
Vrozené hyperbilirubinemie a molekulární mechanizmy žloutenky
Neonatal hyperbilirubinemia and molecular mechanisms of jaundice
The introductory summarises the classical path of heme degradation and classification of jaundice. Subsequently, a description of neonatal types of jaundice is given, known as Crigler ‑ Najjar, Gilbert’s, Dubin‑Johnson and Rotor syndromes, emphasising the explanation of the molecular mechanisms of these metabolic disorders. Special attention is given to a recently discovered molecular mechanism of the Rotor syndrome. The mechanism is based on the inability of the liver to retrospectively uptake the conjugated bilirubin fraction primarily excreted into the blood, not bile. A reduced ability of the liver to uptake the conjugated bilirubin contributes to the development of hyperbilirubinemia in common disorders of the liver and bile ducts and to the toxicity of xenobiotics and drugs using transport proteins for conjugated bilirubin.
Key words:
heme – bilirubin – Gilbert’s syndrome – Dubin‑Johnson syndrome – Rotor syndrome – OATP1B1 – OATP1B3
Authors:
M. Jirsa; E. Sticová
Authors‘ workplace:
Centrum experimentální medicíny IKEM Praha, přednosta prof. MU Dr. Luděk Červenka, CSc., MBA
Published in:
Vnitř Lék 2013; 59(7): 566-571
Category:
Overview
Úvod shrnuje klasickou dráhu degradace hemu a z ní odvozené rozdělení žloutenek. Následuje popis vrozených žloutenek známých jako Crigler ‑ Najjarův, Gilbertův, Dubin‑Johnsonův a Rotorův syndrom s důrazem na vysvětlení molekulární podstaty těchto metabolických poruch. Zvláštní pozornost je věnována nedávno objevenému molekulárnímu mechanizmu Rotorova syndromu spočívajícímu v neschopnosti jater zpětně vychytávat frakci konjugovaného bilirubinu vylučovaného primárně do krve a nikoliv do žluči. Snížená schopnost jater vychytávat konjugovaný bilirubin přispívá i k rozvoji hyperbilirubinemie u běžných onemocnění jater a žlučových cest a k toxicitě xenobiotik a léků využívajících transportní proteiny pro konjugovaný bilirubin.
Klíčová slova:
hem – bilirubin – Gilbertův syndrom – Dubin‑Johnsonův syndrom – Rotorův syndrom – OATP1B1 – OATP1B3
Úvod
Bilirubin je konečným produktem degradace hemu, jehož hlavními zdroji jsou erytrocyty a buňky erytropoetické řady pohlcované makrofágy ve slezině a v krvetvorné kostní dřeni. Méně významným zdrojem jsou cytochromy v jaterních buňkách. Nekonjugovaný, ve vodě prakticky nerozpustný bilirubin, vázaný na albumin, je krví transportován do jater. Po disociaci vazby na albumin je bilirubin importován do hepatocytů, v jejichž cytosolu se váže na proteiny ze skupiny glutation ‑ S ‑ transferáz označované dříve názvem ligandin. Následuje konjugace se dvěma molekulami kyseliny glukuronové, která se odehrává v lumen endoplazmatického retikula za katalýzy uridindifosfátglukosiduronát bilirubin glukuronosyltransferázou UGT1A1 (EC 2.4.1.17) (obr. 1).
Gen UGT1A1 lokalizovaný v komplexním genovém lokusu UGT1A [1] kóduje jediný pro bilirubin specifický konjugační enzym UGT1A1. Konjugovaný, ve vodě rozpustný bilirubin je zatím neznámým mechanizmem exportován z endoplazmatického retikula zpět do cytosolu, odkud migruje ke žlučovému pólu hepatocytu. Ze žlučového pólu je konjugovaný bilirubin aktivně secernován do žluči působením kanalikulárního ABC transportéru ABCC2/ MRP2 fungujícího jako exportní pumpa multivalentních organických aniontů. Vedle této pumpy je pravděpodobná účast dalších transportérů s nižší afinitou ke konjugovanému bilirubinu (např. ABCG2), které při vyřazení genu ABCC2 přejímají úlohu bilirubinové pumpy [2]. Ve střevě je konjugovaný bilirubin dekonjugován, zčásti resorbován a zčásti dále degradován působením bakteriální střevní flóry.
Dědičné poruchy metabolizmu jsou klíčem k poznání metabolických drah. Nejinak je tomu i v případě metabolické dráhy degradace hemu. Na základě biochemického vyšetření je možno rozlišovat převážně nekonjugované žloutenky, vyvolané buď nadprodukcí bilirubinu při hemolýze, nebo sníženou rychlostí jaterní konjugace, a převážně konjugované žloutenky, jejichž vyvolávající příčinou je onemocnění jaterního parenchymu, intrahepatální cholestáza nebo obstrukce žlučových cest (obr. 1).
![Odbourávání hemu na žlučová barviva a rozdělení hyperbilirubinemií podle převažujícího typu bilirubinu v séru [44].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/ef7d07d88f061c5819cd493ead0cfafb.jpg)
Dědičné převážně nekonjugované hyperbilirubinemie jsou vyvolány mutacemi v genu pro UGT1A1 a zahrnují Crigler ‑ Najjarův syndrom I. a II. typu a benigní hyperbilirubinemii Gilbertova typu. Dědičné převážně konjugované hyperbilirubinemie zahrnují Dubin‑Johnsonův a Rotorův syndrom.
Crigler ‑ Najjarův syndrom (CNS)
Crigler ‑ Najjarův syndrom (CNS) [3] je raritní autozomálně recesivní porucha konjugace bilirubinu vyvolaná mutacemi ve strukturálních oblastech genu UGT1A1. Podle koncentrace sérového bilirubinu se rozlišuje CNS I. typu (OMIM #218800) a CNS II. typu (OMIM #606785) [4].
Pro CNS I. typu jsou charakteristické hodnoty bilirubinu nad 350 µmol/ l. Ostatní laboratorní nálezy, chromoexkreční testy, jakož i jaterní histologie jsou v mezích normy. Konjugační aktivita bilirubinu je prakticky nulová a bilirubinemie neklesá po podání fenobarbitalu. Příčinou jsou nonsense a některé missense mutace v exonech či jejich intronickém okolí, jejichž důsledkem je ztráta funkce či prakticky nulová exprese proteinu UGT1A1 [5]. Neléčená choroba je letální, postižení umírají zpravidla v dětském věku na jádrový ikterus. Terapie spočívá v celoživotní intermitentní fototerapii (někdy až po dobu 12 hod denně) nebo v transplantaci jater. Při fototerapii dochází k intenzivnímu vylučování ve vodě rozpustných fotoizomerů nekonjugovaného bilirubinu do žluči. Ve střevě probíhá částečná zpětná izomerace na nekonjugovaný bilirubin IXa,který vstupuje do enterohepatálního cyklu. Zvýšení účinnosti fototerapie je možno dosáhnout adjuvantní terapií, která spočívá v sekvestraci bilirubinu ve střevě (např. vazbou na neresorbovatelný fosforečnan vápenatý) nebo v urychlení střevní degradace bilirubinu. K transplantaci jater se přistupuje kolem 5. roku věku, neboť účinnost fototerapie s věkem postupně klesá a tím roste riziko náhlého poškození mozku [6].
CNS II. typu je benigní formou CNS. Bilirubinemie dosahuje hodnot 100 – 350 µmol/ l. Stejně jako u CNS I. typu jsou ostatní laboratorní nálezy a jaterní morfologie normální. Aktivita UGT1A1 v játrech je snížena na 10 – 25 % normální hodnoty a hlavním produktem bilirubinu je bilirubin monoglukosiduronát. Na rozdíl od většiny případů CNS I. typu dochází u CNS II. typu po podání fenobarbitalu k výrazné indukci transkripce genu UGT1A1, která se projeví poklesem bilirubinemie. CNS II. typu je podmíněn takovými mutacemi v genu UGT1A1, z nichž alespoň jedna nemá za následek kompletní inaktivaci či nulovou expresi kódovaného enzymu [5]. Průběh CNS II. typu je benigní a s výjimkou novorozeneckého období zpravidla nevyžaduje žádnou terapii.
Gilberův syndrom (GS)
Familiární nehemolytická žloutenka, popsaná jako benigní hyperbilirubinemie již v roce 1901 [7], je autozomálně recesivně dědičná metabolická odchylka podmíněná konstitutivním snížením rychlosti konjugace bilirubinu v játrech na hodnoty kolem 30 % [8,9]. Prevalence GS v kavkazské populaci je 5 – 10 % [10,11]. V evropské populaci je GS v převážné většině případů podmíněn homozygotním stavem pro variantu A(TA7)TAA TATA ‑ boxu v promotoru genu UGT1A1 [12]. Normální sekvence TATA ‑ boxu je A(TA6)TAA. Inzerce TA má za následek snížení exprese strukturálně normálního enzymu UGT1A1 [12]. U většiny nositelů GS je exprese UGT1A1 dále snížena přítomností druhé mutace T>G v pozici – 3 279 od začátku translace genu UGT1A1 v tzv. gtPBREM (glucuronosyltransferase phenobarbital response enhancing motif) [13,14]. Tato mutace je příčinou zpomaleného poklesu bilirubinemie po podání fenobarbitalu, čehož bylo v minulosti využíváno diagnosticky v tzv. fenobarbitalovém testu. Vedle uvedených mutací v promotoru genu UGT1A1 mohou GS vyvolat i mutace ve strukturálních oblastech genu. Nejčastější je heterozygotní mutace c.211G>A vyskytující se především v asijských populacích [15]. V homozygotním stavu vyvolává tato mutace CNS II. typu [16]. Fenotyp heterozygotů pro některé mutace vyvolávající CNS odpovídá fenotypu GS [17].
Klinicky se familiární nehemolytická žloutenka Gilbertova typu projevuje jako intermitentní ikterus sklér, popř. mírná žloutenka kůže a sliznic. K tomu se mohou přidružit neurotické symptomy, zvýšená únava, nechutenství bez poklesu tělesné hmotnosti a abdominální dyskomfort. Fyzikální vyšetření je zcela v normě stejně jako laboratorní nález s výjimkou hyperbilirubinemie nekonjugovaného typu zřídka přesahující 100 µmol/ l. Bilirubinemie se zvyšuje při fyzické zátěži, stresu a hladovění, čehož bylo v minulosti využíváno k diagnostickému testu (tzv. test hladověním). U části nositelů GS byla pozorována snížená clearance bromsulfoftaleinu, indocyaninové zeleně a dalších aniontových barviv [4]. V bioptických vzorcích jater bylo pozorováno hromadění lipofuscinu v hepatocytech, a to především v centrilobulární zóně [4]. Ani testy jaterní chromoexkrece, ani výsledky jaterní biopsie nemají žádnu diagnostickou hodnotu, a proto tato vyšetření nejsou indikována. GS dnes není považován za chorobu, nýbrž za metabolickou odchylku, která nevyžaduje žádnou terapii ani dietní či režimová opatření. U nositelů GS byl naopak prokázán nižší výskyt kardiovaskulárních chorob a nádorových onemocnění, což je přičítáno antioxidačním vlastnostem bilirubinu [18].
Dubin‑Johnsonův syndrom (DJS)
Dubin‑Johnsonův syndrom (DJS, OMIM #237500) je vzácná benigní autozomálně recesivní konjugovaná hyperbilirubinemie. Genetickou příčinou DJS jsou mutace v genu ABCC2/ MRP2 [19]. Gen ABCC2 je lokalizován na dlouhém raménku 10. chromozomu (10q24), zaujímá 45 kb a sestává z 32 protein kódujících exonů.
Pro klinický obraz je charakteristický kolísavý ikterus sklér a někdy i kůže bez dalších příznaků. Hladina celkového bilirubinu se pohybuje kolem 100 µmol/ l, přičemž více než 50 % připadá na přímý bilirubin [20,21]. Ostatní laboratorní nálezy včetně odpadu celkových porfyrinů jsou normální, avšak izomer I koproporfyrinu představuje více než 50 % vylučovaného koproporfyrinu [22]. Vylučování bromsulfoftaleinu do žluči po i.v. podání je zpomaleno a v 90. min po podání dochází k opětovnému vzestupu jeho koncentrace [23]. Obdobnou kinetiku jako bromsulfoftalein mají indocyaninová zeleň a další organické anionty včetně radiofarmak používaných k cholescintigrafii, takže vizualizace jater i plnění žlučníku jsou opožděny [24 – 26]. Jaterní histologie je normální s výjimkou akumulace tmavého melanin‑like pigmentu s výraznou autofluorescencí v hepatocytech. Pigment, jehož chemické složení dosud není známo, dává pozitivní PAS reakci a redukuje amoniakální roztok stříbra (Masson ‑ Fontanova reakce), avšak na rozdíl od melaninu neredukuje neutrální roztok stříbra. Byly dokumentovány i případy DJS bez pigmentace jater [27,28]. Imunohistologicky lze ve většině případů prokázat absenci proteinu ABCC2 v kanalikulární membráně hepatocytů [29]. Výjimečně může být mutovaný nefunkční protein ABCC2 normálně exprimován [30].
Na našem pracovišti byly zachyceny nové mutace v genu ABCC2 u jedinců s DJS [31,32] a zdokumentován první případ dosud nepopsané převážně nekonjugované dědičné žloutenky vyvolané přítomností mutací typických pro DJS a GS [31]. Podíl konjugovaného bilirubinu u této tzv. duální hereditární žloutenky činí pouze 20 – 50 % sérové koncentrace celkového bilirubinu.
Rotorův syndrom (RS)
Rotorův syndrom (RS, OMIM #237540) je velmi vzácná autozomálně recesivně dědičná forma převážně konjugované žloutenky, která je klinickým a základním laboratorním vyšetřením neodlišitelná od DJS. Stejně jako u DJS je u RS převažujícím vylučovaným koproporfyrinem izomer I. Na rozdíl od DJS je odpad celkových porfyrinů v moči zvýšen a poměr izomeru I koproporfyrinu k izomeru III bývá nižší než u DJS [33]. Vylučování aniontových barviv játry je pomalejší než u DJS a nedochází k opětovnému vzestupu jejich koncentrace [34]. Při cholescintigrafii se nezobrazí ani játra ani žlučník a zobrazení žlučníku bylo pozorováno teprve po řadě hodin [24,35,36]. Jaterní histologie je zcela normální. Příčina Rotorova syndromu nebyla donedávna známa.
Na našem pracovišti se během 10 let jeho existence podařilo shromáždit klinické a laboratorní nálezy 11 nositelů Rotorova syndromu pocházejících ze 4 středoevropských, 3 arabských a 1 filipínské rodiny. Ve spolupráci se skupinou Ing. S. Kmocha z Ústavu dědičných metabolických poruch 1. lékařské fakulty UK a VFN v Praze se nám kombinací homozygotního mapování a mapování unikátních delecí podařilo identifikovat kandidátní lokus na chromozomu 12p12 obsahující dvojici genů SLCO1B1 a SLCO1B3, jejichž všechny alely byly mutovány u všech 11 jedinců s RS [37]. Naopak všichni rodiče a sourozenci postižených nesli alespoň 1 normální alelu genu SLCO1B1 nebo SLCO1B3.
Geny SLCO1B1 a SLCO1B3 kódují proteiny OATP1B1 a OATP1B3 exprimované v sinusoidální membráně jaterních buněk, které jsou odpovědné za jaterní vychytávání nekonjugovaného i konjugovaného bilirubinu, jakož i řady dalších endogenních látek, xenobiotik a léků. Odpověď na otázku, jak se konjugovaný bilirubin dostane do sinusoidální krve, poskytly experimenty s geneticky modifikovanými myšími liniemi provedené skupinou A. Schinkela v The Netherlands Cancer Institute v Amsterdamu [37]. Výsledky ukázaly, že významná frakce konjugovaného bilirubinu je za fyziologických podmínek secernována do krve prostřednictvím sinusoidální bilirubinové pumpy MRP3 homologní s MRP2. Zvýšená exprese proteinu MRP3 byla již dříve popsána jako hlavní kompenzační mechanizmus vylučování konjugovaného bilirubinu z cytosolu hepatocytů do krve u DJS [38] a při některých formách získané cholestázy, u nichž je blokována sekrece konjugovaného bilirubinu do žluči (sepse, toxiny, obstrukce žlučových cest) [39]. V centrální zóně jaterního lalůčku, kde je exprese rotorovských proteinů OATP1B1 a OATP1B3 nejvyšší, dochází ke zpětnému a téměř kompletnímu vychytávání konjugovaného bilirubinu z krve a jeho následnému vyloučení do žluče (obr. 2).
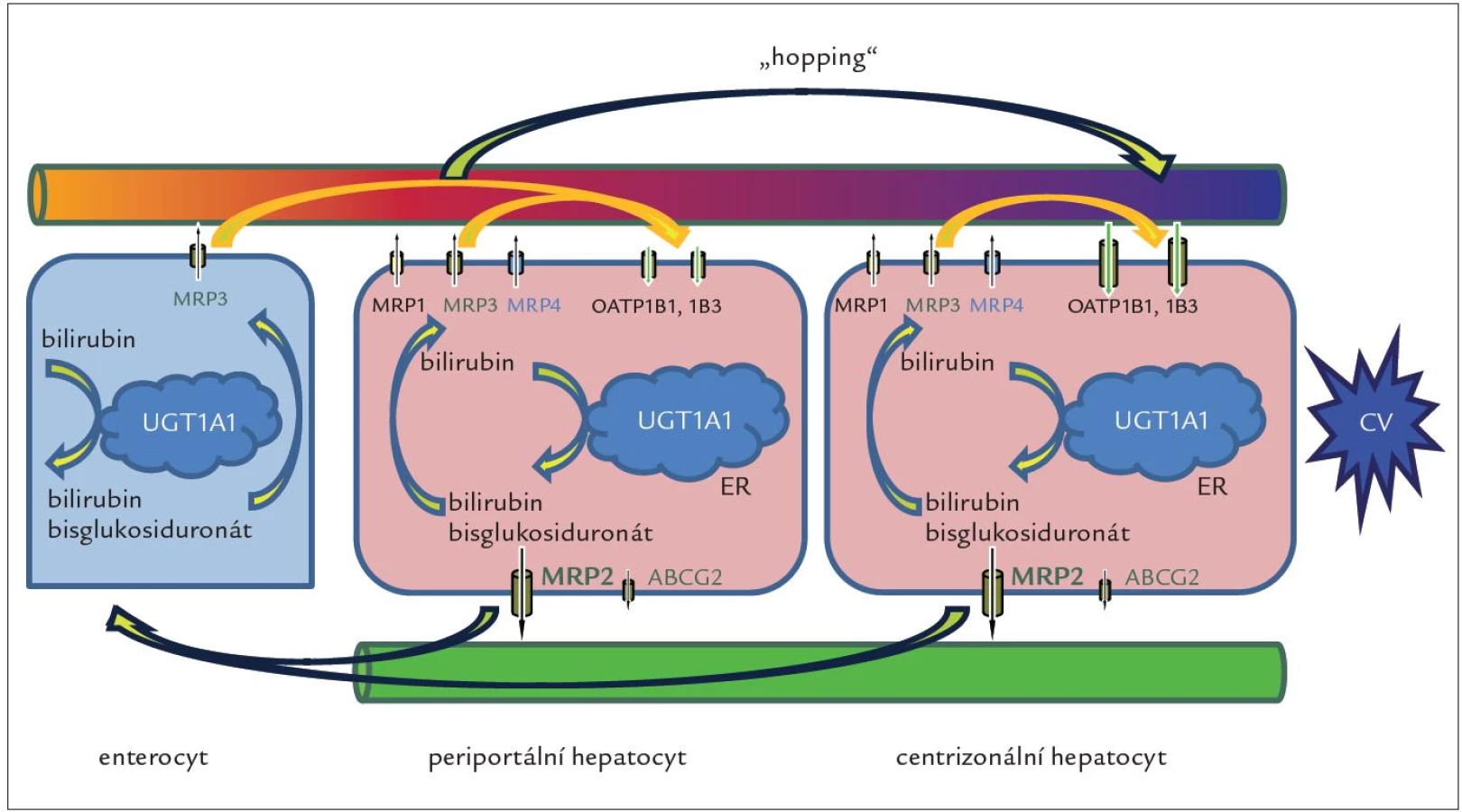
Rotorovské proteiny se podílejí i na vychytávání bilirubinu konjugovaného mimo játra, zejména ve střevě. Pravděpodobně největší význam transportní smyčky tvořené pumpou MRP3 a rotorovskými proteiny (obr. 2) spočívá v ochraně portálních hepatocytů před toxickým působením zvýšených koncentrací látek přicházejících do jater z portálního oběhu a ve zvýšení sekreční kapacity jater na podkladě přesunu části nálože organických aniontů a aniontových konjugátů z přetížených periportálních hepatocytů do méně zatěžovaných hepatocytů v centrální zóně [40].
Molekulární mechanizmy žloutenky a jejich farmakologický význam
Poznání molekulárního mechanizmu žloutenky u RS má význam pro vysvětlení patofyziologie žloutenky u běžných chorob jater a žlučových cest. Při parenchymové žloutence dochází vedle poruchy konjugace a sekrece bilirubinu i k poruše jaterního vychytávání nekonjugovaného a patrně i konjugovaného bilirubinu ze sinusoidální krve. Na snížení vychytávání obou forem bilirubinu se může podílet snížená aktivita či exprese proteinů OATP1B1 a OATP1B3. U cholestázy či obstrukce žlučových cest může být konjugovaná hyperbilirubinemie potencována snížením exprese rotorovských proteinů. Tuto hypotézu podporují imunohistologické nálezy snížené exprese proteinů OATP1B1 a OATP1B3 u některých cho-lestatických jaterních chorob [41].
Jako zásadní se jeví význam jaterního cyklu bilirubinu a dalších organických aniontů pro vychytávání a metabolizmus xenobiotik a léků v jaterních buňkách. Rotorovské proteiny např. zprostředkovávají vychytávání nekonjugovaných žlučových kyselin, konjugovaných steroidů, hormonů štítné žlázy, bromsulfoftaleinu, indocyaninové zeleně, radiofarmak pro cholescintigrafii, benzylpenicilinu, rifampicinu, atorvastatinu, pravastatinu, rosuvastatinu, některých sartanů či metotrexátu [42]. Rovněž bylo prokázáno, že variace v rotorovských genech jsou asociovány se závažnými nežádoucími účinky léků, např. statinů [43].
Závěr
Dráha degradace hemu je považována za jednu z nejjednodušších a nejlépe prostudovaných metabolických drah u člověka. Cesta k poznání molekulární podstaty RS však ukázala, že i v takto detailně prostudované metabolické dráze existuje řada dosud nezodpovězených otázek, mezi něž patří např. dosud neznámý mechanizmus třídění molekul konjugovaných v endoplazmatickém retikulu na sloučeniny vylučované do žluči a do krve či úloha nosičství patogenních mutací v některé z alel genů SLCO1B1 či SLCO1B3 v rozvoji nežádoucích účinků některých běžně používaných léků.
doc. MU Dr. Mgr. Milan Jirsa, CSc.
www.ikem.cz
e‑mail: milan.jirsa@ikem.cz
Doručeno do redakce: 20. 3. 2013
Sources
1. Owens IS, Basu NK, Banerjee R. UDP ‑ glucuronosyltransferases: gene structures of UGT1 and UGT2 families. Methods Enzymol 2005; 400 : 1 – 22.
2. Vlaming ML, Pala Z, van Esch A et al. Functionally overlapping roles of Abcg2 (Bcrp1) and Abcc2 (Mrp2) in the elimination of methotrexate and its main toxic metabolite 7 – hydroxymethotrexate in vivo. Clin Cancer Res 2009; 15 : 3084 – 3093.
3. Crigler JF Jr, Najjar VA. Congenital familial nonhemolytic jaundice with kernicterus; a new clinical entity. AMA Am J Dis Child 1952; 83 : 259 – 260.
4. Chowdhury JR, Wolkoff AW, Chowdhury NR et al. Hereditary jaundice and disorders of bilirubin metabolism. In: Scriver CR, Beaudet AL, Sly WS et al (eds). The metabolic & molecular bases of inherited disease. New York: McGraw Hill 2001 : 3063 – 3101.
5. Kadakol A, Ghosh SS, Sappal BS et al. Genetic lesions of bilirubin uridine ‑ diphosphoglucuronate glucuronosyltransferase (UGT1A1) causing Crigler ‑ Najjar and Gilbert syndromes: correlation of genotype to phenotype. Hum Mutat 2000; 16 : 297 – 306.
6. Jansen PL. Diagnosis and management of Crigler ‑ Najjar syndrome. Eur J Pediatr 1999; 158 (Suppl 2): S89 – S94.
7. Gilbert A, Lereboulet P. La cholemie simple familiale. Sem Med 1901; 21 : 241 – 243.
8. Arias IM, London IM. Bilirubin glucuronide formation in vitro; demonstration of a defect in Gilbert’s disease. Science 1957; 126 : 563 – 564.
9. Black M, Billing BH. Hepatic bilirubin udp ‑ glucuronyl transferase activity in liver disease and gilbert’s syndrome. N Engl J Med 1969; 280 : 1266 – 1271.
10. Owens D, Evans J. Population studies on Gilbert’s syndrome. J Med Genet 1975; 12 : 152 – 156.
11. Sieg A, Arab L, Schlierf G et al. Prevalence of Gilbert’s syndrome in Germany. Dtsch Med Wochenschr 1987; 112 : 1206 – 1208.
12. Bosma PJ, Chowdhury JR, Bakker C et al. The genetic basis of the reduced expression of bilirubin UDP ‑ glucuronosyltransferase 1 in Gilbert’s syndrome. N Engl J Med 1995; 333 : 1171 – 1175.
13. Sugatani J, Kojima H, Ueda A et al. The phenobarbital response enhancer module in the human bilirubin UDP ‑ glucuronosyltransferase UGT1A1 gene and regulation by the nuclear receptor CAR. Hepatology 2001; 33 : 1232 – 1238.
14. Sugatani J, Yamakawa K, Yoshinari K et al. Identification of a defect in the UGT1A1 gene promoter and its association with hyperbilirubinemia. Biochem Biophys Res Commun 2002; 292 : 492 – 497.
15. Koiwai O, Nishizawa M, Hasada K et al. Gilbert’s syndrome is caused by a heterozygous missense mutation in the gene for bilirubin UDP ‑ glucuronosyltransferase. Hum Mol Genet 1995; 4 : 1183 – 1186.
16. Aono S, Yamada Y, Keino H et al. Identification of defect in the genes for bilirubin UDP ‑ glucuronosyl ‑ transferase in a patient with Crigler ‑ Najjar syndrome type II. Biochem Biophys Res Commun 1993; 197 : 1239 – 1244.
17. Aono S, Adachi Y, Uyama E et al. Analysis of genes for bilirubin UDP ‑ glucuronosyltransferase in Gilbert’s syndrome. Lancet 1995; 345 : 958 – 959.
18. Vitek L, Jirsa M, Brodanova M et al. Gilbert syndrome and ischemic heart disease: a protective effect of elevated bilirubin levels. Atherosclerosis 2002; 160 : 449 – 456.
19. Paulusma CC, Oude Elferink RP. The canalicular multispecific organic anion transporter and conjugated hyperbilirubinemia in rat and man. J Mol Med 1997; 75 : 420 – 428.
20. Dubin IN. Chronic idiopathic jaundice; a review of fifty cases. Am J Med 1958; 24 : 268 – 292.
21. Shani M, Seligsohn U, Gilon E et al. Dubin‑Johnson syndrome in Israel. I. Clinical, laboratory, and genetic aspects of 101 cases. Q J Med 1970; 39 : 549 – 567.
22. Wolkoff AW, Cohen LE, Arias IM. Inheritance of the Dubin‑Johnson syndrome. N Engl J Med 1973; 288 : 113 – 117.
23. Erlinger S, Dhumeaux D, Desjeux JF et al. Hepatic handling of unconjugated dyes in the Dubin‑Johnson syndrome. Gastroenterology 1973; 64 : 106 – 110.
24. Bar ‑ Meir S, Baron J, Seligson U et al. 99mTc ‑ HIDA cholescintigraphy in Dubin‑Johnson and Rotor syndromes. Radiology 1982; 142 : 743 – 746.
25. Bujanover Y, Bar ‑ Meir S, Hayman I et al. 99mTc ‑ HIDA cholescintigraphy in children with Dubin‑Johnson syndrome. J Pediatr Gastroenterol Nutr 1983; 2 : 311 – 312.
26. Artiko V, Obradovic VV, Kostic K et al. Cholescintigraphy in Dubin‑Johnson syndrome. Nucl Med Rev Cent East Eur 1999; 2 : 83 – 84.
27. Arias IM. Studies of chronic familial non‑hemolytic jaundice with conjugated bilirubin in the serum with and without an unidentified pigment in the liver cells. Am J Med 1961; 31 : 510 – 518.
28. Shieh CC, Chang MH, Chen CL. Dubin‑Johnson syndrome presenting with neonatal cholestasis. Arch Dis Child 1990; 65 : 898 – 899.
29. Kartenbeck J, Leuschner U, Mayer R et al. Absence of the canalicular isoform of the MRP gene ‑ encoded conjugate export pump from the hepatocytes in Dubin‑Johnson syndrome. Hepatology 1996; 23 : 1061 – 1066.
30. Mor ‑ Cohen R, Zivelin A, Rosenberg N et al.Identification and functional analysis of two novel mutations in the multidrug resistance protein 2 gene in Israeli patients with Dubin‑Johnson syndrome. J Biol Chem 2001; 276 : 36923 – 36930.
31. Cebecauerova D, Jirasek T, Budisova L et al. Dual hereditary jaundice: simultaneous occurrence of mutations causing Gilbert’s and Dubin‑Johnson syndrome. Gastroenterology 2005; 129 : 315 – 320.
32. Sticova E, Elleder M, Hulkova H et al. Dubin‑Johnson syndrome coinciding with colon cancer and atherosclerosis. World J Gastroenterol 2013; 19 : 946 – 950.
33. Wolkoff AW, Wolpert E, Pascasio FN et al. Rotor’s syndrome. A distinct inheritable pathophysiologic entity. Am J Med 1976; 60 : 173 – 179.
34. Wolpert E, Pascasio FM, Wolkoff AW et al. Abnormal sulfobromophthalein metabolism in Rotor’s syndrome and obligate heterozygotes. N Engl J Med 1977; 296 : 1099 – 1101.
35. Fretzayas AM, Garoufi AI, Moutsouris CX et al. Cholescintigraphy in the diagnosis of Rotor syndrome. J Nucl Med 1994; 35 : 1048 – 1050.
36. Fretzayas AM, Stavrinadis CS, Koukoutsakis PM et al. Diagnostic approach of Rotor syndrome with cholescintigraphy. Clin Nucl Med 1997; 22 : 635 – 636.
37. van de Steeg E, Stranecky V, Hartmannova Het al. Complete OATP1B1 and OATP1B3 deficiency causes human Rotor syndrome by interrupting conjugated bilirubin reuptake into the liver. J Clin Invest 2012; 122 : 519 – 528.
38. König J, Rost D, Cui Y et al. Characterization of the human multidrug resistance protein isoform MRP3 localized to the basolateral hepatocyte membrane. Hepatology 1999; 29 : 1156 – 1163.
39. Lee J, Boyer JL. Molecular alterations in hepatocyte transport mechanisms in acquired cholestatic liver disorders. Semin Liver Dis 2000; 20 : 373 – 384.
40. Iusuf D, van de Steeg E, Schinkel AH. Hepatocyte hopping of OATP1B substrates contributes to efficient hepatic detoxification. Clin Pharmacol Ther 2012; 92 : 559 – 562.
41. Keitel V, Burdelski M, Warskulat U et al. Expression and localization of hepatobiliary transport proteins in progressive familial intrahepatic cholestasis. Hepatology 2005; 41 : 1160 – 1172.
42. Niemi M, Pasanen MK, Neuvonen PJ. Organic anion transporting polypeptide 1B1: a genetically polymorphic transporter of major importance for hepatic drug uptake. Pharmacol Rev 2011; 63 : 157 – 181.
43. Link E, Parish S, Armitage J et al. SLCO1B1 variants and statin‑induced myopathy – a genomewide study. N Engl J Med 2008; 359 : 789 – 799.
44. Karlson P, Gerok W, Gross W. Patobiochemie. Praha: Academia 1987.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2013 Issue 7
Most read in this issue
- Vztah bilirubinu k nemocem vyvolaným zvýšeným oxidačním stresem
- Cystické nádory pankreatu – naše zkušenosti
- Onemocnění jater u diabetiků
- Akutní pankreatitida – novinky v léčbě