spanish Network on Mastocytosis (REMA) in a series of 113 patients. Blood 2006; 108 : 2366–2372.
9. Kirshenbaum AS, Goff JP, Semere T et al. Demonstration that human mast cells arise from a progenitor cell population that is CD34(+), c-kit(+), and expresses aminopeptidase (CD13). Blood 1999; 94 : 2333–2342.
10. Kitamura Y, Yokoyama M, Matsuda H et al. Spleen colony-forming cell as common precursor for tissue mast cells and granulocytes. Nature 1981; 291 : 159–160.
11. Kozák T, Doubek M, Polívka J et al. Diagnostika a léčba systémové mastocytózy. XXIV. olomoucké hematologické dny. Sborník abstrakt 2010: abstract 1756.
12. Lennert K, Parwaresch MR. Mast cells and mast cell neoplasia: a review. Histopathology 1979; 3 : 349–365.
13. Lim KH, Tefferi A, Laso TL et al. Systemic mastocytosis in 342 consecutive adults: survival studies and prognostic factors. Blood 2009; 113 : 5727–5736.
14. Ma Y, Zeng S, Metcalfe DD et al. The c-KIT mutation causing human mastocytosis is resistant to STI571 and other KIT kinase inhibitors; kinases with enzymetic site mutations show different inhibitor sensitivity profiles than wild-type kinases and those with regulatory-type mutations. Blood 2002; 99 : 1741–1744.
15. Metcalfe DD, Akin C. Mastocytosis: molecular mechanisms and clinical disease heterogenity. Leuk Res 2001; 25 : 577–582.
16. Nagata H, Worobec AS, Oh CK et al. Identification of a point mutation in the catalytic domain of the protooncogene c-kit in peripheral blood mononuclear cells of patients who have mastocytosis with an associated hematologic disorder. Proc Natl Acad Sci USA 1995; 95 : 10560–10564.
17. Nováková L, Kučera P. Systémová mastocytóza. Transfuze Hematol dnes 2009; 15 : 31–38.
18. Pardanani A, Brockman SR, Páternoster SF et al. FIP1L1PDGFRA fusion: prevalence and clinicopathologic correlates in 89 consecutive patients with moderate to severe eosinophilia. Blood 2004; 104 : 3038–3045.
19. Pardanani A, Elliott M, Reeder T et al. Imatinib for systemic mast-cell disease. Lancet 2003; 362 : 535–536.
20. Pardanani A, Kimlinger T, Reeder T et al. Bone marrow mast cell immunophenotyping in adults with mast cell disease: a prospective study of 33 patients. Leuk Res 2004; 28 : 777–783.
21. Péč M. Mastocytózy. Martin: Osveta 2004.
22. Purtill D, Cooney J, Sinniah R et al. Dasatinib therapy for systemic mastocytosis: four cases. Eur J Haematol 2008; 80 : 456–458.
23. Swerdlow SH, Campo E, Harris NL et al. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon: IARC Press 2008.
24. Tharp MD. Mastocytosis. In: Bolognia JL, Jorizzo JL, Rapini RP (eds). Dermatology. New York: Mosby Elsevier 2009 : 1845–1853.
25. Ustun C, Corless CL, Savage N et al. Chemotherapy and dasatinib induce long-term hematologic and molecular remission in systemic mastocytosis with acute myeloid leukemia with KIT D816V. Leuk Res 2009; 33 : 735–741.
26. Valent P, Akin C, Sperr W et al. Diagnosis and treatment of systemic mastocytosis: state of the art. Br J Haematol 2003; 122 : 695–717.
27. Valent P, Akin C, Sperr W et al. Mastocytosis: pathology, genetics, and current options for therapy. Leuk Lymphoma 2005; 46 : 35–48.
28. Valent P, Spanblöchl E, Bankl HC et al. Kit ligand/mast cell growth factor-independent differentiation of mast cells in myelodysplasia and chronic myeloid leukemic blast crisis. Blood 1994; 84 : 4322–4332.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2010 Issue Supplementum 2
Most read in this issue
- Hemofagocytující lymfohistiocytóza
- Erdheimova-Chesterova nemoc v obrazech
- Systémová mastocytóza
- Histiocytóza z Langerhansových buněk u dětí a dospívajících