
Diagnostika cystické fibrózy u dospělých
Diagnostics of cystic fibrosis in adults
Introduction:
There is an increasing number of cystic fibrosis (CF) patients with the diagnosis established in adulthood worldwide.
Aim:
To give an overview of our experience with the diagnostics of CF in adulthood in the Czech Republic.
Methods:
CF patients with the diagnosis determined at the age ≥ 18 years during 2000–2014 period were selected from the Czech Registry of CF (www.cfregistr.cz). Demographic and clinical data were reported from medical records at the time of diagnosis and as of 31st December 2014. Only those with two CF causing mutation or with one CF causing mutation together with sweat chloride concentration > 60 mmol/l were included in the study. The clinical presentation was compared with a control group consisting of homozygous F508del patients with the diagnosis established in childhood.
Results:
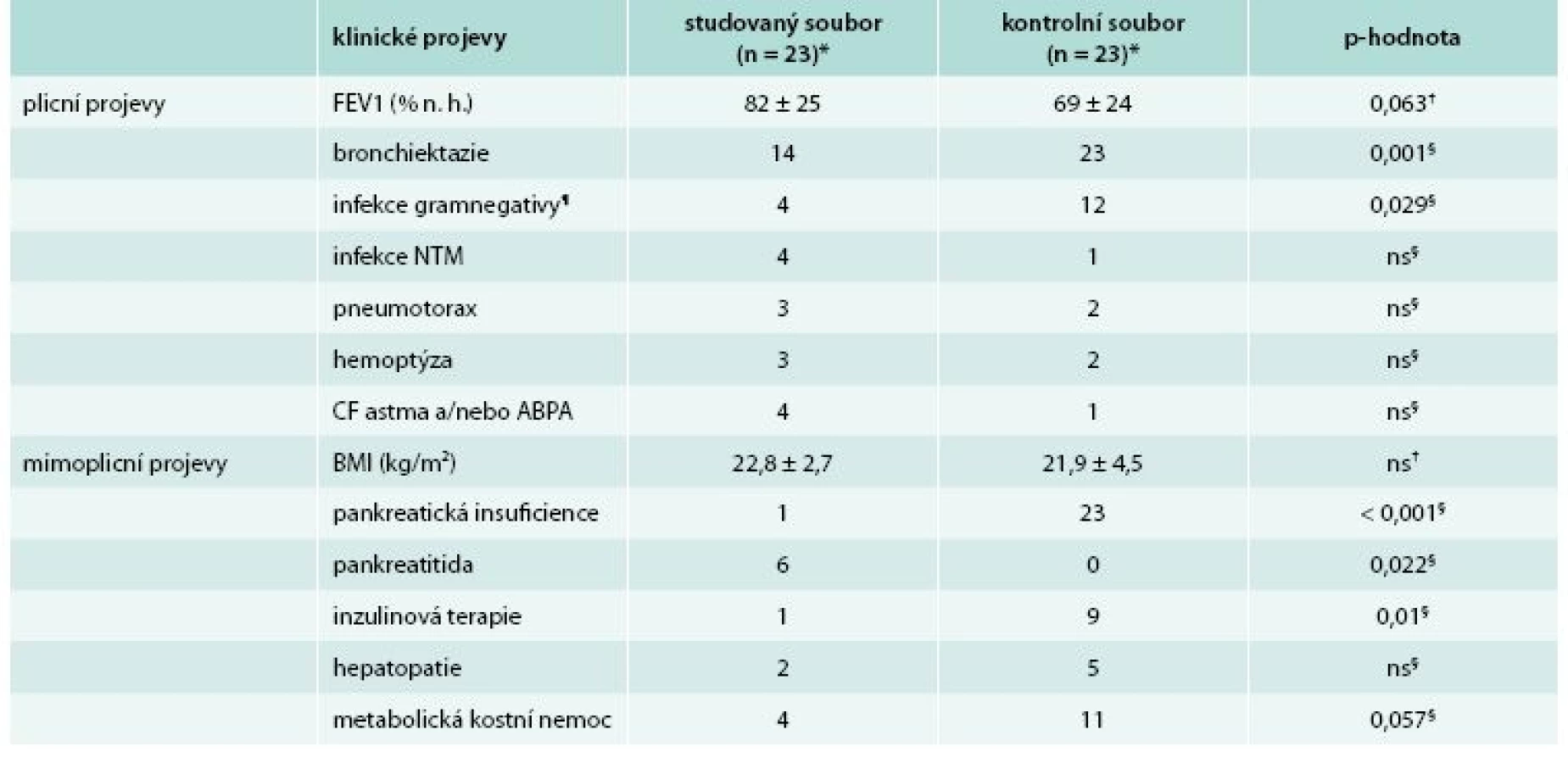
23 patients (16 men and 7 women) with the diagnosis determined at a mean age of 32.9 ± 8.5 years were included in the study. Presenting symptoms included bronchiectasis and/or haemoptysis in 12 cases, obstructive azoospermia in 7 cases and recurrent pancreatitis in 4 cases. When compared with the control group, the patients had higher age (38.6 ± 8.3 vs. 28.3 ± 4.7 years; p < 0.001), a lower concentration of sweat chloride (62 ± 23 vs. 90 ± 12 mmol/l; p < 0.001), less frequent airway infections with Pseudomonas aeruginosa and/or Burkholderia cepacia complex (4 vs. 12; p = 0.029), bronchiectasis (14 vs. 23; p = 0.001), exocrine pancreatic insufficiency (1 vs. 23; p < 0.001) and therapy with insulin (1 vs. 9; p = 0.01); on the contrary, pancreatitis was more frequent (6 vs. 0; p = 0.022).
Conclusion:
Diagnosis of CF in adults should be considered in those with corresponding symptoms in respiratory, digestive and reproductive tract. Clinical presentation differs from classical CF in many parameters.
Key words:
adults – cystic fibrosis – diagnostics
:
Libor Fila 1; Alžběta Grandcourtová 1; prim. MUDr. Lucie Valentová Bartáková 1; Zuzana Antušová 2; Eva Pokojová 3; Vladimír Herout 3; Petr Jakubec 4; Radka Bittenglová 5; Miloslav Marel 1
:
Pneumologická klinika 2. LF UK a FN v Motole, Praha
1; Plicní klinika LF UK a FN Hradec Králové
2; Klinika nemocí plicních a tuberkulózy LF a FN Brno, pracoviště Bohunice
3; Klinika plicních nemocí a tuberkulózy LF UP a FN Olomouc
4; Klinika pneumologie a ftizeologie LF UK a FN Plzeň
5
:
Vnitř Lék 2016; 62(5): 360-364
:
Original Contributions
Práce byla přednesena na XIX. Kongresu České a Slovenské pneumologické a ftizeologické společnosti v Brně 17.–19. 6. 2015.
Úvod:
Celosvětově přibývá nemocných cystickou fibrózou (CF) s diagnózou stanovenou v dospělém věku.
Cíl:
Podání přehledu zkušeností s diagnostikou CF v dospělosti v České republice.
Metody:
Nemocní s CF, u nichž byla diagnóza stanovena ve věku ≥ 18 let v období let 2000–2014, byli vyhledáni v Českém registru CF (www.cfregistr.cz) a z jejich zdravotnické dokumentace byla zaznamenána demografická a klinická data z doby diagnózy a k 31. 12. 2014. Podmínkou zařazení do studie byl záchyt dvou mutací vyvolávajících CF nebo jedné mutace spolu s koncentrací chloridů v potu > 60 mmol/l. Klinický obraz byl porovnán s kontrolním souborem tvořeným F508del homozygotními nemocnými s diagnózou stanovenou v dětství.
Výsledky:
Do studie bylo zařazeno 23 nemocných (16 mužů a 7 žen) s diagnózou stanovenou v průměrném věku 32,9 ± 8,5 roku. K diagnóze vedla přítomnost bronchiektazií nebo hemoptýzy ve 12 případech, obstruktivní azoospermie v 7 případech a recidivující pankreatitidy ve 4 případech. Oproti kontrolnímu souboru měli nemocní vyšší věk (38,6 ± 8,3 vs 28,3 ± 4,7 roku; p < 0,001), nižší koncentraci chloridů v potu (62 ± 23 vs 90 ± 12 mmol/l; p < 0,001), méně často infekci dýchacích cest Pseudomonas aeruginosa nebo komplexem Burkholderia cepacia (4 vs 12; p = 0,029), bronchiektazie (14 vs 23; p = 0,001), insuficienci zevní sekrece pankreatu (1 vs 23; p < 0,001) a léčbu inzulinem (1 vs 9; p = 0,01); častější byly naopak pankreatitidy (6 vs 0; p = 0,022).
Závěr:
Na CF je v dospělosti třeba myslet v případě odpovídající symptomatologie v oblasti dýchacího, trávicího a rozmnožovacího ústrojí. Klinický obraz se od klasických forem CF liší v řadě parametrů.
Klíčová slova:
cystická fibróza – diagnostika – dospělí
Úvod
Cystická fibróza (CF) je nejčastější vrozené, život zkracující onemocnění bělošských populací, jehož výskyt je v České republice udáván mezi 1 : 2 700 a 1 : 4 000 živě narozených [1,2]. Základem diagnostiky je nález relevantního klinického obrazu (event. pozitivní rodinná anamnéza nebo pozitivní novorozenecký screening) spolu s průkazem dysfunkce cystic fibrosis transmembrane conductance regulator (CFTR) proteinu pomocí potního testu nebo molekulárně genetického vyšetření (event. vyšetření rozdílu transepiteliálních potenciálů nosní sliznice) [3]. Se zlepšující se péčí o nemocné s CF dochází k prodlužování přežití, čímž narůstá zastoupení dospělých pacientů. V registru americké CF Foundation v roce 2014 poprvé převýšil počet dospělých s CF se zastoupením 50,7 % počet dětí [4]. V České republice podíl dospělých s CF dosáhl již 43,3 % [5]. Na tomto nárůstu se rovněž podílí zlepšená diagnostika u mírných forem onemocnění, včetně stanovení diagnózy až po dosažení dospělosti. Předkládaná práce má za cíl shrnout patnáctileté zkušenosti s touto problematikou v centrech pro léčbu CF v České republice.
Metody
Do studie byli retrospektivně zařazeni nemocní s diagnózou CF stanovenou v období let 2000–2014 v centrech pro léčbu CF při pneumologických pracovištích ve ve FN v Praze-Motole, FN v Brně – Bohunicích, FN v Hradci Králové, FN Olomouc a FN v Plzni. Diagnóza byla ve všech případech stanovena na základě relevantního klinického obrazu, vyšetření koncentrace chloridů v potu a molekulárně genetického vyšetření. Podmínkou zařazení do studie byl v případě koncentrace chloridů v potu > 60 mmol/l záchyt alespoň jedné mutace genu CFTR, resp. 2 mutací genu CFTR při koncentraci chloridů v potu ≤ 60 mmol/l.
U nemocných byl zaznamenán věk v době diagnózy k 31. 12. 2014, pohlaví, symptomy vedoucí k diagnóze, koncentrace chloridů v potu, mutace genu CFTR, stav plicních funkcí vyjádřený pomocí usilovně vydechnutého objemu za první sekundu (FEV1) v % náležitých hodnot, stav výživy vyjádřený pomocí body mass indexu (BMI) v kg/m2, stav infekce dýchacích cest (záchyt gramnegativních bakterií typických pro CF a netuberkulózních mykobakterií), další projevy z oblasti dýchacího ústrojí (bronchiektazie, pneumotorax, hemoptýza vyžadující hospitalizaci, CF astma a alergická bronchopulmonální aspergilóza – ABPA), manifestace v oblasti pankreatu (pankreatitidy, insuficience zevní sekrece pankreatu a potřeba léčby inzulinem), jater (steatóza, fibróza či cirhóza jater) a skeletu (osteopenie a osteoporóza). Uvedené klinické projevy byly diagnostikovány dle standardních postupů.
Kontrolní soubor byl tvořen nemocnými s CF odpovídajícího pohlaví, homozygoty F508del, s diagnózou CF stanovenou v dětském věku, u kterých byly rovněž zaznamenány výše uvedené parametry (data ke 2. pololetí roku 2014). Statistické vyhodnocení bylo provedeno pomocí statistického software Primer of Biostatistics, 6th ed. (McGraw-Hill, Blacklick, OH, USA, 2005).
Výsledky
Do studie bylo zařazeno celkem 23 nemocných, jejichž demografická data spolu se symptomy vedoucími k diagnóze a výsledky vyšetření koncentrace chloridů v potu a molekulárně genetického vyšetření jsou uvedeny v tab. 1.
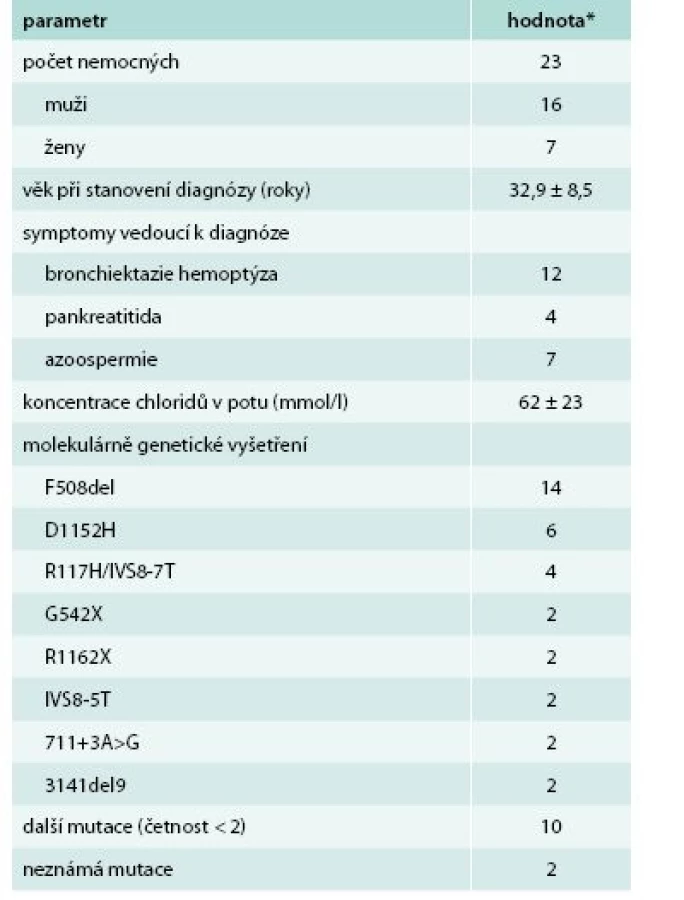
Kontrolní soubor tvořilo 23 nemocných s CF (16 mužů a 7 žen), homozygotů F508del, s mediánem věku při stanovení diagnózy 0,5 (IQR 0,17–1,42) roku a průměrnou (SD) koncentrací chloridů v potu 90 ± 12 mmol/l. U nemocných s CF s diagnózou stanovenou v dospělosti byla oproti kontrolnímu souboru zjištěna signifikantně nižší koncentrace chloridů v potu (p < 0,001; t-test) a statisticky významně vyšší věk k 31. 12. 2014 (38,6 ± 8,3 vs 28,3 ± 4,7 roku; p < 0,001; t-test).
Porovnání klinické manifestace u nemocných s diagnózou CF stanovenou v dospělosti s kontrolním souborem je uvedeno v tab. 2.
Diskuse
Stanovení diagnózy CF u dospělých osob je často náročným úkolem. Relativně snazší je její stanovení u mužů, a to vzhledem k nejkonstantnějšímu klinickému projevu CF, obstruktivní azoospermii, která se nachází u 97–98 % případů [6]. Ani v těchto případech však nemusí být situace zcela jednoduchá, neboť se kromě klasického obrazu CF setkáváme s atypickými (nonclassic) formami CF s variabilním orgánovým postižením a normální nebo hraniční koncentrací chloridů v potu. Stavy, které nesplňují diagnostická kritéria CF, pak označujeme jako CFTR-related disease (starší terminologií CFTR-patie) [7]. Mezi CFTR-related disease řadíme monosymptomatická onemocnění s normální či hraniční koncentrací chloridů v potu a průkazem jen 1 mutace genu CFTR, a to diseminované bronchiektazie, recidivující a chronickou pankreatitidu a obstruktivní azoospermii [8]. Skutečností rovněž je, že klinický obraz se může v čase vyvíjet. Např. u 25letého muže s obstruktivní azoospermií, hraniční koncentrací chloridů v potu a průkazem 1 mutace je iniciálně stanovena diagnóza CFTR-related disease. Diagnóza je však zpochybněna, když pacient s časovým odstupem prodělá 2 epizody pankreatitidy, a atypická forma CF je následně potvrzena při průkazu 2. mutace genu CFTR. Z uvedeného vyplývá nutnost dlouhodobého klinického sledování těchto nemocných a případného přehodnocení diagnostického závěru.
Mírnější klinické formy CF tak u nás v řadě případů nejspíše unikají diagnóze, především u žen. Vždyť v registru britského CF Trustu je mezi nemocnými, u kterých byla diagnóza CF stanovena před zahájením novorozeneckého screeningu (v roce 2007), celkem 741 osob, tj. 13,4 %, se zjištěním tohoto onemocnění v dospělosti (ve Velké Británii je za dospělost považován věk 16 a více let) [9]. Diagnostické možnosti zvyšuje vyšetření bioelektrických potenciálů nosní nebo střevní sliznice, které umožňují posoudit dysfunkci CFTR proteinu [10]. Toto vyšetření však dosud není v České republice dostupné.
Ve kterých klinických situacích tedy na možnost CF u dospělého myslet? Jako vysoce sugestivní jsou uváděny bilaterální bronchiektazie horních laloků, opakovaný záchyt komplexu Burkholderia cepacia nebo mukózních kmenů Pseudomonas aeruginosa v respiračních sekretech, obstruktivní azoospermie při kongenitální bilaterální absenci vas deferens a hypochloremická alkalóza při absenci zvracení. Mezi sugestivní, avšak málo specifické příznaky pak patří chronický produktivní kašel, hemoptýzy při absenci tuberkulózy a vaskulitid, ABPA, chronická pansinusitida, nosní polypóza, recidivující pankreatitidy, insuficience zevní sekrece pankreatu, syndrom obstrukce distálního střeva, osteoporóza u mladších 40 let a paličkovité prsty [11].
V uvedených případech je indikováno vyšetření potního testu a při opakovaném průkazu hraničních či patologických hodnot molekulárně genetické vyšetření. V případě pankreaticky suficientích a atypických forem CF lze očekávat průkaz alespoň jedné tzv. mírné mutace (třída IV nebo V mutací genu CFTR) [12]. V našem souboru se s vyšší četností vyskytly mutace D1152H (v 6 případech) a R117H/IVS8–7T (ve 4 případech).
Mutace D1152H je spojena se sníženou vodivostí CFTR proteinu pro chloridové ionty (třída IV) a má variabilní hodnoty koncentrace chloridů v potu, od normálních až po patologické. Rovněž klinická manifestace je variabilní. Plicní postižení nemusí být vyjádřeno vůbec, bronchiektazie jsou přítomny u asi 2/3 případů, popsány jsou však i stavy s těžkou ventilační poruchou. Chronická infekce dýchacích cest Pseudomonas aeruginosa je méně častá, a to u asi 1/3 případů. Nemocní jsou pankreaticky suficientní s rizikem rozvoje pankreatitidy. U mužů může být zachována plodnost [13,14].
Mutace R117H je nejčastější mutací s reziduální funkcí CFTR proteinu při poruše jeho aktivace a snížené chloridové vodivosti (třída III a IV). Současně syntéza CFTR proteinu závisí na délce polytymidinového traktu intronu 8 (IVS8), která ovlivňuje adekvátní sestřih exonu 9. Jeho pravděpodobnost klesá v pořadí 9T, 7T a 5T. Varianty IVS8–7T a zejména IVS8–5T tak vedou ke snížené syntéze CFTR-proteinu (třída V), přičemž varianta IVS8–5T se může uplatnit jako příčina atypické formy CF i samostatně, tedy bez kombinace s mutací R117H [15–17]. V našem souboru se varianta IVS8–5T uplatnila u 2 nemocných.
V prezentované práci byl studovaný soubor porovnán po stránce plicních i mimoplicních projevů s nemocnými s CF, homozygoty F508del jako reprezentanty klasické formy onemocnění. V oblasti dýchacího ústrojí měli nemocní diagnostikovaní v dospělosti méně často bronchiektazie a infekci dýchacích cest gramnegativními bakteriemi typickými pro CF a rovněž tendenci k lepším hodnotám plicních funkcí. Po stránce mimoplicních projevů byly u nemocných diagnostikovaných v dospělosti častěji přítomny pankreatitidy, naopak méně častá byla insuficience zevní sekrece pankreatu a léčba inzulinem, spolu s tendencí k nižšímu výskytu osteopenie a osteoporózy. Nutno podotknout, že pankreatitidy byly ve 4 případech symptomem vedoucím k diagnóze. V těchto případech šlo o lehčí formy onemocnění s iritačním či edematózním postižením pankreatu zvládnuté konzervativními postupy. Závažné formy pankreatitid s nekrózami, abscesy či šokovým stavem pozorovány nebyly. Kromě uvedených recidivujících pankreatitid došlo ve 2 případech k rozvoji pankreatitidy chronické. Tato forma patří spolu s deficitem α1-antitrypsinu a autoaktivačními mutacemi intrapankreatického kationického trypsinogenu mezi genetické formy chronické pankreatitidy [18].
Uvedené nálezy jsou v souladu s výsledky studií zahraničních autorů. De Gracia et al srovnávali dospělé nemocné s CF s diagnózou stanovenou v dětství a v dospělosti [19]. V této práci měli nemocní s diagnózou stanovenou v dospělosti lepší funkce plic a stav výživy, méně často infekci dýchacích cest Pseudomonas aeruginosa, insuficienci zevní sekrece pankreatu, hepatopatie a diabetes mellitus, naopak častější byl výskyt ABPA a pankreatitid. Diagnostikovaní v dospělosti byli rovněž méně často léčeni za hospitalizace a pomocí transplantace plic. Symptomy vedoucí k diagnóze byly v této studii nejčastěji z oblasti dýchacího ústrojí (40 případů; ve 4 případech vedla k suspekci na CF jako první symptom ABPA), dále to byly pankreatitidy (4 případy), azoospermie (4 případy) a rodinná anamnéza CF (2 případy).
Obdobnou studii provedli Rodman et al u dospělých s CF starších 40 let [20]. I v této práci měli nemocní CF diagnostikovaní v dospělosti lepší funkce plic a méně často infekci dýchacích cest Pseudomonas aeruginosa, insuficienci zevní sekrece pankreatu a diabetes mellitus. Naopak častější byl u nich záchyt netuberkulózních mykobakterií v respiračních sekretech.
Rozsáhlá byla v této oblasti práce Gilljamové et al z let 1960–2001 [21]. Celkem 7 % pacientů s CF bylo diagnostikováno ve věku 16 a více let, přičemž ve 33 % případů bylo k diagnostice využito i vyšetření bioelektrických potenciálů nosní sliznice. U osob diagnostikovaných po roce 1990 byly symptomy vedoucí k diagnóze z oblasti dýchacího ústrojí v 39 % případů, inferitility v 26 % případů, současné plicní a gastrointestinální symptomy v 22 % případů a pankreatitidy v 4 % případů. V ostatních případech (9 %) šlo o diabetes mellitus, biliární cirhózu jater a genetický screening. V uvedeném souboru činila průměrná hodnota FEV1 81 % n. h. a bronchiektazie byly přítomny u 59 % případů (v našem souboru činilo průměrné FEV1 82 % n. h. a bronchiektazie byly přítomny u 61 % případů). Pankreaticky insuficientních bylo 15 % osob (v našem souboru 4 %). Jako 2. mutace byla R117H/IVS8–7T přítomna u 11 % subjektů a koncentrace chloridů v potu byla v 35 % případů < 60 mmol/l (v našem souboru u 18 %, resp. v 57 % případů).
Nemocní, u kterých byla v dospělosti diagnostikována CF, jsou následně dispenzarizováni v centrech pro léčbu CF. Vzhledem k pestrým možnostem klinické manifestace tohoto onemocnění je jejich léčba přísně individualizovaná. Doporučené postupy pro péči o tyto pacienty jsou dány v dokumentech vytvořených mezinárodními panely expertů odborných společností, jako je např. European Cystic Fibrosis Society, bristký CF Trust či americká CF Foundation.
Závěr
Prezentovaná práce ukazuje rozmanitost klinických obrazů CF u nemocných s diagnózou stanovenou v dospělosti spolu s náročností diagnostického procesu. Většina nemocných se rekrutuje z pneumologických pracovišť a z center asistované reprodukce, ale nelze zapomínat ani na gastroenterologické případy. Stanovení diagnózy CF, byť s mírnými projevy, má dopad nejen na pacienta samotného, ale i na jeho pokrevní příbuzné v případě plánování rodiny.
Práce byla podpořena projektem MZ ČR – RVO FN Motol 00064203.
Poděkování autorského kolektivu patří paní Mgr. Aleně Bílkové a panu MUDr. Marku Turnovcovi, kteří spravují Český registr CF.
MUDr. Libor Fila, Ph.D.
libor_fila@seznam.cz
Pneumologická klinika 2. LF UK a FN v Motole, Praha
www.lf2.cuni.cz
Doručeno do redakce 2. 12. 2015
Přijato po recenzi 21. 3. 2016
Sources
1. Houštěk J, Vávrová V. Frequency and forms of mucoviscidosis in Czechoslovakia. Cesk Pediatr 1965; 20(3–5): 412–414.
2. Balaščáková M, Holubová A, Skalická V et al. Pilot newborn screening project for cystic fibrosis in the Czech Republic: defining role of the delay in its symptomatic diagnosis and influence of ultrasound-based prenatal diagnosis on the incidence of the disease. J Cyst Fibros 2009; 8(3): 224–227.
3. Rosenstein BJ, Cutting GR. The diagnosis of cystic fibrosis: a consensus statement. Cystic Fibrosis Foundation Consensus Panel. J Pediatr 1998; 132(4): 589–595.
4. Cystic Fibrosis Foundation Patient Registry. 2014 Annual Data Report. Bethesda (Maryland). Dostupné z WWW: <https://www.cff.org/2014_CFF_Annual_Data_Report_to_the_Center_Directors.pdf/>. [31.10.2015].
5. Český registr cystické fibrózy. Dostupné z WWW: <https://www.cfregistr.cz/index.php?akce=statistika>. [31.10.2015].
6. Chen H, Ruan YC, Xu WM et al. Regulation of male fertility by CFTR and implications in male infertility. Hum Reprod Update 2012; 18(6): 703–713. Erratum in Hum Reprod Update 2012; 18(6): 715.
7. Boyle MP. Nonclassic cystic fibrosis and CFTR-related diseases. Curr Opin Pulm Med 2003; 9(6): 498–503.
8. Bombieri C, Claustres M, De Boeck K et al. Recommendations for the classification of diseases as CFTR-related disorders. J Cyst Fibros 2011; 10:(Suppl 2): S86-S102.
9. Informace dostupné z WWW: <http://www.cysticfibrosis.org.uk/media/1596846/RegistryReport2014.pdf> [31.10.2015].
10. De Boeck K, Derichs N, Fajac I et al. [ECFS Diagnostic Network Working Group; EuroCareCF WP3 Group on CF diagnosis]. New clinical diagnostic procedures for cystic fibrosis in Europe. J Cyst Fibros 2011; 10(Suppl 2): S53-S66.
11. De Boeck K, Wilschanski M, Castellani C et al. Diagnostic Working Group. Cystic fibrosis: terminology and diagnostic algorithms. Thorax 2006; 61(7): 627–635.
12. Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. N Engl J Med 2005; 352(19): 1992–2001.
13. Burgel PR, Fajac I, Hubert D et al. Non-classic cystic fibrosis associated with D1152H CFTR mutation. Clin Genet 2010; 77(4): 355–364.
14. Terlizzi V, Carnovale V, Castaldo G et al. Clinical expression of patients with the D1152H CFTR mutation. J Cyst Fibros 2015; 14(4): 447–452.
15. Castellani C, Cuppens H, Macek M Jr et al. Consensus on the use and interpretation of cystic fibrosis mutation analysis in clinical practice. J Cyst Fibros 2008; 7(3): 179–196.
16. Noone PG, Pue CA, Zhou Z et al. Lung disease associated with the IVS8 5T allele of the CFTR gene. Am J Respir Crit Care Med 2000; 162(5): 1919–1924.
17. Peckham D, Conway SP, Morton A et al. Delayed diagnosis of cystic fibrosis associated with R117H on a background of 7T polythymidine tract at intron 8. J Cyst Fibros 2006; 5(1): 63–65.
18. Dítě P, Trna J, Novotný I et al. Chronická pankreatitida v roce 2011. Vnitř Lék 2011; 57(11): 891–966.
19. de Gracia J, Alvarez A, Mata F et al. Cystic fibrosis in adults: study of 111 patients. Med Clin (Barc) 2002; 119(16): 605–609.
20. Rodman DM, Polis JM, Heltshe SL et al. Late diagnosis defines a unique population of long-term survivors of cystic fibrosis. Am J Respir Crit Care Med 2005; 171(6): 621–626.
21. Gilljam M, Ellis L, Corey M et al. Clinical manifestations of cystic fibrosis among patients with diagnosis in adulthood. Chest 2004; 126(4): 1215–1224.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2016 Issue 5
Most read in this issue
- Hypercalcemia, symptoms, differential diagnostics and treatment, or importance of calcium investigation
- Diagnostics of cystic fibrosis in adults
- Can fish oil improve wound healing in surgery?
- Hepatic transit times and liver elasticity compared with meld in predicting a 1 year adverse clinical outcome of a clinically diagnosed cirrhosis