
Diagnostika poruch hemostázy
Diagnostic strategies in disorders of hemostasis
Hemostasis is a complicated biological system, where the balance between procoagulation and anticoagulation processes maintains fluidity of blood through intact blood vessels and creates thrombi when it is needed to prevent bleeding from the impaired vessels. The modern model of hemostasis is divided into 2 principal phases, the first being defined as primary hemostasis which involves the platelet-vessel interplay, while the second, defined as secondary hemostasis, mainly involves coagulation factors and surfaces of activated cells. The activation and amplification of the coagulation cascade is regulated by natural inhibitors of coagulation. The blood clots which arise to prevent loss of blood must subsequently be broken down and the compact blood vessel wall must be restored. This process is called fibrinolysis. Bleeding and thromboses are manifestations of the impaired hemostatic balance. Detection of its cause is important for efficient treatment and prevention of the condition. This requires a combined evaluation of the family and personal history, a clinical anamnesis along with the evaluation of laboratory results. It is not reasonable or economical to perform all the available tests of hemostasis at the same time. It is recommendable to proceed from the global to the screening and then special tests and, through the process of elimination, obtain an explanation of the bleeding or prothrombotic phenotypes. The purpose of this report is to provide a brief overview of the principal causes of the hemostatic disorders and the practices which facilitate their diagnosis.
Key words:
diagnosis of hemorrhagic manifestations – diagnostics of thrombophilias – DIC diagnostics – hemophilia A – thrombopathy – von Willebrand disease
Authors:
Ingrid Hrachovinová
Authors‘ workplace:
Ústav hematologie a krevní transfuze, Praha
Published in:
Vnitř Lék 2018; 64(5): 537-544
Category:
Overview
Hemostáza je komplikovaný biologický systém, v němž rovnováha prokoagulačních a antikoagulačních procesů udržuje tekutost krve v neporušených cévách a vytváří tromby, pokud je potřeba zabránit krvácení z porušených cév. Moderní model hemostázy se dělí na 2 základní fáze, primární hemostázu, která zahrnuje kontakt cévní stěny a krevních destiček, zatímco druhá fáze, která se nazývá sekundární hemostáza, zahrnuje koagulační faktory a povrchy aktivovaných buněk. Aktivace a amplifikace koagulační kaskády je usměrňována přirozenými inhibitory koagulace. Sraženiny, které vznikají, aby zabránily úniku krve, musí být následně rozrušeny, aby byla obnovena kompaktní cévní stěna. Tento proces se nazývá fibrinolýza. Krvácení a trombózy jsou projevem porušené hemostatické rovnováhy. Nalezení její příčiny je důležité pro účinnou léčbu a prevenci poruchy hemostázy. Vyžaduje to kombinované vyhodnocení rodinné a personální historie, klinickou anamnézu spolu s vyhodnocením laboratorních výsledků. Není rozumné ani ekonomické provést všechna dostupná vyšetření hemostázy najednou. Vhodné je postupovat od globálních přes screeningové po speciální testy a vylučovací metodou dospět k vysvětlení krvácivého nebo trombotického fenotypu. Účelem tohoto sdělení je krátký přehled nejdůležitějších příčin poruch hemostázy a postupů, které ulehčí jejich diagnostiku.
Klíčová slova:
diagnostika DIC – diagnostika krvácivých onemocnění – diagnostika trombofilií – hemofilie A – trombopatie – von Willebrandova choroba
Úvod
Hemostáza je komplikovaný a přesně regulovaný proces, který udržuje tekutost krve v cévách a zároveň zabraňuje jejímu úniku z poškozených cév. Porušení hemostatické rovnováhy může způsobit krvácivé nebo trombotické stavy.
Hemostáza se dělí na hemostázu primární (zahrnuje cévní stěnu a krevní destičky) a sekundární, neboli koagulaci, která je mnohem lépe prozkoumaná, zabývá se plazmatickými koagulačními proteiny a jejich přirozenými inhibitory při tvorbě trombinu a následně fibrinu. Do hemostázy také zahrnujeme fibrinolytické a regenerační procesy.
Diagnostika poruch hemostázy
Základem správné diagnostiky poruch hemostázy je kombinace informací klinických, anamnestických a laboratorních. K diagnostice poruch hemostázy můžeme přistoupit 2 způsoby:
- pacient má krvácivé/trombotické problémy a lékař za pomoci laboratoře musí stanovit příčinu krvácení nebo trombózy, aby byl pacient, pokud možno cíleně a adekvátně léčen
- na základě náhodného koagulačního screeningu (např. předoperační vyšetření) byly zjištěny patologické výsledky koagulačních testů a laboratorní vyšetření by mělo objasnit příčinu této patologie – zjištěný výsledek vede často k důležitým preventivním opatřením ve smyslu prevence krvácení nebo léčby a prevence trombózy
Klinické a anamnestické vyšetření
Řádné klinické a anamnestické vyšetření odliší vrozené formy poruchy hemostázy od získaných a poruchu primární hemostázy od sekundární. Důležité je to zejména u krvácivých stavů.
Primárně je nutné u krvácejícího pacienta zhodnotit, zda se jedná o lokální příčinu krvácení z důvodu úrazu nebo krvácení způsobené vrozenou nemocí, nebo kombinace obojího. Dále je nutné zjistit, zda se jedná o poruchu primární hemostázy nebo sekundární hemostázy (koagulace).
Při rozhodování, zda se jedná o lokální poranění nebo o systémovou poruchu hemostázy, je důležité si uvědomit, že systémová porucha hemostázy se projevuje:
- krvácením na více místech
- epizodami spontánního krvácení
- krvácivými epizodami s časovým odstupem od poranění nebo prodloužené krvácení
- slizničním krvácením s epistaxemi
- hematomy nebo krvácení do kloubů
Přibližné rozlišení, zda se jedná o krvácení z příčiny poruchy primární hemostázy nebo koagulační poruchy, ukazuje tab. 1.
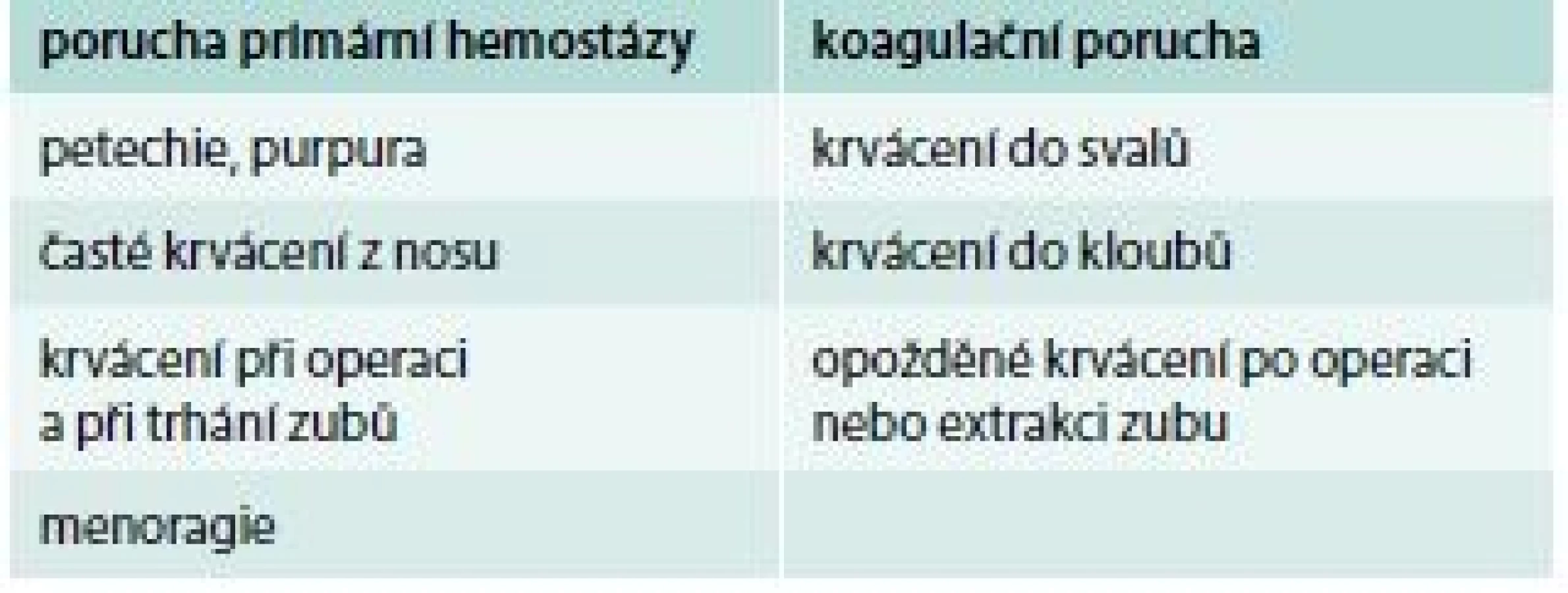
Hlavní znaky pro rozlišení, zda se jedná o vrozenou poruchu krvácení nebo získanou, ukazuje tab. 2. Je ale obtížné odlišit nově vzniklé získané krvácivé onemocnění od lehké formy vrozené krvácivé choroby jen na základě klinické anamnézy.

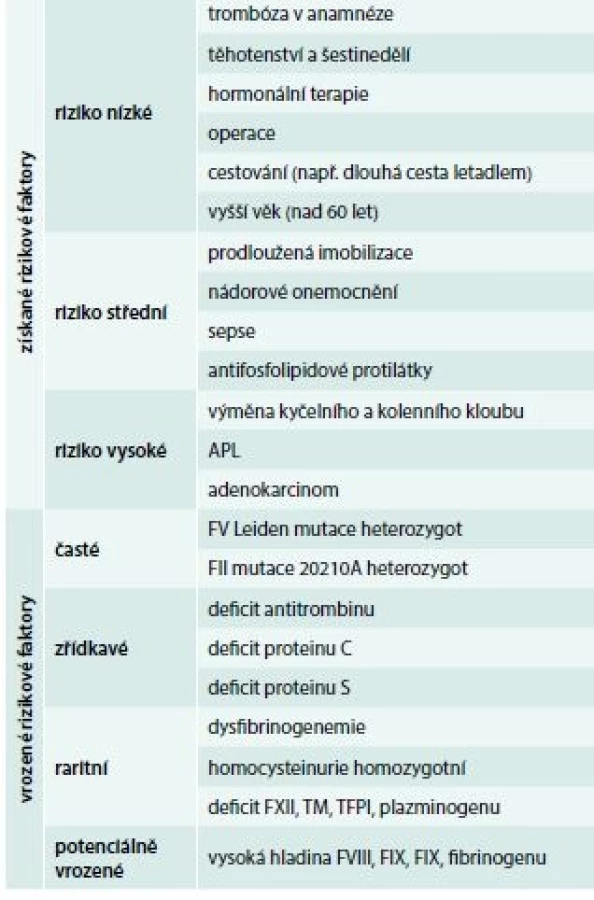
U trombotických stavů je situace jiná, protože vznik trombózy má komplexní charakter a zpravidla je následkem kombinace získané a vrozené příčiny (tab. 3), proto je velmi důležité při anamnéze zjistit, zda existují a jaké jsou získané trombotické rizikové faktory.
Laboratorní diagnostika poruch hemostázy
Laboratorní diagnostika hemostázy není jednoduchá, zahrnuje testy globální, screeningové a speciální. Postupujeme vždy od testů globálních nebo screeningových ke speciálním.
Globální testy: krvácivost, Rumpelův-Leedův test, konsumpce protrombinu, retrakce koagula, PFA, TEG aj
Screeningové testy: APTT, PT, TT, dRVVT, etanol gelifikační test, TGT, agregace krevních destiček v plazmě aj
Speciální testy: stanovení aktivity a množství koagulačních faktorů, přirozených inhibitorů koagulace, molekulárních markerů aktivace hemostázy, D-dimerů a fibrinolytických faktorů a jejich inhibitorů aj
Laboratorní diagnostika krvácivého stavu
Krvácivými stavy rozumíme chorobné stavy, u kterých dochází k neúměrnému krvácení při minimálním poranění, případně i spontánně.
Z etiologického hlediska je dělíme do 3 skupin:
- z poruch plazmatické koagulace – koagulopatie
- z destičkových příčin – trombocytopenie, trombocytopatie
- z poruch cévní stěny – vaskulopatie
Laboratorní vyšetření vždy začíná vyšetřením krevního obrazu (počet krevních destiček) a základním koagulačním screeningem (vyšetření APTT, PT, TT, fibrinogen). Možné příčiny krvácení na základě diferenciální diagnostiky základních koagulačních testů pacientů s normálním počtech krevních destiček uvádí tab. 4.
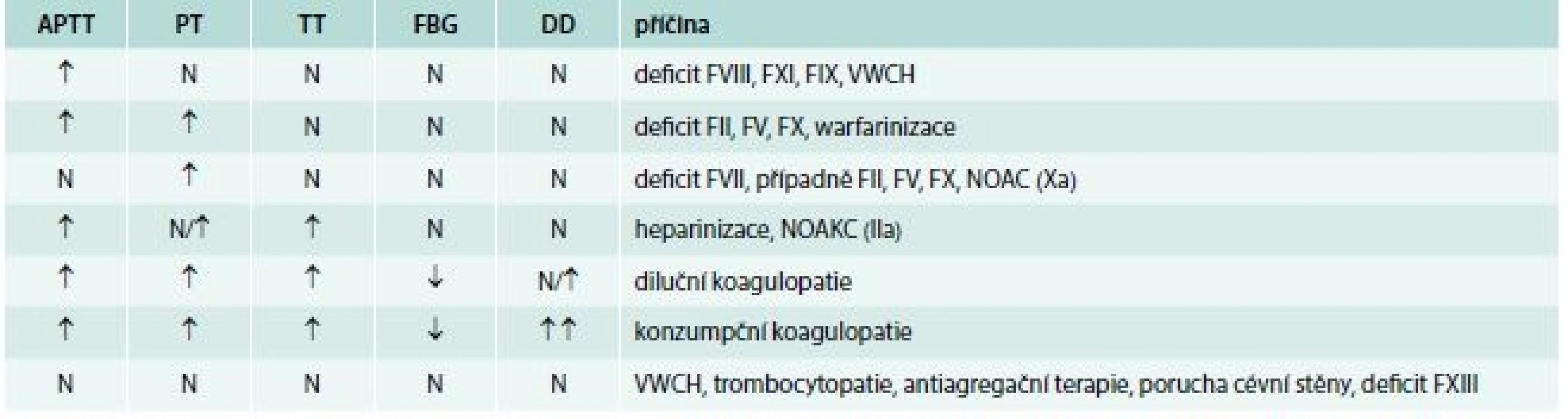
Koagulopatie
Pojmem koagulopatie označujeme krvácivé stavy zapříčiněné nefunkčností nebo deficitem koagulačních faktorů, který může být jak vrozený, tak získaný, izolovaný nebo kombinovaný.
Hemofilie
Hemofilie je vrozené krvácivé koagulační onemocnění, které postihuje přibližně 1 z 10 000 mužů. Vedle hemofilie A, která je 6krát četnější a je způsobena deficitem FVIII v cirkulující krvi, existuje také hemofilie B, která je způsobena deficitem FIX. Stejná klinická manifestace obou onemocněni je způsobena úlohou FVIII a FIX v plazmatické koagulaci, kde spolu tvoří tenázový komplex. Podle hloubky deficitu klasifikujeme hemofilii: na těžkou (< 1 %), středně těžkou (1–5 %) a lehkou (5–40 %) formu. Závažným problémem hemofilie je vznik autoprotilátek inhibujících FVIII (FIX). Vyskytují se u 5–35 % pacientů s těžkou až středně těžkou formou onemocněni. Častěji se objevují u hemofilie. Hemofilie je na chromozom X vázaná choroba, muži mají projevy choroby (krvácejí) a ženy jsou z pravidla bez příznaků krvácení, jsou přenašečky onemocnění. Krvácení u hemofiliků má komplexní patofyziologii. Defekt FVIII/FIX způsobuje nedostatečnou tvorbu trombinu, následkem toho vzniká nestabilní krevní sraženina. Nedostatečné množství trombinu také způsobuje časnou a zvýšenou fibrinolýzu (snížení funkce TAFI).
Laboratorní diagnostika – hemofilie A:
- screeningový test: prodloužené APTT, které se koriguje ve směsi s normální plazmou
- hlavní specifický test: aktivita FVIII (FVIII : C, FVIII : Cr)
- dodatečné testy: inhibitor FVIII, FVIII : Ag a genetické stanovení kauzální mutace
Diferenciální diagnostika – nutno odlišit lehkou formu hemofilie A od Von Willebrandovy choroby 2N vyšetřením vazebné kapacity FVIII k VWF (VWF : FVIIIB).
Laboratorní diagnostika – hemofilie B:
- screeningový test: prodloužené APTT, které se koriguje ve směsi s normální plazmou
- hlavní specifický test: aktivita FIX (FIX : C)
- dodatečné testy: inhibitor FIX, FIX : Ag a genetické stanovení kauzální mutace
Von Willebrandova choroba
Von Willebrandova choroba (VWCH) je nejčastější vrozené krvácivé onemocnění. Na základě populační studie je zastoupena v 1 %, ale symptomatický výskyt VWCH je popisován jen v 0,01 % populace. Je způsobena defektem nebo deficitem von Willebrandova faktoru. Přenáší se mezi generacemi oběma pohlavími autosomálně dominantně i recesivně, závisí na subtypu VWCH. Funkce VWF je:
- v primární hemostáze – adheze trombocytů k subendoteliálním strukturám prostřednictvím GPIb a kolagenu v endoteliální matrix, adheze a agregace trombocytů vazbou na receptory trombocytů
- v koagulaci – ochrana FVIII před proteolytickou degradací
Laboratorní diagnostika VWCH:
- základní metody: APTT, PFA100, krvácivost, konsumpce protrombinu
- hlavní specifický test: vyšetření funkce VWF (VWF : RCo, VWF : CB), VWF : Ag, FVIII : C
- dodatečné testy k vyšetření subtypů VWCH: RIPA, vyšetření multimerů VWF, VWF : FVIIIB, VWFpp, genetické stanovení kauzální mutace
Rozdělení VWCH do typů a subtypů na základě výsledků jednotlivých vyšetření ukazuje tab. 5.

K dalšímu rozlišení subtypu 2A na terciální subtypy (IIA, IIC, IID, IIE) a odlišení subtypu 2M od 2A je nutná znalost multimerní struktury VWF.
Deficity dalších koagulačních faktorů
Prevalence vzácných vrozených krvácivých onemocnění (deficit FII, FV, FVII, FX, FXI, FXIII, dys/hypo/afibrinogenemie) je 1 pacient na 500 000 až 2 miliony lidí.
Laboratorní diagnostika vzácných koagulopatií:
- hlavní metody: APTT, PT, TT, fibrinogen
- hlavní specifický test: funkční aktivita jednotlivých koagulačních faktorů
- doplňkové testy: kvantitativní stanovení faktorů (:Ag), a případně stanovení inhibitoru, genetické vyšetření kauzální mutace
Vliv jednotlivých deficitů koagulačních faktorů na prodloužení screeningových testů uvádí tab. 6.
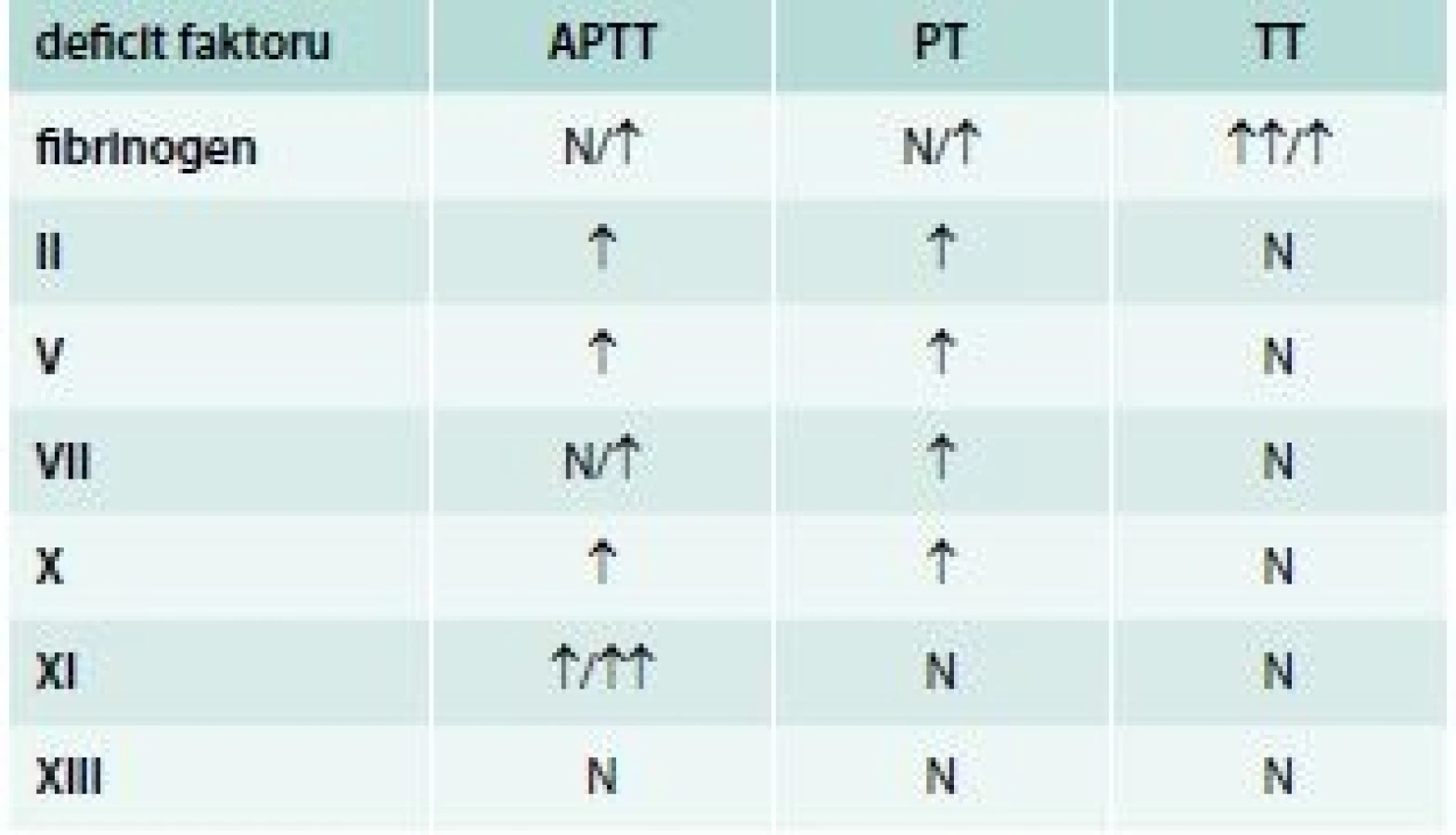
Problémy s laboratorní diagnostikou vzácných krvácivých koagulopatií
Zásadním problémem je, že změřená aktivita koagulačního faktoru nemusí odpovídat tíži krvácení pacienta. U některých pacientů s funkčním deficitem fibrinogenu a normální kvantitou fibrinogenu (dysfibrinogenemie) dochází místo krvácení k tromboembolickým stavům.
Specifické inhibitory koagulačních faktorů
Pojmem specifický inhibitor koagulačního faktoru označujeme přítomnost protilátky proti koagulačnímu faktoru, která výrazným způsobem snižuje jeho prokoagulační aktivitu. V klinické praxi se nejčastěji vyskytují specifické inhibitory proti FVIII, mnohem méně proti FIX nebo VWF a raritně proti dalším koagulačním faktorům (FV, FII, FX, FXIII, FXI aj).
Laboratorní diagnostika inhibitoru u pacientů s hemofilií A nebo B:
screeningový test: prodloužené APTT, korekční testy APTT s normální plazmou – typickým znakem specifického inhibitoru je, že korekční testy jsou časově a teplotně závislé, proto je nutné měřit APTT hned po smíchání plazem a pak po 2 hod inkubace při 37 ºC
- hlavní specifický test: FVIII : C/FXI : C
- dodatečné testy: inhibitor FVIII/FIX Bethesda metodou nebo její modifikací Nijmegen
Laboratorní diagnostika inhibitorů u pacientů se získanou hemofilií A:
- screeningový test: prodloužené APTT, korekční testy APTT s normální plazmou – APTT je nutné měřit hned po smíchání plazem a pak po 2 hod inkubace při 37 ºC
- hlavní specifický test: FVIII : C nebo FVIII : Cr
- dodatečné testy: inhibitor FVIII Bethesda metodou nebo její modifikací Nijmegen – je nutno striktně dodržet odečet inhibitoru co nejblíže 50% inhibici FVIII, protože se jedná zpravidla o nekompletně neutralizující inhibitor s kinetikou II. řádu a může dojít u nižších titrů k podhodnocení a u vysokých k nadhodnocení výsledku
Diferenciální diagnostika – nutno odlišit protilátky typu lupus antikoagulans, které mohou u některých APTT reagencií způsobovat falešně pozitivní deficit FVIII a pozitivní inhibitor FVIII.
Laboratorní diagnostika pacientů se získaným inhibitorem VWF:
- základní metody: APTT, PFA100, krvácivost, konsumpce protrombinu
- hlavní specifický test: vyšetření funkce VWF (VWF : RCo, VWF : CB), VWF : Ag, FVIII : C
- dodatečné testy: vyšetření multimerů VWF, VWFpp, inhibitoru VWF modifikovanou Bethesda metodou (volný inhibitor se vyskytuje jen u 20 % pacientů)
Diferenciální diagnostika – odlišení získané formy VWCH od vrozené VWCH je obtížné. Velmi pomáhá důkladná anamnéza a historie krvácení pacienta i rodinných příslušníků. Patofyziologie získané formy je různorodá a nemusí být na rozdíl od hemofilie A a B způsobena přítomností inhibujících protilátek. Pro získanou formu VWCH svědčí patologické nebo chybějící multimery VWF, vysoká koncentrace VWpp a zkrácená kinetika podávání koncentrátu VWF.
Laboratorní diagnostika pacientů se získaným inhibitorem dalších koagulačních faktorů:
- screeningové metody: APTT, PT, TT, fibrinogen, směsné testy na principu APTT nebo PT, případně TT nemusí být průkazné, protože volný inhibitor se vyskytuje zřídka
- hlavní specifický test: vyšetření funkce jednotlivých koagulačních faktorů
- dodatečné testy: vyšetření inhibitoru (ale volný inhibitor se vyskytuje jen u malého procenta pacientů)
Při podezření na inhibitor dalších koagulačních faktorů je důležitá anamnéza probíhajících onemocnění, protože získaný deficit dalších koagulačních faktorů může být vázán na určité onemocnění (např. získaný deficit FX u amyloidózy). Patofyziologie získané formy je různorodá a stejně tak jako u získané VWCH nemusí být způsobena přítomností inhibujících protilátek.
Trombocytopenie, trombocytopatie
Porucha hemostatické funkce trombocytů je způsobena jejich sníženým počtem (< 150 × 109/l) nebo poruchou jejich funkce nebo kombinací obou možností. Snížený počet trombocytů je způsoben:
- sníženou produkcí v kostní dřeni z různých příčin, někdy jsou spojené s morfologickými poruchami – mikrotrombocyty (Wiskottův-Aldrichův syndrom) nebo makrocyty (Mayovy-Hegglinovy anomálie, Fechnerův syndrom, Sebastianův syndrom)
- zvýšená sekvestrace trombocytů ve slezině
- trombocytopenické purpury ze zvýšené destrukce, která může být na základě imunitní destrukce (viz článek MUDr. Červinky v tomto čísle časopisu Vnitřní lékařství, s. 526n) nebo neimunitní destrukce (např. DIC, TTP, HUS), při níž zpravidla dochází k periferní konsumpci v mikrocirkulaci
Trombocytopatie rozlišujeme vrozené s poruchou trombocytárních glykoproteinů (Bernardův-Soulierův syndrom – BSS, Glanzmannova trombastenie – GT), s poruchou granul trombocytů (syndrom šedých trombocytů – DGD), s poruchou signální transdukce nebo poruchou prokoagulační aktivity (Skottův syndrom). Získané trombocytopatie nacházíme u myeloproliferativních onemocnění, chronických ledvinových onemocnění, monoklonálních gamapatií, u léčby léky inhibujícími destičkové funkce.
Laboratorní diagnostika pacientů s vrozenými trombocytopatiemi
Vyšetření zahrnuje stanovení počtu destiček, stanovení jejich tvaru a struktury, funkce destiček pomocí PFA-100 a agregace krevních destiček po jednotlivých agonistech (ADP, epinefrin, collagen, trombin nebo ristocetin). Při podezření na deficit určitého receptoru se využívá průtokové cytometrie s monoklonálními protilátkami proti receptorům. Při prokázaném vrozeném deficitu nebo funkci receptoru pokračuje vyšetření hledáním kauzální mutace.
Diseminovaná intravaskulární koagulace
Diseminovaná intravaskulární koagulace (DIC) je závažný krvácivý stav, který vzniká dysbalancí mezi koagulační aktivitou a fibrinolýzou. Výsledkem jsou mikrotromby v mikrocirkulaci a krvácení v důsledku spotřeby koagulačních faktorů a destrukce krevních destiček.
Laboratorní diagnostika DIC – diagnostika je nepřímá, neexistuje jeden test na potvrzení/vyloučení DIC:
- screeningové testy: snížený počet trombocytů, AT, fibrinogenu, prodloužené APTT, PT
- konfirmační testy: zvýšené fibrinové monomery (FM), fragmenty trombinu
Bodový systém pro diagnostiku DIC podle ISTH ukazuje tab. 7. Používá se, pouze pokud je přítomno onemocnění, které je asociováno s DIC.

Laboratorní diagnostika trombotického stavu
Trombotické stavy jsou vrozené nebo získané poruchy hemostázy, které jsou spojeny se zvýšeným výskytem nebo rizikem trombózy.
Trombofilie je definována jako defekt hemostázy, který je pravděpodobně příčinou zvýšeného sklonu k trombózám. Častěji hovoříme o trombofilních rizikových faktorech, protože se na vzniku trombóz podílejí s určitou mírou pravděpodobnosti. Velmi často se na vzniku trombózy podílejí dva i více trombofilních rizikových faktorů, proto je vždy nutné vyšetřit kompletní dostupný panel rizikových faktorů.
Rizikové faktory dělíme do dvou skupin (tab. 3): rizikové faktory vrozené a rizikové faktory získané.
Laboratorní vyšetření vždy začínáme základním koagulačním screeningem (vyšetření APTT, PT, TT, fibrinogen) a vyšetřením D-dimerů. Zvýšená hladina D-dimerů je jediným laboratorním uznávaným markrem ukazujícím na přítomnost žilní trombózy. Trombózu detekuje přibližně s 95% senzitivitou, ale bohužel jen s asi 50% specifitou. Specifitu může negativně ovlivnit přítomnost DIC a zánětlivých stavů.
Pokračujeme specifickými testy, které by měly zachytit jednotlivé nejdůležitější trombofilní rizikové faktory.
Specifické testy: FVIII : C, aktivita AT, aktivita PC, aktivita PS, APCR, hladina homocysteinu, molekulárně genetické markery mutace FV Leiden, FII 20210A.
Vrozené trombofilní rizikové faktory
Deficit antitrombinu
Antitrombin (AT) je účinný inhibitor trombinu, aktivovaného FX (FXa) a dalších faktorů hemostázy (např. FIXa, FXIa, FVIIa, tPA). Je nejdůležitějším přirozeným inhibitorem koagulace. Patří do rodiny serpinů (Serine Protease Inhibitors), AT má název SERPINC1. Při fyziologických podmínkách má AT malou inhibiční aktivitu, která se zvětší 1 000krát vazbou na heparin nebo jiné heparinové glykosylaminoglykany (heparansulfát). Heparin se váže do AT specifickou pentasacharidovou sekvencí, jejíž vazba vyvolá konformační změny v AT, které akcelerují inhibiční funkci.
Vrozený deficit AT je vzácný, vyskytuje se v populaci v rozmezí od 1 : 2 000 do 1 : 5 000. Incidence deficitu je pravděpodobně podhodnocena, protože funkční testy jsou nedostatečně senzitivní na nejčastější mutace, které se vyskytují v české populaci (např. Leu99Phe, Arg47His). Diagnóza vrozeného deficitu musí být založena na opakovaném funkčním stanovení AT, rodinném screeningu a dohledání kauzální mutace. Vrozený deficit AT dělíme na kvantitativní (typ I) nebo kvalitativní (typ II). Typ II je dále rozdělen na velmi častý a méně trombogenní typ IIb (IIHBS – heparin binding site), který je způsoben defektem v místě, v němž se váže heparin, na typ IIa (IIRS – reactive site), který je způsoben mutacemi v místě vazby trombinu.
Laboratorní diagnostika:
- hlavní specifický test: vyšetření funkce AT (na principu FXa)
- dodatečné testy k vyšetření subtypu AT: vyšetření funkce AT (na principu FIIa) a AT : Ag, genetické stanovení kauzální mutace
Deficit proteinu C
Protein C (PC) je vitamin K dependentní serinová proteáza. Po aktivaci trombinem na aktivovaný protein C (APC) funguje jako přirozený inhibitor koagulace. APC má relativně dlouhý poločas v cirkulaci (20 min). Inhibuje koagulační kaskádu spolu se svým kofaktorem proteinem S tím, že specificky štěpí v několika místech FVIIIa a FVa, čímž degradují jejich aktivitu v koagulaci.
Incidence deficitu PC se pohybuje v normální populaci od 1 : 200 do 1 : 500. Přibližně u 1–4 narozených dětí na milion se vyskytuje homozygotní nebo složený heterozygotní deficit, který se projevuje po narození závažnými trombózami, které jsou bez léčby (koncentrát PC, APC) většinou fatální. Vrozený deficit PC dělíme na typ kvantitativní (typ I) a kvalitativní (typ II), přičemž typ I je nejčastější (85 % případů).
Laboratorní diagnostika:
- hlavní specifický test: vyšetření funkce PC (chromogenní metodika)
- dodatečné testy k vyšetření subtypu PC: genetické stanovení kauzální mutace a PC : AG
Deficit proteinu S
Protein S (PS) je vitamin K dependentní glykoprotein. Jako kofaktor APC se podílí na přirozené inhibici generace trombinu v koagulaci. Vyskytuje se ve volné formě a též vázaný do komplexu s C4BP. Předpokládá se, že funkci inhibitoru koagulace má volný PS. Nedávno bylo zjištěno, že PS zasahuje též do inhibice iniciace koagulace. PS slouží též jako kofaktor TFPI (tissue factor pathway inhibitor) při interakci TFPI a FXa. Incidence deficitu PS se pohybuje v normální populaci od 1 : 500 do 1 : 1 000. Velmi vzácně se vyskytuje homozygotní nebo složený heterozygotní deficit, který se projevuje závažnými trombózami po narození, které jsou bez léčby většinou fatální. Tento stav je často doprovázen závažnou retinopatií. Vrozený deficit PS dělíme na 3 subtypy: typ I (deficit PS-C4BP i PS) a III (deficit PS) kvantitativní defekty a typ II kvalitativní defekt.
Laboratorní diagnostika:
- hlavní specifický test: vyšetření funkce PS (koagulační metodika)
- dodatečné testy k vyšetření subtypu PS: volný PS (LIA metoda), PS : Ag a genetické stanovení kauzální mutace
Molekulárně genetické trombofilní markery
Faktor V Leiden (rs6025) je záměnná mutace jediného nukleotidu G za A v pozici 1 691 genu pro FV, která má za následek záměnu Arg506Gln. Způsobuje rezistenci k aktivovanému proteinu C (APC R) tím, že dochází k pomalejší degradaci FVa a FVIIIa. Prevalence heterozygotních nosičů této mutace v Evropě je mezi zdravou populací mezi 2–15 %. V asijské a africké populaci je to jen 0–2 %. V české populaci jsme nalezli 7 % heterozygotních nosičů u zdravé populace a 34 % nosičů mutace u nemocných s žilní trombózou ve věku do 45 let. FV Leiden je nejčastějším rizikovým faktorem pro vznik žilní trombózy. Riziko vzniku trombózy se zvyšuje 10krát u homozygotních nosičů mutace.
Laboratorní diagnostika:
hlavní specifický test: vyšetření APCR (koagulační nebo chromogenní metodika), genetické stanovení mutace – důležitá je informace, zda se jedná o heterozygota nebo homozygota pro mutaci.
Mutace v genu pro protrombin FII20210A (rs1799963) se nachází v 3’ nepřepisované části genu pro trombin. Vede k větší stabilitě mRNA a mírně zvýšené hladině protrombinu v krvi. Heterozygotní nosiči mají 2–3krát zvýšené riziko vzniku žilní trombózy, jejich zastoupení ve zdravé populaci je velmi rozdílné, podobně jako je to u FV Leiden. V české populaci jsme nalezli 3 % heterozygotních nosičů u zdravé populace a 10 % nosičů mutace u nemocných s žilní trombózou ve věku do 45 let.
Laboratorní diagnostika:
- hlavní specifický test: genetické stanovení mutace – důležitá je informace, zda se jedná o heterozygota nebo homozygota pro mutaci.
Získané trombotické rizikové faktory
Nejdůležitější získaným trombotickým rizikovým faktorem jsou antifosfolipidové protilátky.
Antifosfolipidové protilátky
Antifosfolipidové protilátky (APA) jsou heterogenní skupina protilátek, která je zaměřena proti proteinům vázaným na negativně nabité fosfolipidy. Dlouhodobá přítomnost APA je průkazem antifosfolipidového syndromu. Laboratorní kritéria antifosfolipidového syndromu jsou: přítomnost lupus antikoagulans (LA), přítomnost antikardiolipinových protilátek (ACLA) a přítomnost anti-β2-glykoproteinu I (anti-β2-GPI). Pro potvrzení APS stačí opakovaná pozitivní přítomnost 2 z těchto 3 markerů. LA postihuje 1–2 % populace, ale pouze u 15–20 % protilátky přetrvávají delší dobu. Je silně spjat s rizikem trombózy a reprodukčními ztrátami.
Laboratorní diagnostika lupus antikoagulans:
- screeningový test: prodloužené APTT citlivé na LA, korekční testy APTT s normální plazmou.
Typickým znakem LA je, že korekční směsné testy nejsou časově a teplotně závislé na rozdíl od přímého inhibitoru koagulačního faktoru.
Doporučený postup vyšetření probíhá ve 3 krocích:
- screeningový test citlivý na LA (APTT, dRVVT) má prodloužený čas
- směsné testy s normální plazmou měřené sreeningovými testy (APTT, dRVVT) zůstávají prodloužené
- přidání fosfolipidů saturuje protilátky a časy testů se zkracují, až normalizují
Vyšetření ACL a anti-β2GPI se provádějí ELISA metodami.
Seznam užitých zkratek:
ACLA – antikardiolipinové protilátky ADP – adenozindifosfát APA – antifosfolipidové protilátky APC – aktivovaný protein C APCR – rezistence k aktivovanému proteinu C APTT – aktivovaný parciální tromboplastinový čas, test na vnitřní cestu koagulace AT – antitrombin AT : Ag – kvantitativní množství antitrombinu C4BP – vazebný protein komplementárního faktoru 4 DD – D-dimery DIC – diseminovaná intravaskulární koagulace DOAC – nová antikoagulancia na principu přímé inaktivace aktivovaných proteinů dRVVT – test s hadím jedem Russellovy zmije FBG – fibrinogen FII-FXIII – koagulační faktory 2–13 FIX : Ag – kvantitativní množství koagulačního faktoru 9 FIX : C – aktivita koagulačního faktoru 9, měřeno koagulační metodou FM – fibrinové monomery FVIII : C – aktivita koagulačního faktoru 8, měřeno koagulační metodou FVIII : Cr – aktivita koagulačního faktoru 8, měřeno chromogenní metodou HUS – hemolyticko-uremický syndrom ISTH – Mezinárodní společnost pro trombózu a hemostázu/International Society on Thrombosis and Hemostasis LA – lupus antikoagulans LIA – latexově zesílená imunochemická metoda PC – protein C PC : Ag – kvantitativní množství PC PFA 100 – analyzátor funkce krevních destiček (výrobní číslo 100) PS – protein S PS : Ag – kvantitativní množství PS PT – protrombinový čas, test na vnější cestu koagulace TFPI – inhibitor cesty tkáňového faktoru TGT – trombinový generační čas TM – trombomodulin tPA – tkáňový plazminogen aktivátor TT – trombinový čas TTP – trombotická trombocytopenická purpura VWF – von Willebrandův faktor VWF : Ag – kvantitativní množství VWF VWF : CB – aktivita VWF vzhledem k vazbě na kolagen VWF : FVIIIB – vazebná schopnost VWF vázat FVIII VWF : RCo – aktivita VWF vzhledem k vazbě na Ib glykoprotein na krevních destičkách VWF : RIPA – vazba VWF na krevní destičky indukovaná ristocetinem VWFpp – propeptid VWF VWCH – von Willebrandova choroba
RNDr. Ingrid Hrachovinová, Ph.D.
Ústav hematologie a krevní transfuze, Praha
Doručeno do redakce 7. 3. 2018
Přijato po recenzi 17. 3. 2018
Sources
- Kitchens KS, Konkle BA, Kessler CM. Consultative Haemostais and Thrombosis. 2nd ed. Saunders: 2007: Chapter 1 : 3–15. ISBN 978–1416024019.
- Key N, Makris M, Lillicrap D (eds). Practical haemostasis and thrombosis. 3rd ed. Wiley-Blackwell: 2017: Chapter 2 : 7–16. ISBN 978–1118344712.
- Colman RW, Marder VJ, Clowes AW (eds) et al. Haemostasis and Thrombosis: Basic Principles and Clinical Practice. 5th ed. LWW: 2005: Chapter 27 : 1147–1158. ISBN 978–0781749961.
- Sadler JE,Budde U, Eikenboom JC et al. Update on the pathophysiology and classification of von Willebrand disease: a report of the Subcommittee on von Willebrand Factor. J Thromb Haemost 2006; 4(10): 2103–2114. Dostupné z DOI: <http://dx.doi.org/10.1111/j.1538–7836.2006.02146.x>.
- Ng C, Motto DG, Di Paola J. Diagnostic approach to von Willebrand disease. Blood 2015; 125(13): 2029–2037. Dostupné z DOI: <http://dx.doi.org/10.1182/blood-2014–08–528398>.
- Tosetto A, Castaman G, Plug I et al. Prospective evaluation of the clinical utility of quantitative bleeding severity assessment in patients referred for hemostatic evaluation. J Thromb Haemost 2011; 9(6): 1143–1148. Dostupné z DOI: <http://dx.doi.org/10.1111/j.1538–7836.2011.04265.x>.
- Favaloro JE, Bonar RA, Meiring M et al. Evaluating errors in the laboratory identification of von Willebrand disease in the real world. Thromb Res 2014; 134(2): 393–403. Dostupné z DOI: <http://dx.doi.org/10.1016/j.thromres.2014.05.020>.
- Peyvandi F, Palla R, Menegatti M et al. Coagulation factor activity and clinical bleeding severity in rare bleeding disorders: results from the European Network of Rare Bleeding Disorders. J Thromb Haemost 2012; 10(4): 615–621. Dostupné z DOI: <http://dx.doi.org/10.1111/j.1538–7836.2012.04653.x>.
- Castellone DD, Adcock DM. Factor VIII activity and inhibitor assays in the diagnosis and treatment of hemophilia A. Semin Thromb Hemost 2017; 43(3): 320–330. Dostupné z DOI: <http://dx.doi.org/10.1055/s-0036–1581127>.
- Goodeve AC, Pavlova A, Oldenburg J. Genomics of bleeding disorders. Haemophilia 2014; 20(Suppl 4): S50-S53. Dotupné z DOI: <http://dx.doi.org/10.1111/hae.12424>.
- Montagnana M, Lippi G, Danese E. An overview of thrombophilia and associated laboratory testing. Methods Mol Biol 2017; 1646 : 113–135. Dostupné z DOI: <http://dx.doi.org/10.1007/978–1-4939–7196–1_9>.
- Thachil J, Lippi G, Favaloro EJ. D-Dimer testing: laboratory aspects and current issues. Methods Mol Biol 2017; 1646 : 91–104. Dostupné z DOI: <http://dx.doi.org/10.1007/978–1-4939–7196–1_7>.
- Wada H, Matsumoto T, Yamashita Y. Diagnosis and treatment of disseminated intravascular coagulation (DIC) according to four DIC guidelines. J Intensive Care 2014; 2(1): 15. Dostupné z DOI: <http:// dx.doi.org/10.1186/2052–0492–2-15>.
- Moll S. Thrombophilia: clinical-practical aspects. J Thromb Thrombolysis 2015; 39(3): 367–378. Dostupné z DOI: <http://dx.doi.org/10.1007/s11239–015–1197–3>.
- Pengo V, Tripodi A, Reber et al. Update of the guidelines for lupus anticoagulant detection. Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the International Society on Thrombosis and Haemostasis. J Thromb Haemost. 2009; 7(10): 1737–1740. Dostupné z DOI: <http://doi: 10.1111/j.1538–7836.2009.03555.x>.
- Stevens SM, Woller SC, Bauer KA et al. Guidance for the evaluation and treatment of hereditary and acquired thrombophilia. J Thromb Thrombolysis 2016; 41(1): 154–164. Dostupné z DOI: <http://dx.doi.org/10.1007/s11239–015–1316–1>.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2018 Issue 5
Most read in this issue
- Autoimunitní hemolytická anémie
- Diferenciální diagnostika anémií
- Vrozené a získané krvácivé stavy
- Hemoglobinopatie