
Granulocytopenie
Granulocytopenia
Granulocytopenia is defined as a decrease of peripheral blood granulocytes below lower limit of normal range. Patients with severe granulocytopenia – agranulocytosis exhibit < 0.5 × 109/l granulocytes in peipheral blood. Granulocytopenia may result from congenital or acquired defective production of granulocyte precursors or it may be a consequence of increased destruction of mature granulocytes, most frequently caused by immune mechanisms. Investigation of origin of granulocytopenia must be connected with exclusion of etiological agents causing secondary neutropenia (infections, autoimmune disorders, drugs, LGL syndrome). Patients with > 0.5 × 109/l of granulocytes usually do not exhibit clinical symptoms unless they do not suffer from a concomitant disease (especially immunodeficiency). Patients with severe granulocytopenia are indicated for supportive treatment and for administration of G-CSF. Children with severe congenital neutropenia (SCN) are at risk of later development of MDS or AML and are candidates for SCT when signs of disease progression appears.
Key words:
diagnosis – granulocytopenia – growth factors – pathogenesis – transplantation –treatment
Authors:
Jaroslav Čermák 1,2
Authors‘ workplace:
Ústav klinické a experimentální hematologie 1. LF UK, Praha
1; Ústav hematologie a krevní transfuze, Praha
2
Published in:
Vnitř Lék 2018; 64(5): 520-525
Category:
Overview
Granulocytopenie je definována jako snížení počtu granulocytů v periferní krvi pod dolní hranici normy. Při poklesu počtu neutrofilů < 0,5 × 109/l mluvíme o těžké granulocytopenii – agranulocytóze. Nedostatek granulocytů může být důsledkem vrozené či získané poruchy tvorby granulocytárních prekurzorů, zvýšený zánik granulocytů vzniká nejčastěji na podkladě imunitních mechanizmů. Při hledání příčiny granulocytopenie je třeba odlišit celou řadu příčin vedoucích k sekundární granulocytopenii (infekce, systémová onemocnění, léky, LGL syndrom). Nemocní s počtem neutrofilů > 0,5 × 109/l většinou nemají klinické příznaky a nevyžadují léčbu, pokud není přítomno komplikující onemocnění, zejména imunodeficit. U těžké granulocytopenie je indikováno krom podpůrné léčby podání G-CSF. Syndromy těžké kongenitální neutropenie (severe congenital neutropenia – SCN) jsou spojeny s rizikem rozvoje MDS a AML v pozdějším věku a nemocní jsou při známkách progrese onemocnění indikováni k transplantaci krvetvorných buněk (stem cell transplantation – SCT).
Klíčová slova:
diagnostika – granulocytopenie – léčba – patogeneze – růstové faktory – transplantace
Úvod
Granulocytopenii definujeme jako snížení počtu granulocytů (neutrofilů, eozinofilů, bazofilů) pod dolní hranici normy, jako neutropenii označujeme snížený počet neutrofilů (segmentů, tyčí). Nemocní s mírnou neutropenií mají počty neutrofilů mezi 1,0–1,5 × 109/l, středně těžká neutropenie je spojena s počty neutrofilů mezi 0,5–1,0 × 109/l, těžká neutropenie je definována poklesem počtu neutrofilů < 0,5 × 109/l [1]. Nemocní s počtem neutrofilů > 0,5 × 109/l většinou nemají klinické příznaky vyplývající z neutropenie, pokud není přítomno jiné komplikující onemocnění, zejména imunodeficit či celkově nepříznivý klinický stav.
Etiologie a patogeneze
Nedostatek granulocytů může být dán snížením tvorby či zvýšeným zánikem. Snížená tvorba bývá způsobena absolutním poklesem produkce mladých forem granulocytů či tvorbou defektních prekurzorů podléhajících zvýšené apoptóze.
Vrozené poruchy tvorby granulocytů vznikají na podkladě mutace genů vedoucí k poruše proliferace granulocytární řady a k předčasnému zániku granulocytárních prekurzorů. Nejčastější příčinou těžké vrozené neutropenie (severe congenital neutropenia – SCN) bývá mutace genu pro elastázu neutrofilů – ELANE gen, vzácněji o mutace jiných genů (HAX1, WAS). Mutace těchto genů vede k těžké poruše proliferace granulocytů se zástavou zrání na úrovní promyelocytů. Mechanizmus vzniku granulocytopenie není zcela objasněn, předpokládá se, že kumulace volných proteinů v buňce vede k abnormálnímu buněčnému stresu, jenž indukuje apoptózu prekurzorů [2]. Klinické projevy mutací ELANE a HAX1 genu odpovídají tzv. Kostmanovu syndromu [3] s recidivujícími infekty postihujícími kůži a sliznice (dutina ústní, nos, uši). Kromě klasické chronicky probíhající těžké vrozené neutropenie mohou mutace ELANE genu způsobovat i tzv. cyklickou neutropenii projevující se cykly neutropenie v intervalu asi 21 dnů. Shwachmanův-Diamondův syndrom vzniká v důsledku mutace SBDS genu. Tento gen je nutný pro normální metabolizmus RNA, funkci a vazbu ribosomálních podjednotek, což jsou základní procesy nutné pro přežití granulocytárních prekurzorů [4]. Mutace SBDS genu je spojena se zvýšeným zánikem granulocytárních prekurzorů, selháním kostní dřeně s pancytopenií v krevním obraze, atrofií pankreatu s lipomatózou a retardací růstu s deformitami skeletu. K nepříznivé prognóze nemocných přispívá častý rozvoj myelodysplastického syndromu (MDS) a následný přechod do akutní myeloidní leukemie (AML) [5]. Ne zcela jasným mechanizmem může vznikat neutropenie u některých syndromů spojených s imunodeficitem, např. u běžného variabilního deficitu imunity (common variable immunodeficiency – CVID) či agamagblobulinemie vázané na X chromozom (XLA) [6].
Získané poruchy tvorby granulocytů jsou nejčastěji součástí syndromů selhání kostní dřeně – aplastické anémie (AA), myelodysplastického syndromu (MDS) či paroxyzmální noční hemoglobinurie (PNH). Na vzniku granulocytopenie se může podílet přímá toxicita způsobená určitou noxou, která vede k zániku granulocytárních prekurzorů, nebo může poškození vést k expresi abnormálních antigenů na povrchu granulocytů. To vede k aktivaci imunitních mechanizmů, sekreci cytokinů aktivujících apoptózu a k aktivaci cytotoxických T-lymfocytů namířených proti prekurzorům granulocytů (obr). Obdobným mechanizmem může vznikat granulocytopenie při některých infekcích (hepatitidy, HIV infekce) či po podání některých léků. Granulocytopenie může být i projevem některých dřeňových lymfoproliferací, zvláště jsou-li spojeny s hypoplazií a fibrózou dřeně (např. leukemie z vlasatých lymfocytů).

Zvýšený zánik granulocytů vzniká nejčastěji působením imunitních mechanizmů. Předpokládá se, že na vzniku autoimunitní granulocytopenie se podílejí obdobné faktory jako u autoimunitní hemolýzy, tj. porucha prezentace autoantigenů imunokompetentním buňkám a porucha tolerance autoreaktivních T-lymfocytů. Důsledkem je indukce přeměny Th0 na Th1-lymfocyty a stimulace tvorby protilátek B-lymfocyty [7]. Protilátky se váží zejména na HNA1 (méně často na HN2 či HN5) antigen na povrchu granulocytů [8]. Následně dochází k fagocytóze granulocytů s navázanou protilátkou makrofágem po vazbě protilátky s Fcγ3b receptorem makrofágů. Komplex antigen-protilátka může vést i k aktivaci komplementu spojené s opsonizací s adherencí neutrofilů [9]. Aktivace imunitního systému rovněž vede k sekreci cytokinů aktivujících apoptózu a k aktivaci cytotoxických T-lymfocytů (viz výše). Granulocytopenie může vznikat i v důsledku zvýšeného odsunu z cirkulace (např. v důsledku zvýšeného vychytávání v místě zánětu) či marginace v orgánech (např. ve slezině). Níže je znázorněno v současnosti nejčastěji používané dělení granulocytopenií/neutropenií.
Základní dělení granulocytopenií/neutropenií podle příčiny
- kongenitální neutropenie (Kostmannův syndrom, Shwachmanův-Diamondův syndrom, vrozené imunodeficitní stavy aj)
- získané neutropenie při postižení kmenové buňky (AA, MDS, PNH)
- imunitně zprostředkované neutropenie (aloimunitní neutropenie novorozenců, autoimunitní neutropenie – systémová onemocnění)
- neutropenie způsobená léky
- neutropenie při infekci
- neutropenie při poruchách metabolizmu (deficit vitaminu B12)
- neutropenie při LGL leukemii
- chronická idiopatická neutropenie
Diagnostika a diferenciální diagnostika
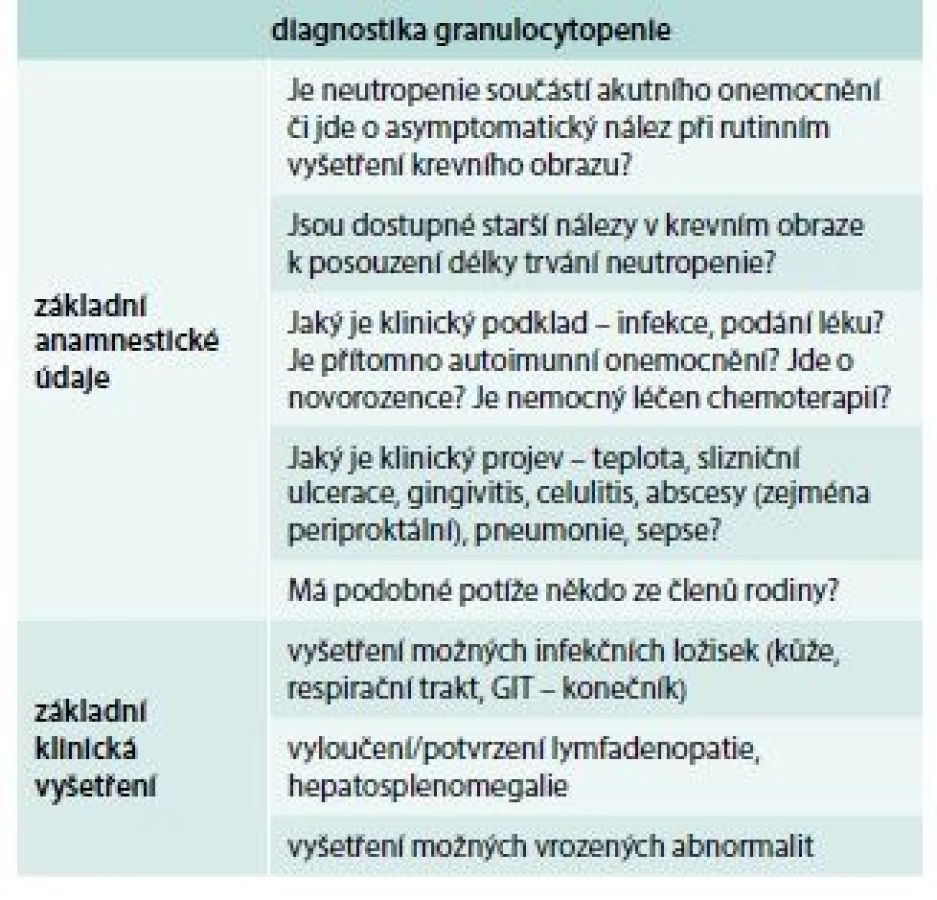
Schéma znázorňuje algoritmus vyšetřování nemocných s granulocytopenií [1]. V prvé fázi se vždy snažíme zjistit, zda granulocytopenie není projevem nějakého nehematologického onemocnění (systémová onemocnění, infekce – hepatitida, CMV infekce, HIV infekce, podání léku) [10]. Pokud se některá z těchto příčin nepotvrdí, je třeba provést vyšetření kostní dřeně (sternální punkce a trepanobiopsie), toto vyšetření nám může pomoci diagnostikovat některou z poruch kmenové krvetvorné buňky (AA, MDS), infiltraci dřeně lymfoproliferací (leukemie z vlasatých lymfocytů), u dětí je třeba se zaměřit zejména u dlouhotrvající neutropenie s hypoplazií granulopoézy v kostní dřeni na některý z vrozených syndromů spojených s granulocytopenií (rodinná anamnéza, molekulárně genetické vyšetření). Pokud je nález v kostní dřeni nesignifikantní, je vhodné znovu pátrat po možné spojitosti s infekcí či s podáváním léku a vyšetřit možnou přítomnost protilátek proti granulocytům. Přítomnost granulocytárních protilátek potvrzuje diagnózu autoimunitní granulocytopenie, negativita testu ale autoimunitní příčinu zcela nevylučuje. V případě nepřítomnosti granulocytárních protilátek je vhodné vyšetření periferní krve pomocí průtokové cytometrie k vyloučení tzv. leukemie z velkých granulárních lymfocytů (LGL leukemie). V tab. 1 jsou uvedeny základní anamnestické údaje a klinická vyšetření, která by neměla být opominuta při podezření na granulocytopenii, v tab. 2 je pak souhrn základních laboratorních vyšetření užívaných v diagnostice a diferenciální diagnostice granulocytopenie


K vyšetření granulocytárních protilátek je používána řada testů: granulocytární cytotoxický test (GCT), granulocytární aglutinační test (GAT), granulocytární imunofluorescenční test (GIFT) přímý či nepřímý s detekcí pomocí mikroskopu či průtokového cytometru a MAIGA test (imobilizace granulocytárních antigenů pomocí specifické monoklonální protilátky), žádný z testů však není schopen identifikovat všechny klinicky relevantní protilátky.
Základní klinické jednotky a léčba
Léčba granulocytopenie se odvíjí od klinických projevů a průběhu onemocnění. Určujícími faktory jsou hloubka granulocytopenie, rychlost jejího vzniku a délka trvání, vyvolávající příčina a přítomnost komorbidit (zejména imunodeficitu). Při mírné a středně těžké neutropenii (počet neutrofilů > 0,5 × 109/l) většinou chybí klinické příznaky, pokud není přítomen současný imunodeficit. Příznaky a průběh bývají závažnější při stavech se sníženou tvorbou granulocytů než u stavů, v nichž je přítomen jejich zvýšený zánik.
Nemocní s počtem neutrofilů > 0,5 × 109/l většinou nevyžadují léčbu, pokud nejsou přítomny komplikace. Pro nemocné s počtem neutrofilů < 0,5 x 109/l je lékem volby granulocyty stimulující faktor (G-CSF), zejména pokud je přítomna komplikující infekce. Iniciální dávka většinou činí 1–3 μg/kg/den, tuto dávku lze zvyšovat až na 20–30 μg/kg/den, při dobrém efektu je nutno klesat na minimální udržovací dávku. Pokud není trvalejší efekt dávek 10 μg/kg/den a vyšších, je nutná jiná léčba, zejména u kongenitální neutropenie pak alogenní transplantace krvetvorných buněk (SCT). G-CSF má efekt i u autoimunitní neutropenie. Podpůrná léčba spočívá v podávání širokospektrých antibiotik při komplikující infekci, podávání imunosupresiv s doporučuje v rámci léčby vyvolávajícího onemocnění, pokud vyžaduje imunosupresi, paušální podávání kortikosteroidů se nedoporučuje.
Vrozené syndromy selhání krvetvorby spojené s granulocytopenií
Těžká kongenitální neutropenie (SCN) se projevuje recidivujícími infekty postihujícími kůži a sliznice (dutina ústní, nos, uši). Metodou volby pro nemocné je podávání G-CSF, dávkování a frekvence podávání by měly vést k trvalému vzestupu počtu granulocytů > 1,0 × 109/l, tento počet by měl být dostatečný pro prevenci vzniku recidivujících infektů. Jako rezistence na léčbu je považována nutnost zvyšování dávky G-CSF nad 120 μg/kg/den [3]. Část těchto nemocných může odpovědět na kombinaci G-CSF a SCF (stem cell faktor). Podávání G-CSF prodloužilo délku života nemocných se SCN o 20 let [11], nicméně, analýza dlouhodobého podávání G-CSF 374 nemocným se SCN prokázala zvýšenou incidenci AML (rozvoj u 22 % nemocných během 15 let) [12]. Incidence byla vyšší u nemocných, kteří dostávali > 8 μg G-CSF/kg/den – 34 % vs 15 % u nemocných s nižší denní dávkou. Nebyl pozorován rozdíl mezi typy mutace ELANE genu, analýza nemocných z německého SCN registru však ukázala vysokou incidenci mutace receptoru pro G-CSF (CSF3R) u nemocných se SCN léčených G-CSF, u kterých se rozvinul MDS/AML (78 % nemocných vs 35 % nemocných, u nichž nedošlo k progresi směrem k AML) [13]. Negativní význam přítomnosti mutace CSF3R je zřejmě dán schopností autoaktivace a hyperproliferace progenitorů nezávisle na růstových faktorech díky poruše endocytózy receptoru, k prolongované aktivaci STAT5 a zvýšené produkci volných kyslíkových radikálů [11]. Recentní mutační analýza nemocných z Mezinárodního SCN registru [14] ukázala vysokou incidenci mutace RUNX1 genu (64,5 %) u nemocných se SCN, 80,5 % těchto nemocných mělo současně mutaci CSF3R. Mechanizmus možné synergie obou mutací je zkoumán, mutace RUNX1 je zřejmě následnou mutací aktivující funkci tohoto genu jako transkripčního faktoru. Alogenní transplantace krvetvorných buněk (SCT) je alternativním léčebným přístupem k nemocným nereagujícím na podávání G-CSF, je však třeba vždy zvážit rizika spojená se SCT (i když je dnes doporučován redukovaný přípravný režim) s riziky vyplývajícími z konkrétního stavu nemocného. Nemocní se Shwachmanovým-Diamondovým syndromem nemusí mít výraznější klinické potíže, při těžší formě granulocytopenie je indikováno intermitentní podávání G-CSF. Nemocné je však třeba pečlivě sledovat včetně opakovaného vyšetření kostní dřeně, progredující cytopenie se známkami dysplazie v kostní dřeni je indikací k alogenní SCT, zejména pokud dochází k nárůstu počtu blastů ve dřeni a jsou přítomny nepříznivé změny karyotypu (monosomie 7, delece 7q) [4]. Alogenní SCT je metodou volby i u SCN při známkách progrese směrem k AML.
Autoimunitní granulocytopenie
Jak již bylo výše zmíněno, autoimunitní granulocytopenie je způsobena tvorbou protilátek proti granulocytům, na jejím vzniku se podílí i zvýšená sekrece cytokinů indukujících apoptózu cílových buněk (Fas ligand) a tvorbu cytotoxických T-lymfocytů. Autoimunitní granulocytopenie se nejčastěji vyskytuje u systémových onemocnění, může být přítomna až u 50 % nemocných se systémovým lupus erythematodes, při němž bývá často spojena s cytopenií v jiných řadách (anémie, trombocytopenie, lymfopenie). Granulocytopenie na autoimunitním podkladě může být přítomna až u 30 % nemocných s revmatoidní artritidou a Sjøgrenovým syndromem, hloubka cytopenie často souvisí s tíží choroby [15]. Kromě léčby základního onemocnění je možno u těžších forem granulocytopenie podávat G-CSF, metotrexát a cyklosporin A ovlivňují tvorbu Fas ligand. U některých nemocných s revmatoidní artritidou však může podávání G-CSF zhoršovat kloubní obtíže.
Neutropenie při infekci
Neutropenie může vznikat u infekce toxickým poškozením prekurzorů, v důsledku inkorporace viru do DNA dojde k apoptóze postižení buňky. Tímto způsobem vzniká neutropenie u infekční hepatitidy, infekce EBV či HIV. Zvýšená sekrece cytokinů u bakteriálních infekcí vede k aktivaci komplementu s indukcí migrace neutrofilů do místa zánětu, u některých jiných infekcí (Rickettsie, Bartonella, některé viry) může aktivace komplementu indukovat adhezi neutrofilů k endotelu poškozenému infekcí. U chronických infekcí (tyfus, brucelóza, malárie) může docházet k marginaci neutrofilů ve slezině. Léčba granulocytopenie souvisí s léčbou vyvolávajícího infektu, u těžké granulocytopenie lze podávat G-CSF [8].
Neutropenie indukovaná léky
Incidence neutropenie vyvolané léky se pohybuje mezi 0,3–1,2/100 000 obyvatel. Toxický účinek léku, který vede k poškození proteosyntézy nebo buněčného dělení, je závislý na jeho dávce a vlastní mechanizmem je nejčastěji indukce tvorby volných hydroxylových radikálů či produkce toxických metabolitů s následnou apoptózou poškozené buňky. V případě idiosynkrazie jde o alergickou reakci, která není závislá na dávce léku a většinou se jedná o indukci tvorby protilátek v důsledku modifikace povrchových antigenů granulocytů či o tvorbu imunitních komplexů. Nejčastěji bývá těžká forma neutropenie popisována po podání některých antibiotik a chemoterapeutik (chloramfenikol, trimetoprim-sulfametoxazol, vankomycin), sulfasalazinu, tiklopidinu, prokainamidu, klozapinu a některých tyreostatik [10]. Neutropenie odeznívá většinou za 4–7 dní po vysazení léku, současně je třeba podávat podpůrnou léčbu a v těžších případech G-CSF.
Neutropenie u LGL syndromu
LGL syndrom se dříve nazýval leukemie z velkých granulárních lymfocytů. Jde o primárně reaktivní polyklonální či oligoklonální expanzi cytotoxických T-lymfocytů, která vzniká jako reakce na noxu (antigenní podnět), následná monoklonální expanze je zřejmě dána overexpresí STAT3, jenž má antiapoptotický efekt a aktivací Src kináz [16]. Neutropenie u LGL syndromu je dána jednak přímou cytotoxicitou zprostředkovanou cytotoxickými T-lymfocyty či indukcí apoptózy neutrofilů produkovanými cytokiny (Fas-L, IFN, TNF). Onemocnění se často sdružuje s revmatoidní artritidou a je významná asociace s přítomností HLA-DR4. Léčba se odvíjí od stupně klinických příznaků, při těžším průběhu je ke korekci neutropenie indikován G-CSF, těžší formy LGL syndromu se léčí podáváním imunosupresiv (metotrexát, cyklofosfamid, cyklosporin A), zkouší se podávání inhibitorů STAT3 či Fas-L [10].
Chronická idiopatická neutropenie
Chronická idiopatická neutropenie postihuje většinou ženy mezi 20.–40. rokem věku. Jedná se o izolovanou neutropenii různého stupně, někdy společně s přítomností lymfopenie. Klinické příznaky závisí na stupni neutropenie, většinou jde o opakované epizody zvýšené teploty, bolesti v krku, únavu a bolesti ve svalech. Příčinou je zvýšená sekrece cytokinů (IFNγ, Fas) indukujících apoptózu neutrofilů [10]. Průběh je většinou benigní, u nemocných s výraznou granulocytopenií a známkami opakovaných infekcí je možno aplikovat G-CSF. Je nutno zdůraznit, že diagnózu chronické idiopatické neutropenie můžeme stanovit až po pečlivém a opakovaném vyloučení jiné příčiny chronické neutropenie, zejména systémového nemocnění, MDS či LGL syndromu.
doc. MUDr. Jaroslav Čermák, CSc.
Ústav hematologie a krevní transfuze, Praha
Doručeno do redakce 3. 12. 2017
Přijato po recenzi 15. 3 2018
Sources
- Palmblad J, Dufour C, Papadaki H. How we diagnose neutropenia in the adult and elderly patient. Haematologica 2014; 99(7): 1130–1133. Dostupné z DOI: <http://dx.oi.org/dx.doi.org/10.3324/haematol.2014.110288>.
- Horwitz MS, Corey SJ, Grimes HL et al. ELANE mutations in cyclic and severe congenital neutropenia: genetics and pathophysiology. Hematol Oncol Clin North Am 2013; 27 : 19–41. vii. Dostupné z DOI: <http://dx.doi.org/10.1016/j.hoc.2012.10.004>.
- Zeidler C, Welte K. Kostmann syndrome and severe congenital neutropenia. Semin Hematol 2002; 39(2): 82–88.
- Smith OP. Shwachman-Diamond syndrome. Semin Hematol 2002; 39(2): 95–102.
- Freedman M, Alter BP. Risk of myelodysplastic syndrome and acute myeloid leukemia in congenital neutropenias. Semin Hematol 2002; 39(2): 128–133.
- Cham B, Bonilla MA, Winkelstein J. Neutropenia associated with primary immunodeficiency syndromes. Semin Hematol 2002; 39(2): 107–112.
- Papadaki H, Eliopoulos AG, Kosteas T et al. Impaired granulocytopoiesis in patients with chronic idiopathic neutropenia is associated with increased apoptosis of bone marrow myeloid progenitor cells. Blood 2003; 101(7): 2591–2600. Dostupné z DOI: <http://dx.doi.org/10.1182/blood-2002–09–2898>.
- Palmblad JEW, von dem Borne AE. Idiopathic, immune, infectious, and idiosyncratic neutropenias. Semin Hematol 2002; 39(2): 113–120.
- Newburger PE, Dale DC. Evaluation and management of patients with isolated neutropenia. Semin Hematol 2013; 50(3): 198–206. Dostupné z DOI: <http://dx.doi.org/10.1053/j.seminhematol.2013.06.010>.
- Gibson C, Berliner N. How we evaluate and treat neutropenia in adults. Blood 2014; 124 : 1251–1258; quiz 1378. Dostupné z DOI: <http://dx.doi.org/10.1182/blood-2014–02–482612>.
- Touw IP. Game of clones: the genomic evolution of severe congenital neutropenia. Hematology Am Soc Hematol Educ Program 2015; 2015 : 1–7. Dostupné z DOI: <http://dx.doi.org/10.1182/asheducation-2015.1.1>.
- Rosenberg PS, Zeidler C, Bolyard AA et al. Stable long-term risk of leukaemia in patients with severe congenital neutropenia maintained on G-CSF therapy. Br J Haematol 2010; 150(2): 196–199. Dostupné z DOI: <http://dx.doi.org/10.1111/j.1365–2141.2010.08216.x>.
- Germeshausen M, Ballmaier M, Welte K. Incidence of CSF3R mutations in severe congenital neutropenia and relevance for leukemogenesis: results of a long-term study. Blood 2007; 109(1): 93–99. Dostupné z DOI: <http://dx.doi.org/10.1182/blood-2006–02–004275>.
- Skokowa J, Steinemann D, Katsman-Kuipers H et al. Cooperativity of RUNX1 and CSF3R mutations in severe congenital neutropenia: a unique pathway in myeloid leukemogenesis. Blood 2014; 123(14): 2229–2237. Dostupné z DOI: <http://dx.doi.org/10.1182/blood-2013–11–538025>.
- Starkebaum G. Chronic neutropenia associated with autoimmune disease. Semin Hematol 2002; 39(2): 121–127.
- Zhang D, Loughran TP Jr. Large granular lymphocytic leukemia: molecular pathogenesis, clinical manifestations, and treatment. Hematology Am Soc Hematol Educ Program 2012; 2012 : 652–659. Dostupné z DOI: <http://dx.doi.org/10.1182/asheducation-2012.1.652>.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2018 Issue 5
Most read in this issue
- Autoimunitní hemolytická anémie
- Diferenciální diagnostika anémií
- Vrozené a získané krvácivé stavy
- Hemoglobinopatie