
Vysoce riziková chronická lymfocytární leukemie – charakteristika a léčebné možnosti
Ultra ‑ high‑risk chronic lymphocytic leukemia – characteristics and treatment options
Chronic lymphocytic leukemia (CLL) is the most common form adult leukemia in western world. The disease is typically characterized by heterogeneous clinical behavior ranging from indolent course to rapidly progressive disease. Using clinical and biological factors we can stratify patients with CLL and prospectively identify those who can be expected unfavorable course. There is a special group known as ultra high‑risk chronic lymphocytic leukemia with an extremely poor prognosis. These are about 10 – 15% of all patients with CLL. They do not respond to standard treatment and their survival is short with a median of 2 – 3 years. For high‑risk patients are considered: patients with a proven TP53 defect, refractory to purine analogues or with early relapse after chemoimmunotherapy based on fludarabine (≤ 24 months). While the standard 1st line treatment protocol in younger patients is chemoimmunotherapy FCR, in case of ultra ‑ high‑risk CLL other methods like allogeneic hematopoietic stem cell transplantation or clinical trials testing the new drugs should be considered. In particular, allogeneic hematopoietic stem cell transplantation is a very promising treatment modality that offers long‑term disease control and cure regardless of the unfavorable CLL subtype. Transplantation treatment should be therefore considered in all younger patients with ultra ‑ high‑risk CLL, who should be without delay referred to a center for intensive hematological treatment.
Key words:
chronic lymphocytic leukemia – high‑risk cytogenetics – allogeneic transplantation
Authors:
D. Lysák; P. Jindra
Authors‘ workplace:
Hematologicko‑onkologické oddělení FN Plzeň, přednosta prim. MU Dr. Pavel Jindra, Ph. D.
Published in:
Vnitř Lék 2013; 59(10): 887-894
Category:
Review
Overview
Chronická lymfocytární leukemie (CLL) představuje v našich podmínkách nejčastější leukemii dospělého věku. Je charakterizována typicky heterogenním chováním pohybujícím se od indolentního průběhu až po rychle progredující onemocnění. Pomocí klinických a biologických faktorů je možné nemocné s CLL stratifikovat a prospektivně identifikovat pacienty, u kterých lze očekávat nepříznivý průběh. Zvláštní skupinu tvoří tzv. vysoce riziková chronická lymfocytární leukemie s extrémně nepříznivou prognózou. Jedná se asi o 10 – 15 % všech pacientů s CLL, kteří nedostatečně reagují na standardní léčbu a jejichž přežití je krátké s mediánem do 2 – 3 let. Za vysoce rizikové se považují pacienti: s prokázaným defektem TP53, refrakterní na purinová analoga nebo s časným relapsem po chemoimunoterapii založené na fludarabinu (≤ 24 měsíců). Zatímco standardním léčebným protokolem 1. linie je u mladších nemocných chemoimunoterapie FCR, v případě vysoce rizikové CLL by měly být zvažovány jiné postupy využívající alogenní transplantaci hematopoetických kmenových buněk nebo klinické studie testující nové preparáty. Zejména alogenní transplantace krvetvorných kmenových buněk představuje velmi slibnou léčebnou modalitu, která nabízí dlouhodobou kontrolu nemoci a možnost vyléčení bez ohledu na nepříznivý subtyp CLL. Transplantační léčba by proto měla být diskutována u všech mladších nemocných s vysoce rizikovou CLL, kteří by měli být včas konzultováni nebo odesláni do centra pro intenzivní hematologickou léčbu.
Klíčová slova:
chronická lymfocytární leukemie – vysoké cytogenetické riziko – alogenní transplantace
Úvod
Chronická lymfocytární leukemie (CLL) představuje nejfrekventnější hematologické maligní onemocnění ve vyspělém světě s incidencí kolem 5 nových případů na 100 000 obyvatel a rok [1]. Výskyt onemocnění roste s věkem a medián věku v době diagnózy se pohybuje kolem 70 let. Nejedná se však zdaleka pouze o onemocnění starých lidí, minimálně 30 % pacientů je diagnostikováno ve věku do 65 let a 12 % ve věku do 55 let [2].
Léčba CLL
Léčba chronické lymfocytární leukemie zaznamenala v posledních 2 dekádách výrazný vývoj a je dnes nejen významně efektivnější, ale také více individualizovaná než tomu bylo v dřívějších letech. Od monoterapie alkylačními cytostatiky se posunula k purinovým analogům, posléze ke kombinační chemoterapii a konečně k současné chemoimunoterapii (nejčastěji protokolem FCR – fludarabin, cyklofosfamid, rituximab). Tento vývoj umožnil zvýšit celkový počet dosažených odpovědí (ORR) z 30 – 70 % po chlorambucilu na 60 – 80 % po fludarabinu a posléze až na více než 90 % po FCR. Stejným způsobem narostl počet dosažených kompletních remisí z méně než 10 % až na téměř 70 % [3 – 5].
Oproti historické léčbě chlorambucilem má chemoimunoterapie příznivý dopad nejen na kvalitu léčebné odpovědi, ale také na parametry přežití [6,7]. Přidání rituximabu k chemoterapii (FCR vs FC) vedlo k oddálení progrese onemocnění (medián 52 vs 33 měsíců). Moderní léčba chemoimunoterapií tak ovlivňuje přirozený vývoj onemocnění, prodlužuje dobu přežití bez progrese a přináší prospěch i s ohledem na celkové přežití. Riziko úmrtí je s režimem FCR o 33 % nižší ve srovnání s chemoterapií FC [8]. Dokonce i u nemocných léčených pro relaps CLL může FCR oproti FC prodloužit celkové přežití až 2násobně (47 – 49 vs 21 – 31 měsíců) [9,10]. FCR nabízí ve 2. linii léčby stále asi 70 % odpovědí a 30 % kompletních remisí. Předpokladem úspěchu je ovšem senzitivita na fludarabin a absence nepříznivých cytogenetických změn (zejména delece 17p) [9].
Moderní léčba pomocí chemoimunoterapie je vysoce efektivní u většiny pacientů s chronickou lymfocytární leukemií. Nicméně nejedná se o léčbu kurativní a všichni pacienti postupně relabují. Část nemocných navíc recidivuje velmi časně po úvodní léčbě a pro tyto nemocné je nezbytné hledat jednak vhodné biologické a klinické ukazatele, které je mohou předem identifikovat jako vysoce rizikové pacienty, a jednak nové léčebné postupy, které mohou prodloužit jejich očekávané přežití.
Rizikové faktory u CLL
V současné době neexistuje léčba, která by byla vhodná pro všechny nemocné s CLL. Při volbě terapie se musí co nejlépe vybalancovat celkový stav, věk a kondice pacienta na jedné straně a potřebná intenzita chemoterapie diktovaná genetickými a dalšími rizikovými faktory na straně druhé. Chronická lymfocytární leukemie je onemocnění s výrazně heterogenním průběhem. Zatímco variabilita klinického chování CLL je známá již velmi dlouho, definice rizikové, event. vysoce rizikové CLL je záležitostí několika posledních let. V uplynulých 2 dekádách bylo identifikováno množství tzv. rizikových faktorů, které umožňují definovat podskupiny pacientů s odlišnou prognózou. Volba vhodné léčby se tak u pacienta s CLL opírá o komplex laboratorních a klinických rizikových faktorů, díky kterým lze v době diagnózy i v dalším průběhu onemocnění predikovat odpověď na léčbu, riziko progrese onemocnění i očekávané přežití. Rizikové faktory jsou tedy východiskem pro individuální plánování léčby u konkrétního pacienta [11,12].
Díky lepšímu porozumění biologii CLL je možné na základě rizikových faktorů nalézt malou skupinu pacientů s tzv. vysoce rizikovou CLL, u kterých má onemocnění výrazně agresivnější průběh. Tito pacienti hůře odpovídají na léčbu, zažívají rychlou progresi onemocnění a mají při použití standardních léčebných protokolů výrazně zkrácené očekávané přežití s mediánem kratším, než jsou 2 – 3 roky. Rizikové skupiny u CLL uvádí tab. 1.
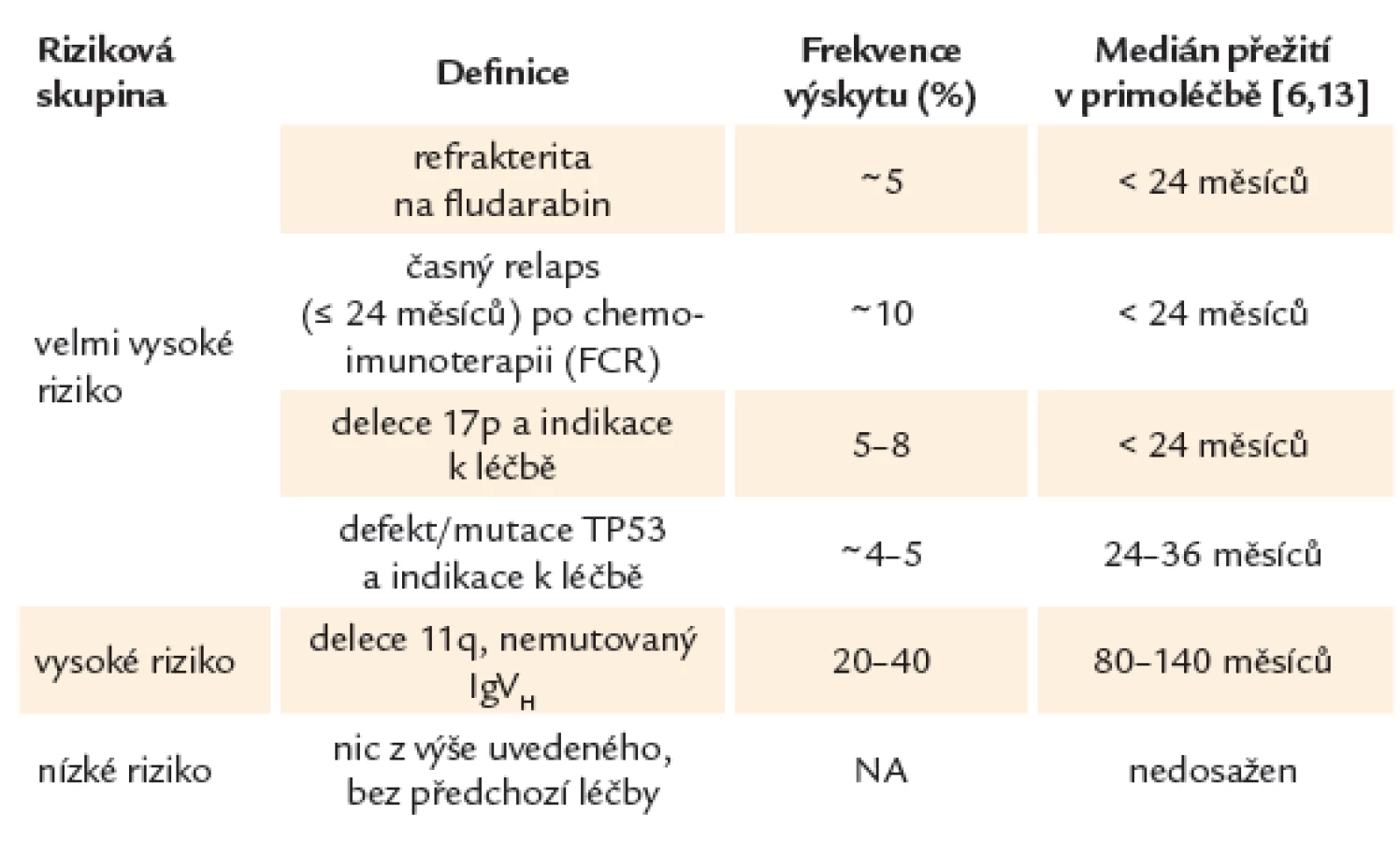
Nalezení rizikového faktoru není samo o sobě indikací k zahájení léčby. Tou stále zůstává symptomatické onemocnění. Příliš časná intervence může vést k selekci rezistentních klonů, které mohou komplikovat budoucí léčbu. Navíc u velmi malé podskupiny vysoce rizikových nemocných může onemocnění vykazovat dlouhodobě benigní klinický průběh.
Genetické aberace a delece 17. chromozomu
Nejvýznamnějšími rizikovými faktory jsou u chronické lymfocytární leukemie různé genetické aberace, které lze pomocí FISH nalézt asi u 80 % pacientů. Na jejich základě byl vytvořen hierarchický model 5 genetických rizikových skupin (17p ‑ , 11q ‑ , trizomie 12q, samostatná 13q ‑ a normální karyotyp), které se signifikantně odlišují v době do progrese onemocnění a v celkovém přežití [13].
Za nejvíce nepříznivou změnu se považuje delece 17. chromozomu (del17p, 17p ‑ , 17p13), kterou lze při FISH vyšetření nalézt u asi 5 – 10 % nově diagnostikovaných pacientů. Delece vede ke ztrátě jedné alely genu TP53 (tumor protein 53). Tento nález je obvykle spojený s dysfunkční mutací zbývající TP53 alely (ve více než 80 % případů). Výsledkem je velmi špatná odpověď na konvenční chemoimunoterapii (ORR a CR po FC 34 % a 0 %; ORR a CR po FCR 68 % a 5 %), rychlá progrese onemocnění (medián 11 měsíců) a krátký medián přežití do 2 – 3 let [8,14 – 17]. Méně často (asi 5 %) se vyskytuje defekt TP53 nezpůsobený delecí 17. chromozomu. Prognóza těchto pacientů je stejně nepříznivá jako v případě del17p [15,18]. Riziko defektní funkce TP53 navíc roste s dobou trvání onemocnění a také v důsledku selekčního tlaku cytotoxické léčby, zejména při použití purinových analogů [19].
TP53 hraje důležitou roli v onkogenezi a odpovědi na chemoterapii u řady humánních solidních i hematologických nádorů. TP53 je umístěn na krátkém raménku chromozomu 17 (17p13) a je suprimován nebo mutován u více než 50 % humánních nádorů. Při poškození DNA cytostatiky nebo radioterapií je TP53 upregulován. Aktivovaný TP53 podporuje transkripci genů zodpovědných za obnovu DNA. Pokud mechanizmus obnovy DNA selže, spouští TP53 buněčnou apoptózu. TP53 tak indukuje reparační buněčné mechanizmy nebo apoptózu při nenapravitelném poškození DNA, resp. buňky, a je důležitý pro kontrolu maligní transformace buněk a jejich odpověď na cytostatika [18].
Při monoterapii chlorambucilem se dosahuje odpovědi maximálně u 20 – 30 % pacientů s del17p a po 2 – 3 měsících pacienti znovu relabují. Také chemoimunoterapie protokolem FCR, který je jinak u CLL velmi efektivní a přináší vysoké, v minulosti nedosažitelné procento kompletních remisí, má u pacientů s del17p menší účinnost s asi 70 % odpovědí a 5 % kompletních remisí [8,18]. Přítomnost del17p je nejdůležitějším negativně prognostickým faktorem určujícím kratší dobu do progrese a kratší celkové přežití [8,20].
Obecně se vliv prognostických markerů, jako např. del17p, s dostupností moderní léčby spíše zvyšuje. Zatímco osud většiny nemocných se pomocí nových léčebných postupů výrazně zlepšil, situace pacientů s vysoce rizikovou CLL se změnila jen omezeně.
Refrakterita na fludarabin
Jako refrakterní CLL se zpravidla označuje onemocnění, které neodpovídá na léčbu purinovými analogy (nebylo dosaženo ani parciální remise) nebo u kterého odpověď trvá méně než 6 měsíců od ukončení terapie. Na rozdíl od povzbudivých výsledků léčby u pacientů senzitivních na fludarabin je prognóza refrakterních nemocných velmi nepříznivá s mediánem celkového přežití 1 – 2 roky [5]. V této skupině nemocných je vysoká pravděpodobnost výskytu mutace TP53. Zatímco v primoléčbě je del17p/ TP53 přítomná maximálně u asi 10 % nemocných, při relapsu onemocnění je to již kolem 20 % nemocných, a u refrakterních onemocnění může záchyt stoupat až na 50 % [18,21]. Zbylá 1/ 2 refrakterních případů je pravděpodobně asociována s dalšími defekty TP53 dráhy nebo se mohou uplatňovat některé nově objevené mutace postihující např. geny SF3B1, NOTCH1 nebo BIRC3. Tyto změny se vyskytují asi u 5 – 10 % nově diagnostikovaných CLL a dostupná data potvrzují jejich prognostický význam a vliv na parametry přežití. NOTCH1 a SF3B1 zkracují celkové přežití na asi 50 měsíců, 10leté přežití se pohybuje mezi 30 – 40 % [22,23]. Nové mutace tak doplňují změny TP53 při identifikaci (vysoce) rizikových CLL. Mutace genu BIRC3 se objevuje asi u 40 % chemorezistentních TP53 wild‑type CLL, naopak její přítomnost nebyla potvrzena u chemosenzitivních případů [24,25]. Metody genetického sekvenování odhalují tímto způsobem další molekulární komplexitu CLL. Nově identifikované genetické léze mají prognostický vliv nezávislý na tradičních klinických a cytogenetických rizikových faktorech. Jejich výskyt a klinický význam se v současné době validuje v rámci studií a lze očekávat, že budou v budoucnu využívány ke zpřesnění prognostické stratifikace pacientů s CLL.
Časná progrese CLL
Nepříznivý průběh onemocnění lze očekávat i u pacientů, kteří sice na léčbu reagují, ale časně relabují – do 24 – 36 měsíců. Chemoimunoterapie u nich relaps pouze oddálí, nemění však nic na nepříznivých biologických vlastnostech onemocnění [16,26,27].
Minimální reziduální nemoc
Důležitým rizikovým faktorem je u CLL také kvalita dosažené léčebné odpovědi. Nemocní, kteří dosáhnou kompletní remise nebo dokonce negativity minimální reziduální nemoci (MRD negativita), mají lepší přežití ve srovnání s pacienty s přetrvávajícím významným leukemickým klonem [9,28]. Dokonce i pacienti léčení 2. linií léčby mají lepší výsledky, pokud dosáhli po předchozí terapii kompletní remise [6,29]. Nízké hodnoty minimální reziduální nemoci tak korelují se snížením rizika progrese onemocnění jak po standardní chemoterapii (FCR), tak po alogenní transplantaci. Negativita reziduální nemoci po alogenní transplantaci je jedním ze silných prediktorů dlouhodobé klinické remise [30]. Vyšetřování minimální reziduální nemoci pomocí citlivých metod, jako jsou vícebarevná průtoková cytometrie nebo kvantitativní ASO IgH PCR s citlivostmi 10 – 4 – 10 – 6, se tak pravděpodobně v blízké budoucnosti přesune z oblasti klinických studií do běžné léčebné praxe.
Vysoce riziková CLL
Z hlediska prognostické stratifikace můžeme u CLL na základě současných léčebných postupů rozeznávat 3 prognostické kategorie. Do skupiny CLL s tzv. velmi vysokým rizikem řadíme pacienty s prokázaným defektem TP53, refrakterní na purinová analoga (fludarabin) nebo s časným relapsem po chemoimunoterapii (≤ 24 měsíců). Tito nemocní představují 10 – 15 % všech léčených pacientů. CLL s tzv. vysokým rizikem většinou zahrnuje změny jako delece 11. chromozomu nebo nemutovaný gen pro variabilní region těžkého řetězce imunoglobulinu (IgVH). Nemocní bez uvedených změn patří do nízce rizikové skupiny [5,16,31].
Režim FCR, který je v současné době standardem v 1. linii léčby u mladších pacientů v dobrém celkovém stavu, bohužel nepřináší uspokojivou odpověď a kontrolu onemocnění u pacientů, kteří jsou zatíženi nepříznivými biologickými vlastnostmi, jako je právě refrakterita na fludarabin nebo del17p. Těmto vysoce rizikovým nemocným by měly být nabídnuty alternativní léčebné postupy v rámci klinických studií a právě tito pacienti mohou nejvíce profitovat z léčby modifikované na základě prognostických faktorů.
Léčba vysoce rizikové CLL
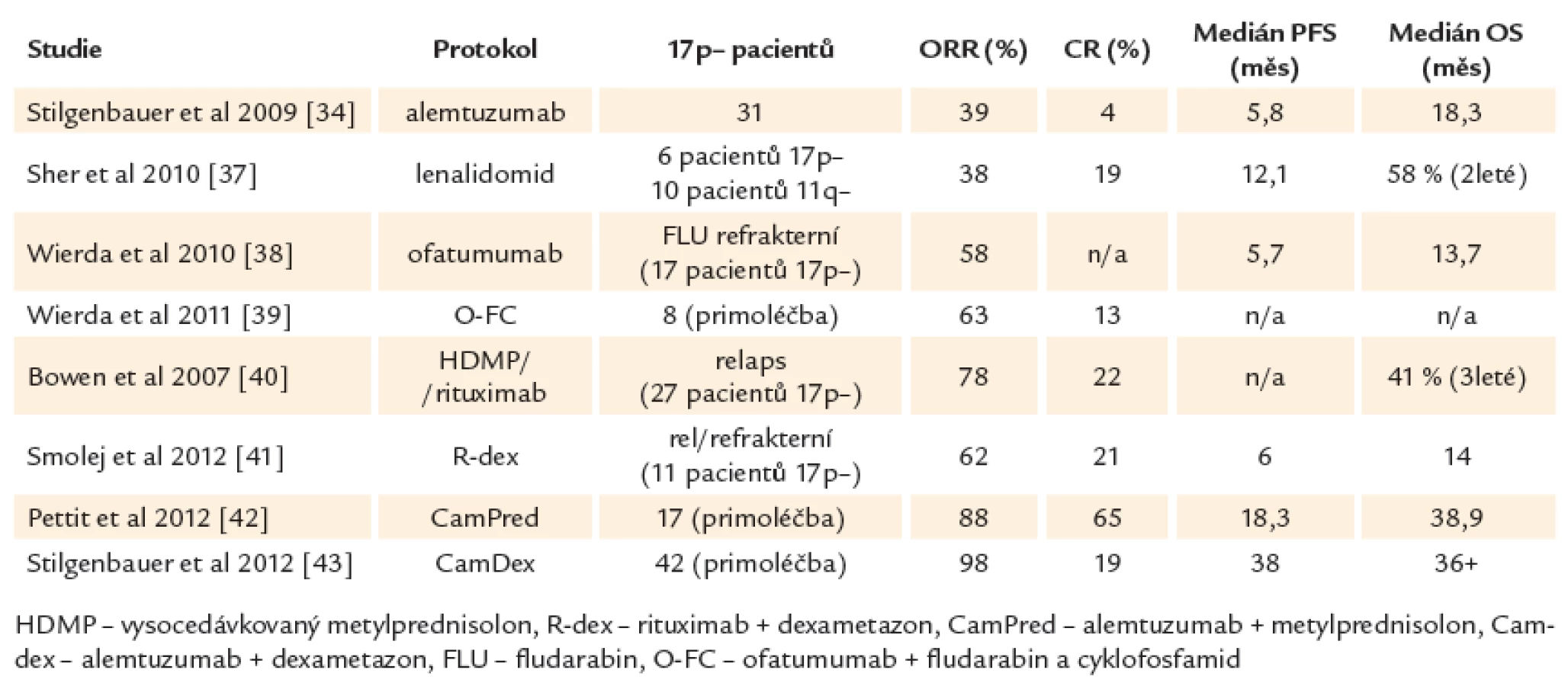
Léčebné možnosti vysoce rizikové CLL zahrnují jak netransplantační postupy různými protokoly, tak transplantaci hematopoetických kmenových buněk (tab. 2).

Netransplantační léčba
Je důležité si uvědomit, že ve většině dostupných studií jsou počty vysoce rizikových pacientů velmi malé a také že neexistují randomizované studie specifické pro tuto podskupinu CLL. Léčba 1. linie u vysoce rizikové CLL je v současné době stále založena především na chemoimunoterapii (FCR). Při léčbě pacientů s defektem TP53 nebo špatnou odpovědí na chemoimunoterapii se dále využívají preparáty, jejichž mechanizmus účinku je na TP53 nezávislý, především alemtuzumab [32,33], kortikosteroidy nebo lenalidomid. Alemtuzumab v monoterapii nabízí odpověď asi u 30 % pacientů (2 – 6 % CR). Odpověď je spíše krátkodobá (medián 8 – 10 měsíců), navíc s nemalým rizikem infekčních komplikací [34 – 36]. Při použití lenalidomidu bylo dosaženo 38 % odpovědí s mediánem doby do progrese pouhých 12 měsíců [37]. Pacienti rezistentní na fludarabin i alemtuzumab mohou profitovat z nové anti‑CD20 protilátky, která se oproti rituximabu váže na velkou i malou smyčku CD20 antigenu. Ofatumumab je u onemocnění s nepříznivou genetikou efektivní v monoterapii i v kombinaci s fludarabinem [38,39]. Slibné jsou také protokoly kombinující vysoko dávkované kortikosteroidy s rituximabem [40,41] nebo s alemtuzumabem [42,43]. Lze jimi dosáhnout až kolem 80 % celkových odpovědí a asi 20 – 36 % kompletních remisí. Jejich trvání je ovšem v porovnání s méně rizikovými genetickými skupinami krátké a ani tyto alternativní protokoly tedy nenabízejí výrazné zlepšení léčebných výsledků (souhrn studií ukazuje tab. 3). Je také nutné počítat s vyšším výskytem infekčních komplikací (asi 50 % infekcí stupně III/ IV oproti 25 % po režimu FCR) a reaktivací cytomegaloviru (asi 50 %) [42]. Hlavními příčinami úmrtí je progrese onemocnění nereagující na konvenční léčbu, infekce, selhání kostní dřeně s pancytopenií a transformace do agresivního nehodgkinovského lymfomu.
Určitou změnu mohou přinést některé nové molekuly dostupné v klinických studiích, které nezřídka nabízejí nadějné výsledky i v případě nepříznivých variant CLL. Bendamustin, jehož cytotoxický efekt obsahuje TP53 dependentní i TP53 nezávislý mechanizmus, prokázal aktivitu v klinických studiích zaměřených na relabující a refrakterní CLL. Při kombinaci s cytostatikem (mitoxantron) nebo cytostatikem a protilátkou (cytosin‑arabinosid, rituximab) nabízí 51 %, resp. 84 % celkových odpovědí (78 % pro del17p) s mediánem doby do progrese až 22, resp. 16 měsíců [44,45]. Další skupinou nových léků jsou inhibitory signálních drah receptoru B‑lymfocytů (BCR). Je známo, že mikroprostředí obklopující CLL lymfocyty a antigenní signalizace jejich receptorů mají zásadní význam pro proliferaci a přežívání nádorových lymfocytů a celkový vývoj onemocnění. Představitelem této skupiny léků je ibrutinib (inhibitor Brutonovy kinázy), který podporuje apoptózu, inhibici proliferace a migrace CLL buněk. Léčba ibrutinibem vede k odpovědi u asi 50 % vysoce rizikových pacientů v monoterapii, při kombinaci s rituximabem se účinnost zvyšuje až na 85 % celkových odpovědí. Podobnou účinnost prokázal i idelalisib (GS ‑ 1101, PI3Kd inhibitor), v kombinaci s bendamustinem a rituximabem přes 80 % odpovědí a pravděpodobnost přežití bez progrese 63 % ve 2 letech [46,47].
Aktuálně dostupné klinické studie s ibrutinibem nejsou zatím pro pacienty s defektem TP53 určeny. Nicméně příznivý bezpečnostní profil BCR inhibitorů je předurčuje k léčbě relabujících nebo refrakterních či geneticky nepříznivých CLL. Terapeutické kombinace s moderními léky typu BCR inhibitorů nebo bendamustinu mohou být účinnou a dobře tolerovanou léčebnou možností u vysoce rizikové CLL nebo sloužit jako cytoredukční léčba před alogenní transplantací. Jejich efektivita však musí být dále ověřena na větších souborech nemocných. Při nedostupnosti klinické studie zůstávají 1. volbou v úvodní léčbě chemoimunoterapie (FCR) nebo alemtuzumab.
Transplantační léčba
Autologní transplantace je u pacientů s CLL sice dobře proveditelná, spojená s nízkou transplantační mortalitou, bohužel se nejedná o léčbu kurativní a nemocní po ní relabují. Provedení autologní transplantace sice může oddálit progresi onemocnění, celkové přežití se však mezi transplantovanými a dispenzarizovanými pacienty neliší (86 % vs 84 % v 5 letech) [48]. Své místo tedy může autologní transplantace mít pouze v určitých velmi specifických situacích, jako je např. transformace CLL do agresivní formy nehodgkinovského lymfomu (nejčastěji difuzní velkobuněčný lymfom) v rámci tzv. Richterova syndromu. Celkově je však tento koncept léčby u CLL prakticky opuštěn.
Alogenní transplantace hematopoetických kmenových buněk může u chronické lymfocytární leukemie nastolit dlouhotrvající remisi a představuje v současné době jedinou potenciálně kurativní modalitu tohoto onemocnění. Pravděpodobnost přežití bez progrese a celkového přežití ve 4 letech je 42 % a 65 % pro všechny nemocné bez ohledu na genetické riziko (45 % a 59 % pro del17p). Oproti standardní léčbě nemají pacienti s delecí 17. chromozomu nebo refrakteritou na fludarabin po alogenní transplantaci horší výsledky [30,34]. V retrospektivní studii Evropské skupiny pro transplantace kostní dřeně (EBMT) byl potvrzen přínos alogenní transplantace u CLL s del17p. Po 3 letech přežívalo 44 % pacientů, 37 % v trvající remisi onemocnění [49]. Souhrn studií s alogenní transplantací u CLL uvádí tab. 4.
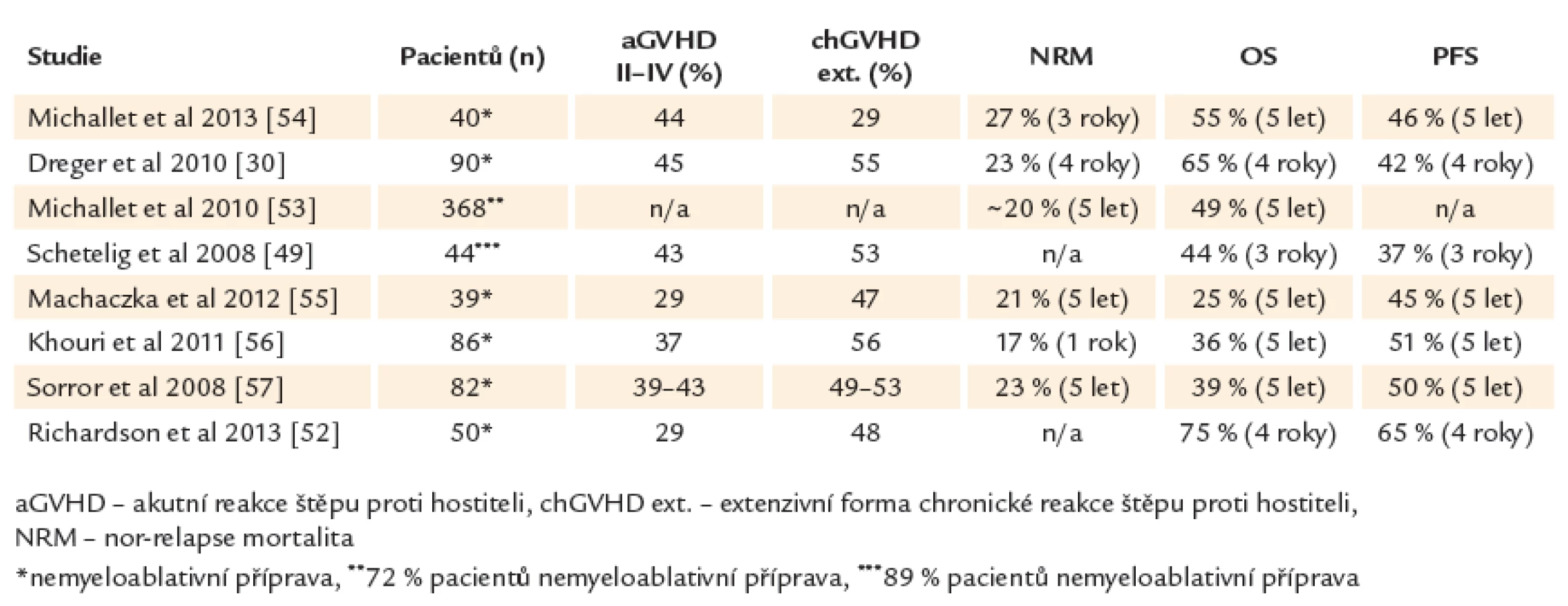
Řada studií prokázala přítomnost efektu reakce štěpu proti leukemii (GVL) u chronické lymfocytární leukemie. Rozvoj reakce štěpu proti hostiteli (GVHD) snižuje pravděpodobnost relapsu nebo progrese onemocnění po alogenní transplantaci. Kompletní eradikace leukemického klonu (MRD negativita) je dosažena pravděpodobněji u pacientů s GVHD nebo ji lze vyvolat pomocí imunoterapie infuzí dárcovských lymfocytů [31,50,51]. Význam GVHD, resp. GVL efektu dokládá i zjištění, že deplece T‑lymfocytů před transplantací pomocí alemtuzumabu nebo ex vivo T deplece transplantátu, které jsou efektivní prevencí GVHD, vedou podle některých studií ke zhoršení výsledků transplantační léčby s vyšší incidencí relapsů [30]. Reakce štěpu proti leukemii je tak hlavním faktorem zajišťujícím dlouhodobou kontrolu nemoci a vyléčení po alogenní transplantaci. Alogenní transplantace mění zažitý prognostický model a nabízí minimálně u 1/ 2 nemocných dlouhodobé přežití i u vysoce rizikové CLL a tedy bez ohledu na tradiční rizikové skupiny. Vhodným indikováním potransplantační imunoterapie u pacientů s vyšším rizikem relapsu onemocnění (přetrvávající reziduální nemoc, přetrvávající smíšený dárcovský chimérizmus) je možné výsledky transplantační léčby dále zlepšit a dosáhnout dlouhodobého (ve 4 letech) celkového přežití 75 % a přežití bez progrese 65 % (61 % a 60 % u del17p) [52].
Jako nejvhodnější cesta se jeví transplantace s využitím nemyeloablativních přípravných protokolů, u které lze očekávat mortalitu spojenou s transplantací do asi 25 %. Ukazuje se také, že využití příbuzného nebo nepříbuzného dárce poskytuje stejné výsledky s podobným dlouhodobým přežitím (55 % vs 59 % v 5 letech). Transplantace s HLA kompatibilním nepříbuzným dárcem je tedy zcela ekvivalentní transplantaci příbuzenecké s HLA kompatibilním sourozencem [30,49,53]. Výsledek transplantace je spíše ovlivněn stavem onemocnění v době transplantace, kdy nejlepšího dlouhodobého přežití dosahují nemocní transplantovaní v kompletní remisi. Naopak nepříznivý vliv má progresivní charakter onemocnění a výrazné předléčení pacienta více než 4 liniemi chemoterapie. Důležitou roli pochopitelně hraje i celkový stav nemocného a přežití se snižuje s rostoucím počtem komorbidit a transplantačním rizikem [30,53].
Kolem 30 % nemocných indikovaných k alogenní transplantaci by mělo nalézt vhodného dárce v rodině. Úspěšnost nalezení nepříbuzného dárce dosahuje asi 80 %. Nedostupnost dárce by proto neměla u většiny pacientů být důvodem neprovedení transplantace, pokud je tato procedura s ohledem na nepříznivý genetický subtyp CLL indikovaná a transplantační riziko u daného nemocného je akceptovatelné.
Minimálně části pacientů s vysoce rizikovou CLL může být nabídnuta alogenní transplantace kostní dřeně. Indikace by měla být jednoznačně zvažována u všech biologicky mladých nemocných ve věku do cca 65 let (výjimečně 70 let), kteří:
- jsou refrakterní na purinová analoga nebo
- relabují do 12 měsíců po monoterapii purinovými analogy či do 24 měsíců po kombinačních režimech založených na purinových analozích nebo
- mají nepříznivý molekulární subtyp s poruchou signální dráhy TP53 a vyžadují léčbu [31].
Výsledky transplantační léčby jsou obecně příznivější, pokud je procedura provedena včas, a naopak se zhoršují s rostoucím počtem chemoterapeutických linií, kterými jsou pacienti předléčeni. Správné načasování je důležité i v případě vysoce nepříznivé CLL. Jakmile jsou naplněna kritéria vysoce rizikové CLL, měli by být nemocní konzultováni v některém z center intenzivní hematologické péče, které může řídit další strategii léčby, obratem zahájit proces vyhledávání vhodného dárce a zvažovat provedení alogenní transplantace krvetvorných buněk. Pacienti s vysoce rizikovou CLL, kteří nejsou kandidáty alogenní transplantace z důvodu věku, celkového stavu či nedostupnosti vhodného dárce, mají omezené možnosti léčby a je vhodné je zařazovat do klinických studií ověřujících aktivitu některých nových léků.
Závěr
V příštích letech se pravděpodobně budou prognostické modely u CLL dále vyvíjet a budou se dále upřesňovat rizikové faktory identifikované před léčbou (molekulárně‑genetické aberace, klinický stav atd.) a po léčbě (kvalita a trvání odpovědi, minimální reziduální nemoc). Porozumění novým mutacím přítomným u CLL umožní hledat nové biologické léky zasahující do její patofyziologie. V současné době představuje poškození signální dráhy TP53 jedinou genetickou abnormalitu, která predikuje léčebnou odpověď a zároveň ovlivňuje v běžné klinické praxi volbu léčby nebo léčebného přístupu u CLL.
Stanovení biologických rizikových faktorů by mělo být standardní součástí vyšetřovacího postupu u všech pacientů vhodných k intenzivní terapii. Vysoce riziková CLL identifikovaná na základě rizikových faktorů je spojená s velmi nepříznivou prognózou a její léčba představuje významnou klinickou výzvu. Implementace nových záchranných protokolů na bázi monoklonálních protilátek a dalších cíleně působících molekul a také rostoucí možnosti využití alogenní transplantace kostní dřeně mohou zlepšit osud pacientů s touto agresivní formou chronické lymfocytární leukemie.
Práce byla podpořena Programem rozvoje vědních oborů Karlovy Univerzity (projekt P36) a projektem Ministerstva zdravotnictví koncepčního rozvoje výzkumné organizace 00669806 – FN Plzeň.
doc. MU Dr. Daniel Lysák, Ph.D.
www.hematologie ‑ onkologie.cz
e‑mail: lysak@fnplzen.cz
Doručeno do redakce: 3. 4. 2013
Přijato po recenzi: 15. 5. 2013
Sources
1. Dores GM, Anderson WF, Curtis RE et al. Chronic lymphocytic leukaemia and small lymphocytic lymphoma: overview of the descriptive epidemiology. Br J Haematol 2007; 139 : 809 – 819.
2. Eichhorst B, Dreyling M, Robak T et al. ESMO Guidelines Working Group. Chronic lymphocytic leukemia: ESMO clinical practice guidelines for diagnosis, treatment and follow‑up. Ann Oncol 2011; 22 (Suppl 6): vi50 – vi54.
3. Keating MJ, O’Brien S, Albitar M et al. Early results of a chemoimmunotherapy regimen of fludarabine, cyclophosphamide, and rituximab as initial therapy for chronic lymphocytic leukemia. J Clin Oncol 2005; 23 : 4079 – 4088.
4. Eichhorst BF, Busch R, Hopfinger G et al. Fludarabine plus cyclophosphamide versus fludarabine alone in first‑line therapy of younger patients with chronic lymphocytic leukemia. Blood 2006; 107 : 885 – 891.
5. Zenz T, Gribben JG, Hallek M et al. Risk categories and refractory CLL in the era of chemoimmunotherapy. Blood 2012; 119 : 4101 – 4107.
6. Tam CS, O’Brien S, Wierda W et al. Long‑term results of the fludarabine, cyclophosphamide, and rituximab regimen as initial therapy of chronic lymphocytic leukemia. Blood 2008; 112 : 975 – 980.
7. Abrisqueta P, Pereira A, Rozman C et al. Improving survival in patients with chronic lymphocytic leukemia (1980 – 2008): The hospital clinic of Barcelona experience. Blood 2009; 114 : 2044 – 2050.
8. Hallek M, Fischer K, Fingerle ‑ Rowson G et al. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: A randomised, open ‑ label, phase 3 trial. Lancet 2010; 376 : 1164 – 1174.
9. Badoux XC, Keating MJ, Wang X et al. Fludarabine, cyclophosphamide, and rituximab chemoimmunotherapy is highly effective treatment for relapsed patients with CLL. Blood 2011; 117 : 3016 – 3024.
10. Wierda W, O’Brien S, Faderl S et al. A retrospective comparison of three sequential groups of patients with Recurrent/ Refractory chronic lymphocytic leukemia treated with fludarabine‑based regimens. Cancer 2006; 106 : 337 – 345.
11. Motyckova M, Zak P, Vroblova V et al. Prognostic markers in chronic lymphocytic leukemia. Vnitř Lék 2011; 57 : 847 – 857.
12. Smolej L, Saudkova L, Spacek M et al. ZAP ‑ 70 in B ‑ cell chronic lymphocytic leukemia: Clinical significance and methods of detection. Vnitř Lék 2006; 52 : 1194 – 1199.
13. Dohner H, Stilgenbauer S, Benner A et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med 2000; 343 : 1910 – 1916.
14. Zent CS, Kay NE. Management of patients with chronic lymphocytic leukemia with a high risk of adverse outcome: The mayo clinic approach. Leuk Lymphoma 2011; 52 : 1425 – 1434.
15. Zenz T, Eichhorst B, Busch R et al. TP53 mutation and survival in chronic lymphocytic leukemia. J Clin Oncol 2010; 28 : 4473 – 4479.
16. Stilgenbauer S, Zenz T. Understanding and managing ultra high‑risk chronic lymphocytic leukemia. Hematology Am Soc Hematol Educ Program 2010; 2010 : 481 – 488.
17. Hallek M, Fingerle ‑ Rowson G, Fink A et al. First‑line treatment with fludarabine (F), cyclophosphamide (C), and rituximab (R) (FCR) improves overall survival (OS) in previously untreated patients (pts) with advanced chronic lymphocytic leukemia (CLL): Results of a randomized phase III trial on behalf of an international group of investigators and the German CLL study group. ASH Annual Meeting Abstracts 2009; 114: Abstract 535.
18. Badoux XC, Keating MJ, Wierda WG. What is the best frontline therapy for patients with CLL and 17p deletion? Curr Hematol Malig Rep 2011; 6 : 36 – 46.
19. Shanafelt TD, Witzig TE, Fink SR et al. Prospective evaluation of clonal evolution during long‑term follow‑up of patients with untreated early‑stage chronic lymphocytic leukemia. J Clin Oncol 2006; 24 : 4634 – 4641.
20. Pospisilova S, Gonzales D, Malcikova J et al. ERIC recommendations on TP53 mutation analysis in chronic lymphocytic leukemia. Leukemia 2012; 26 : 1458 – 1461.
21. Zenz T, Mertens D, Kuppers R et al. From pathogenesis to treatment of chronic lymphocytic leukaemia. Nat Rev Cancer 2010; 10 : 37 – 50.
22. Rossi D, Rasi S, Fabbri G et al. Mutations of NOTCH1 are an independent predictor of survival in chronic lymphocytic leukemia. Blood 2012; 119 : 521 – 529.
23. Oscier DG, Rose‑Zerilli MJ, Winkelmann N et al. The clinical significance of NOTCH1 and SF3B1 mutations in the UK LRF CLL4 trial. Blood 2013; 121 : 468 – 475.
24. Rossi D, Rasi S, Spina V et al. Integrated mutational and cytogenetic analysis identifies new prognostic subgroups in chronic lymphocytic leukemia. Blood 2013; 121 : 1403 – 1412.
25. Rossi D, Fangazio M, Rasi S et al. Disruption of BIRC3 associates with fludarabine chemorefractoriness in TP53 wild‑type chronic lymphocytic leukemia. Blood 2012; 119 : 2854 – 2862.
26. Zenz T, Bush R, Fink A et al. Genetics of Patients with F ‑ Refractory CLL or Early Relapse After FC or FCR: Results From the CLL8 Trial of the GCLLSG ASH Annual Meeting Abstracts 2010; 116 : 2427.
27. Panovská A, Smolej L, Lysák D et al. The outcome of chronic lymphocytic leukemia patients who relapsed after fludarabine, cyclophosphamide, and rituximab. Eur J Haematol 2013; 90 : 479 – 485.
28. Boettcher S, Fischer K, Stilgenbauer S et al. Quantitative MRD assessments predict progression free survival in CLL patients treated with fludarabine and cyclophosphamide with or without rituximab – a prospective analysis in 471 patients from the randomized GCLLSG CLL8 trial. ASH Annual Meeting Abstracts 2008; 112: Abstract 326.
29. Keating MJ, O‘Brien S, Albitar M et al. Early results of a chemoimmunotherapy regimen of fludarabine, cyclophosphamide, and rituximab as initial therapy for chronic lymphocytic leukemia. J Clin Oncol 2005; 23 : 4079 – 4088.
30. Dreger P, Dohner H, Ritgen M et al. Allogeneic stem cell transplantation provides durable disease control in poor ‑ risk chronic lymphocytic leukemia: Long‑term clinical and MRD results of the german CLL study group CLL3X trial. Blood 2010; 116 : 2438 – 2447.
31. Dreger P, Corradini P, Kimby E et al. Indications for allogeneic stem cell transplantation in chronic lymphocytic leukemia: The EBMT transplant consensus. Leukemia 2007; 21 : 12 – 17.
32. Smolej L, Prochazka V, Spacek M et al. Guidelines for alemtuzumab treatment in chronic lymphocytic leukaemia (CLL). Vnitř Lék 2012; 58 : 232 – 236.
33. Doubek M, Jungova A, Brejcha M et al. Alemtuzumab in chronic lymphocytic leukemia treatment: Retrospective analysis of outcome according to cytogenetics. Vnitř Lék 2009; 55 : 549 – 554.
34. Stilgenbauer S, Zenz T, Winkler D et al. Subcutaneous alemtuzumab in fludarabine ‑ refractory chronic lymphocytic leukemia: Clinical results and prognostic marker analyses from the CLL2H study of the german chronic lymphocytic leukemia study group. J Clin Oncol 2009; 27 : 3994 – 4001.
35. Lozanski G, Heerema N, Flinn I et al. Alemtuzumab is an effective therapy for chronic lymphocytic leukemia with p53 mutations and deletions. Blood 2004; 103 : 3278 – 3281.
36. Keating MJ, Flinn I, Jain V et al. Therapeutic role of alemtuzumab (CAMPATH ‑ 1H) in patients who have failed fludarabine: results of a large international study. Blood 2002; 99 : 3554 – 3561.
37. Sher T, Miller KC, Lawrence D et al. Efficacy of lenalidomide in patients with chronic lymphocytic leukemia with high‑risk cytogenetics. Leuk Lymphoma 2010; 51 : 85 – 88.
38. Wierda WG, Kipps TJ, Mayer J et al. Hx ‑ CD20 – 406 Study Investigators. Ofatumumab as single‑agent CD20 immunotherapy in fludarabine ‑ refractory chronic lymphocytic leukemia. J Clin Oncol 2010; 28 : 1749 – 1755.
39. Wierda WG, Kipps TJ, Dürig J et al. 407 Study Investigators. Chemoimmunotherapy with O ‑ FC in previously untreated patients with chronic lymphocytic leukemia. Blood 2011; 117 : 6450 – 6458.
40. Bowen DA, Call TG, Jenkins GD et al. Methylprednisolone ‑ rituximab is an effective salvage therapy for patients with relapsed chronic lymphocytic leukemia including those with unfavorable cytogenetic features. Leuk Lymphoma 2007; 48 : 2412 – 2417.
41. Smolej L, Doubek M, Panovská A et al. Rituximab in combination with high ‑ dose dexamethasone for the treatment of relapsed/ refractory chronic lymphocytic leukemia. Leuk Res 2012; 36 : 1278 – 1282.
42. Pettitt AR, Jackson R, Carruthers S et al. Alemtuzumab in combination with methylprednisolone is a highly effective induction regimen for patients with chronic lymphocytic leukemia and deletion of TP53: final results of the national cancer research institute CLL206 trial. J Clin Oncol 2012; 30 : 1647 – 1655.
43. Stilgenbauer S, Cymbalista F, Leblond V et al. Alemtuzumab Plus Oral Dexamethasone, Followed by Alemtuzumab Maintenance or Allogeneic Transplantation in Ultra High‑Risk CLL: Updated Results From a Phase II Study of the Gcllsg and fcgcll/ MW. ASH Annual Meeting Abstracts 2012; 120: Abstract 716.
44. Köppler H, Fuss H, Hurtz HJ et al. Bendamustine plus mitoxantrone for relapsed/ refractory chronic lymphocytic leukaemia (CLL): results of a multicentre phase II study of the German CLL Study Group (GCLLSG). Br J Hematol 2012; 158 : 238 – 241.
45. Visco C, Finotto S, Pomponi F et al. The combination of rituximab, bendamustine, and cytarabine for heavily pretreated relapsed/ refractory cytogenetically high‑risk patients with chronic lymphocytic leukemia. Am J Hematol 2013; 88 : 289 – 293.
46. Courtre SE, Leonard JP, Furman RR et al. Combinations of the Selective Phosphatidylinositol 3 - Kinase ‑ Delta (PI3Kdelta) Inhibitor GS ‑ 1101 (CAL ‑ 101) with Rituximab and/ or Bendamustine Are Tolerable and Highly Active in Patients with Relapsed or Refractory Chronic Lymphocytic Leukemia (CLL): Results From a Phase I Study. ASH Annual Meeting Abstracts 2012; 120: Abstract 191.
47. Burger JA, Keating MJ, Wierda WG et al. The Btk Inhibitor Ibrutinib (PCI ‑ 32765) in Combination with Rituximab Is Well Tolerated and Displays Profound Activity in High‑Risk Chronic Lymphocytic Leukemia (CLL) Patients. ASH Annual Meeting Abstracts 2012; 120: Abstract 187.
48. Michallet M, Dreger P, Sutton L et al. Autologous hematopoietic stem cell transplantation in chronic lymphocytic leukemia: Results of european intergroup randomized trial comparing autografting versus observation. Blood 2011; 117 : 1516 – 1521.
49. Schetelig J, van Biezen A, Brand R et al. Allogeneic hematopoietic stem ‑ cell transplantation for chronic lymphocytic leukemia with 17p deletion: A retrospective european group for blood and marrow transplantation analysis. J Clin Oncol 2008; 26 : 5094 – 5100.
50. Dreger P, Brand R, Milligan D et al. Reduced ‑ intensity conditioning lowers treatment‑related mortality of allogeneic stem cell transplantation for chronic lymphocytic leukemia: A population ‑ matched analysis. Leukemia 2005; 19 : 1029 – 1033.
51. Ritgen M, Stilgenbauer S, von Neuhoff N et al. Graft ‑ versus ‑ leukemia activity may overcome therapeutic resistance of chronic lymphocytic leukemia with unmutated immunoglobulin variable heavy‑chain gene status: Implications of minimal residual disease measurement with quantitative PCR. Blood 2004; 104 : 2600 – 2602.
52. Richardson SE, Khan I, Eawstron A et al. Risk‑stratified adoptive cellular therapy following allogeneic hematopoietic stem cell transplantation for advanced chronic lymphocytic leukemia. Br J Haematol 2013; 160 : 640 – 648.
53. Michallet M, Sobh M, Milligan D et al. The impact of HLA matching on long‑term transplant outcome after allogeneic hematopoietic stem cell transplantation for CLL: A retrospective study from the EBMT registry. Leukemia 2010; 24 : 1725 – 1731.
54. Michallet M, Socié G, Mohty M et al. Rituximab, fludarabine, and total body irradiation as conditioning regimen before allogeneic hematopoietic stem cell transplantation for advanced chronic lymphocytic leukemia: long‑term prospective multicenter study. Exp Hematol 2013; 41 : 127 – 133.
55. Machaczka M, Johansson JE, Remberger M et al. Allogeneic hematopoietic stem cell transplant with reduced ‑ intensity conditioning for chronic lymphocytic leukemia in Sweden: does donor T ‑ cell engraftment 3 months after transplant predict survival? Leuk Lymphoma 2012; 53 : 1699 – 1705.
56. Khouri IF, Bassett R, Poindexter N et al.Nonmyeloablative allogeneic stem cell transplantation in relapsed/ refractory chronic lymphocytic leukemia: long‑term follow‑up, prognostic factors, and effect of human leukocyte histocompatibility antigen subtype on outcome. Cancer 2011; 117 : 4679 – 4688.
57. Sorror ML, Storer BE, Sandmaier BM et al. Five‑year follow‑up of patients with advanced chronic lymphocytic leukemia treated with allogeneic hematopoietic cell transplantation after nonmyeloablative conditioning. J Clin Oncol 2008; 26 : 4912 – 4920.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2013 Issue 10
Most read in this issue
- Metody vyšetřování mikrocirkulace kůže
- Infekční endokarditida na trikuspidální chlopni u intravenózní narkomanky
- Současný přístup k léčbě pacientů s metastatickým kolorektálním karcinomem
- Manažment dyslipidémií – prítomnosť a budúcnosť. Odporúčania Angiologickej sekcie Slovenskej lekárskej komory (2013)