
Vrozené a získané krvácivé stavy
Congenital and acquired bleeding disorders
Hemostasis can be characterized as an array of physiological mechanisms providing both blood fluidity in the intact blood vessels and hemostasis in the event of impaired continuity of the blood vessel wall. The impaired hemostatic balance may lead on the one hand to an increased tendency to bleed, either spontaneously or only in response to an external stimulus. At the opposite end of bleeding are thrombophilic conditions characterized by an increased tendency to blood coagulation and thereby to the development of venous or arterial thrombosis. The hemostatic balance is the result of normal functioning of the blood vessel wall, platelets and plasma agents which include the coagulation and fibrinolytic systems and their inhibitors. By coagulation we understand the process leading to the formation of fibrin networks including the controlled interaction of coagulation factors. It is a physiological process as opposed to thrombosis which can be defined as increased coagulation under pathological conditions.
Key words:
coagulation – coagulation factors – coagulopathy – hemophilia – inhibitors of coagulation factors deficiencies – purpura – thrombocytopathy – thrombocytopenia
Authors:
Peter Salaj
Authors‘ workplace:
Ústav hematologie a krevní transfuze, Praha
Published in:
Vnitř Lék 2018; 64(5): 547-558
Category:
Overview
Hemostázu lze charakterizovat jako soubor fyziologických mechanizmů zajišťujících jak fluiditu krve v intaktním cévním řečišti, tak i zástavu krvácení v případě poruchy kontinuity cévní stěny. Narušení hemostatické rovnováhy může vyústit na jedné straně ve zvýšený sklon ke krvácení, a to spontánně nebo jenom po vnějším podnětu. Protipólem krvácení jsou trombofilní stavy charakterizované zvýšenou tendencí ke krevnímu srážení, a tím ke vzniku žilních, nebo tepenních trombóz. Hemostatická rovnováha je výsledkem normální funkce cévní stěny, krevních destiček a činitelů plazmatických, zahrnujících systém koagulační, fibrinolytický a jejich inhibitory. Pojmem koagulace rozumíme proces vedoucí k formování fibrinových sítí, zahrnující kontrolovanou interakci koagulačních faktorů. Jedná se o fyziologický proces na rozdíl od trombózy, kterou lze definovat jako zvýšenou koagulaci za patologických podmínek.
Klíčová slova:
deficity koagulačních faktorů – hemofilie – inhibitory koagulačních faktorů – koagulace – koagulopatie – purpura – trombocytopatie – trombocytopenie
Úvod
Krvácivými stavy rozumíme chorobné stavy, u kterých dochází už po minimálním poranění, případně i spontánně k neúměrnému krvácení nebo k jeho protrahování. Porucha kteréhokoliv z hemostatických mechanizmů může vyústit v krvácivý stav. Z etiologického hlediska lze proto krvácivé stavy rozdělit do 3 základních skupin, a to na krvácivé stavy:
- z destičkových příčin – trombocytopenie a trombocytopatie
- z poruch plazmatické koagulace – koagulopatie
- z poruch cévní stěny – vaskulopatie
Trombocytopenie a trombocytopatie
S poruchou hemostatické funkce trombocytů se setkáváme jak při jejich poklesu (trombocytopenie je obecně definovaná jako snížený počet trombocytů < 140 × 109/l v periferní krvi), tak i při jejich funkční poruše (trombocytopatie). Někdy může být krvácivý stav výsledkem kombinace obou těchto chorobných mechanizmů (trombocytopenické trombocytopatie).
U většiny chorobných stavů spojených se sníženým počtem trombocytů je trombocytopenie jenom jedním z laboratorních nálezů, který je nutno ozřejmit z hlediska etiologie, klinického významu a rizika pro pacienta. Z etiopatogenetického hlediska k trombocytopeniím dochází v důsledku snížené produkce trombocytů, nebo při jejich zvýšené periferní destrukci, případně sekvestraci trombocytů v retikulo-endoteliálním systému.
Trombocytopenické purpury představují etiologicky nesourodou skupinu nemocí charakterizovaných krvácivou diatézou při různě hlubokém poklesu počtu trombocytů. I když obecně platí, že rozsah krvácivé diatézy narůstá s poklesem počtu trombocytů, prognóza pacienta a riziko krvácení jsou závislejší na typu onemocnění než na absolutním počtu trombocytů.
Trombocytopenie ze snížené trombocytární produkce
Představují nesourodou skupinu onemocnění charakterizovanou nedostatečnou nebo neefektivní trombopoézou. Příčinou insuficientní produkce trombocytů může být aplazie nebo infiltrace kostní dřeně, případně podávání léků s myelotoxickým účinkem (např. cytostatika, tiazidová diuretika, estrogeny). Tíže trombocytopenie, její délka a rozsah krvácení u pacientů jsou variabilní. Kompletní vyšetření kostní dřeně je nezbytnou součástí diferenciální diagnostiky těchto poruch.
Do této skupiny patří i velice raritní hereditární trombocytopenie, které jsou často falešně diagnostikovány a léčeny jako imunitní trombocytopenie. Většinou se jedná o izolované trombocytopenie, které bývají asociovány s morfologickými poruchami trombocytů. Záchyt mikrotrombocytů je např. typický pro Wiskottův-Aldrichův syndrom a trombocytopenii vázanou na chromozom X a makrotrombocyty nacházíme u Mayovy-Hegglinovy anomálie, Fechtnerova syndromu a Sebastianova syndromu. U těchto pacientů pozorujeme typický rodinný výskyt poruchy, celoživotní anamnézu trombocytopenie se zvýšeným sklonem ke krvácení.
Trombocytopenické purpury ze zvýšené sekvestrace trombocytů
U dospělého člověka je denní produkce trombocytů přibližně 150 × 109/l, přičemž až jedna třetina z tohoto množství je fyziologicky sekvestrována ve slezině a dvě třetiny kolují v periferní krvi. Vlivem splenomegalie může dojít k takové změně poměru sekvestrovaných a volně cirkulujících trombocytů, že ve slezině je kumulováno až 90 % celkového objemu. Kombinovaný záchyt sníženého počtu trombocytů a zvětšené sleziny je nutné objasnit z hlediska původu hypersplenizmu. Příčinou splenomegalie bývá nejčastěji nádorová infiltrace, infekce, venostáza nebo hemolýza.
Trombocytopenické purpury ze zvýšené trombocytární destrukce
Rozlišujeme 2 základní mechanizmy zvýšené trombocytární destrukce, imunitní a neimunitní.
K imunitní destrukci trombocytů dochází v důsledku vazby specifických nebo nespecifických protilátek na povrch krevních trombocytů, která vede k jejich zvýšenému imunologickému odstranění. Do této skupiny kromě idiopatické trombocytopenické purpury (ITP) patří i potransfuzní purpura, novorozenecká aloimunitní trombocytopenie a heparinem indukovaná trombocytopenie II. typu (HIT II). Trombocytopenie v důsledku tvorby antitrombocytárních protilátek může být součástí kteréhokoliv jiného systémového autoimunitního procesu.
K neimunitním trombocytopeniím ze zvýšené destrukce zařazujeme diseminovanou intravaskulární koagulaci (DIC), trombotickou trombocytopenickou purpuru (TTP) a hemolyticko-uremický syndrom (HUS). U těchto stavů je trombocytopenie následkem periferní konzumpce, hlavně v oblasti mikrocirkulace.
Trombotická trombocytopenická purpura
Trombotická trombocytopenická purpura (TTP) je těžké multisystémové onemocnění charakterizované vzájemnou kombinací pentády příznaků: mikroagiopatické hemolytické anémie, konzumpční trombocytopenie, fluktuujícího neurologického nálezu, teploty a poruchy renálních funkcí.
Onemocnění je spojeno s tvorbou okluzí v terminálních arteriolách a kapilárách, ke kterým dochází v důsledku deficitu proteázy (depolymerázy) štěpící multimery vWF (von Willebrandův faktor). Následkem toho se v plazmě pacientů objevují nenaštípané neobvykle velké multimery vWF (ULvWFM – Unusually Large vWF Multimers), které za podmínek zvýšeného smykového stresu (shear stress) indukují trombocytární hyperagregaci s následnou konzumpční trombocytopenií, hemolýzou a ischemizací postižených orgánů. Příčinou deficitu je buď vrozený nedostatek (familiární forma TTP), nebo specifický inhibitor (autoprotilátka) štěpící proteázy (získané formy TTP). Z tohoto hlediska TTP můžeme dělit na idiopatickou, familiární a sekundární formu. Zvýšený výskyt TTP je udáván v souvislosti s infekcí, těhotenstvím, u nádorových a autoimunitních nemocí, po cyklosporinu A, tiklopidinu, mitomycinu C a chininu.
Trombotické okluze mohou postihnout kterýkoliv orgán lidského těla, s predominantní alterací mozku, ledvin a srdce. Klasická pentáda příznaků se vyskytuje asi u 40 % pacientů. Ale kombinaci anémie a trombocytopenie nacházíme u všech nemocných.
Klinický obraz pacientů je variabilní, v závislosti na rozsahu a lokalizaci mikroangiopatické ischemizace. Hemoragická diatéza různé intenzity a rozsahu je typickým nálezem TTP. Neurologické abnormality mohou mít charakter bolestí hlavy a lehkých poruch chování přes zjevné motorické a senzorické deficity, epileptiformní stavy až po hluboké komatózní stavy. Postižení má tranzitorní charakter a obvykle je vlivem terapie kompletně reverzibilní. Nefrologické změny mohou mít charakter proteinurie, mikroskopické hematurie a azotemie. Těžká renální insuficience s potřebou dialýzy je vzácná. V případě rozsáhlejšího mikrotrombotického procesu může dojít k postižení kteréhokoliv orgánu s příslušnou klinickou symptomatologií.
V laboratorním nálezu pacientů dominuje hematologické postižení: trombocytopenie a Coombs-negativní mikroangiopatická hemolytická anémie se záchytem schistocytů. Hladina laktátdehydrogenázy (LDH) v séru je zvýšená nejen z důvodu hemolýzy, ale i v důsledku tkáňové ischemie. Základní koagulační nález v kontrastu s klinickým obrazem je v normě, nebo vykazuje jenom mírné odchylky. Onemocnění může probíhat akutně, s jednou fulminantní atakou. U jedné třetiny pacientů dochází po indukční terapii k recidivám TTP. Chronické formy s přetrvávajícími známkami onemocnění jsou často spojeny s jiným systémovým postižením.
Lékem volby u pacientů s TTP je plazmaferéza, kterou je nutno provádět denně až do normalizace počtu trombocytů, hemoglobinu, LDH a vymizení neurologického postižení. Ústup renálního postižení je pozvolnější. Iniciální terapie plazmou je indikovaná jenom na přechodnou dobu v případě nedostupnosti plazmaferéz. U získaných forem TTP, s pravděpodobnou autoimunitní patogenezí se terapie plazmou kombinuje s podáváním kortikoidů. V případě rezistence na daný terapeutický postup lze zintenzivnit plazmatické výměny (provádění 2krát denně, změna náhradního roztoku z plazmy na K-plazmu) nebo imunosupresivní terapii (podávaní vinkristinu, cyklofosfamidu, nebo anti CD20).
Závažnost správné diagnostiky a terapie TTP zvyšuje i fakt, že mortalita neléčených pacientů je 90 %. V současnosti při správném terapeutickém postupu u 70–85 % pacientů dochází k navození kompletní remise.
Hemolyticko-uremický syndrom
Hemolyticko-uremický syndrom (HUS) je onemocnění klinicky podobné TTP, charakterizované kombinací konzumpční trombocytopenie, mikroagiopatické hemolytické anémie s dominantním renálním postižením.
Získané formy HUS se predilekčně objevují u dětí, u nichž představují nejčastější příčinu akutního selhání ledvin. Vyskytuje se ve 3 základních formách. Pro endemickou formu onemocnění jsou typické krvavé průjmy v předchorobí spojené s infekcí bakterií Escherichia coli 0157:H7 nebo jinou bakterií produkující verotoxin. Jejich vlivem dochází k endoteliálnímu postižení hlavně v oblasti ledvin. K přenosu infekce dochází přímým kontaktem s infikovanými lidmi a zvířaty nebo potravinami. Tato forma představuje až 90 % případů HUS u dětí.
Sporadická forma je klinicky podobná TTP, bez krvavých průjmů a s obrazem multiorgánového postižení. Často se objevuje sekundárně – např. po porodu, po užívání některých léků (mitomycin C, cyklosporin A, takrolimus) nebo po transplantaci kmenových buněk.
Familiární forma představuje 5–10 % případů HUS a je provázena ve srovnání s 3–5% mortalitou u získaných forem vysokou mortalitou (54%). V klinickém obrazu dominují recidivující ataky onemocnění v různých intervalech.
Laboratorní nález je u HUS velice podobný nálezu u TTP – hluboká trombocytopenie, hemolytická anémie s dominujícími známkami renálního postižení. Na rozdíl od TTP jsou u pacientů s HUS hladiny depolymerázy vWF normální. U familiárních forem HUS byl opakovaně prokázán deficit H faktoru svědčící pro poruchu regulace komplementu.
Diseminovaná intravaskulární koagulopatie
Diseminovanou intravaskulární koagulopatii (DIC) lze charakterizovat jako koagulační dysbalanci mezi prokoagulační aktivitou trombinu a fibrinolytickou aktivitou plazminu, rezultující v intravaskulární tvorbu fibrinu s trombotickými okluzemi v mikrocirkulaci, s následnou orgánovou ischemizací, konzumpcí koagulačních faktorů a krvácením.
Nejedná se o samostatné onemocnění, ale o syndrom, v jehož přítomnosti musíme vždy hledat a řešit vyvolávající příčinu. Ke stavům nejčastěji asociovaným s DIC patří hlavně sepse, trauma, nádory (solidní tumory, myeloproliferativní a lymfoproliferativní nemoci), porodní komplikace (embolizace plodovou vodou, abrupce placenty), cévní abnormality (Kasabachův-Merritové syndrom, velké cévní aneuryzma), těžké jaterní poškození, těžká toxická a imunologická reakce (hadí uštknutí, transfuzní reakce, rejekce transplantátu).
Klíčovou roli v patogenezi DIC hraje zvýšená tvorba fibrinu, ke které dochází v důsledku zvýšené generace trombinu, suprese fyziologických antikoagulačních mechanizmů a inhibice fibrinolýzy.
Klinická manifestace DIC je variabilní, závislá na typu primárního onemocnění, rozsahu postižení (generalizovaný versus lokalizovaný) a časovém průběhu – tempu koagulační konzumpce (akutní versus chronický).
Akutní forma DIC
Přestože jsou spouštěcím patologickým mechanizmem DIC depozice fibrinu v mikrocirkulaci, nejčastějším iniciálním klinickým projevem jeho akutní formy je krvácení, rezultující z konzumpce trombocytů a trombin senzitivních koagulačních faktorů. Současně v důsledku cirkulační obstrukce dochází k hypoperfuzi orgánů, jejich ischemizaci, infarktům a nekrózám. Predilekčně jsou postiženy plíce, centrální nervová soustava, ledviny, gastrointestinální trakt a kůže (tab. 1), ale potenciálně může dojít k alteraci kteréhokoliv orgánu. Bez adekvátního terapeutického zásahu dochází k šokovému stavu a úmrtí pacienta.
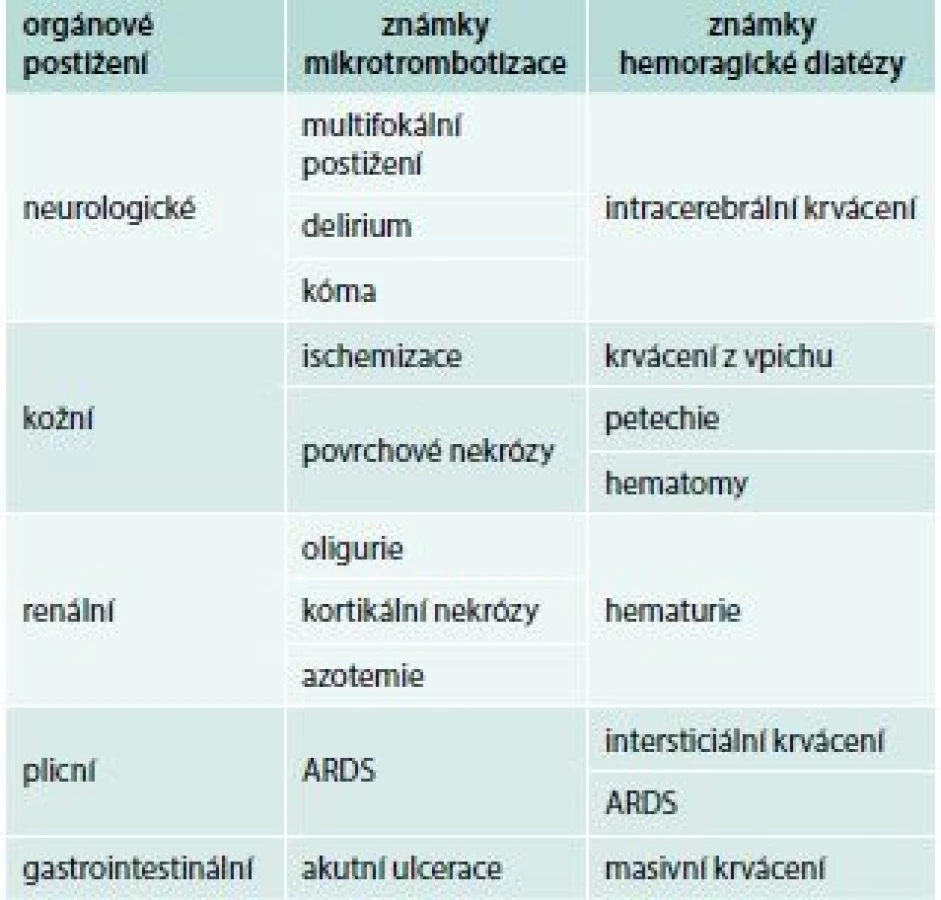
Chronická forma DIC
Jedná se o subakutně probíhající, kompenzovanou formu s protrahovanou klinickou nebo jenom laboratorní manifestací. Na rozdíl od akutní DIC jsou častěji v popředí klinického obrazu tromboembolické projevy (žilní a arteriální trombózy, infarkty, nebakteriální trombotická endokarditida). Krvácení je lehkého až středně těžkého stupně, hlavně v oblasti kůže a sliznic. Vyskytuje se u maligních onemocnění (specificky u mucin produkujících adenokarcinomů), často u syndromu mrtvého plodu a jaterních onemocnění.
Lokalizovaná forma DIC
Je charakterizovaná striktně anatomicky lokalizovanou konzumpcí trombocytů a koagulačních faktorů. Jenom ojediněle je spojená s krvácivými projevy. K ischemizaci a orgánové ischemii mimo ložisko postižení nedochází. K této formě DIC dochází hlavně u aneuryzmatu aorty, velkých hemangiomů (Kasabachův-Merrittové syndrom) a při rejekci transplantovaných ledvin.
Laboratorní test, na základě kterého lze potvrdit nebo vyloučit diagnózu DIC, neexistuje. Navíc je důležité si uvědomit, že jak klinická, tak i laboratorní variabilita obrazu DIC je veliká. S cílem zjednodušit a unifikovat diagnostiku DIC bylo vypracováno několik algoritmů vycházejících z bodových systémů. K nejznámějším patří algoritmus doporučovaný Mezinárodní společností pro trombózu a hemostázu (International Society on Thrombosis and Haemostasis – ISTH), tab. 2.

Terapie DIC
Základem terapie pacientů s DIC je identifikace a terapie primárního onemocnění, bez toho jsou všechny léčebné postupy odsouzeny k neúspěchu. Dekompenzované formy DIC jsou často spojeny s alterací celkového stavu pacienta s bezprostředním ohrožením jeho života, proto monitorování základních životních funkcí, korekce hypovolemie a anémie jsou nezbytnou součástí péče o pacienty s DIC.
Specifická terapie DIC zahrnuje antikoagulační a substituční terapii. Její rozsah a intenzita ve velké míře závisí na celkovém stavu pacienta a klinické symptomatologii.
Antikoagulační terapie je podávána s cílem přerušit tvorbu solubilního fibrinu a zamezit mikrotrombotizaci. Lékem volby v indikovaných případech jsou nadále nefrakciované hepariny, přičemž doporučená dávka se výrazně liší v závislosti na základním onemocnění a zkušenostech pracoviště. Heparinová terapie je kontraindikována u stavů spojených s rozsáhlým krvácením do anatomicky uzavřených prostorů (intrakraniální, intraspinální, perikardiální a paratracheální krvácení).
Součástí substituční terapie je podávaní trombocytárních koncentrátů, normalizace hladin antitrombinu (AT) a koagulačních faktorů. Hlavní indikací substituce koagulačních faktorů a trombocytů u DIC je akutní krvácení, případně příprava před chirurgickým zákrokem. Nejčastějším náhradním roztokem s obsahem koagulačních faktorů je plazma obsahující dostatečné hladiny většiny koagulačních faktorů, včetně fibrinogenu a koagulačních inhibitorů. K substituci koncentráty fibrinogenu přistupujeme u pacientů s DIC až při poklesu jeho hladiny < 1 g/l. U stavů spojených s predilekčním deficitem faktorů protrombinového komplexu (warfarinizace pacienta, deficit vitaminu K) lze aplikovat i koncentráty protrombinového komplexu. Bezprostřední ohrožení života a rezistence na konzervativní postup je indikací k podání rekombinantního aktivovaného faktoru VIIa (NovoSeven®). K substituci trombocytů přistupujeme u krvácejících pacientů při poklesu počtu krevních destiček < 50 × 109/l.
Hereditární trombocytopatie
Hereditární trombocytopatie jsou raritní, převážně autosomálně recesivně dědičné choroby, se sklonem ke zvýšenému krvácení (hlavně s lokalizací do sliznic a kůže) v klinickém obrazu.
Podle místa primárního poškození lze kvalitativní poruchy trombocytů rozdělit do několika skupin:
- poruchy trombocytárních glykoproteinů (GP)
- poruchy granul trombocytů
- poruchy signální transdukce
- poruchy trombocytární prokoagulační aktivity
K podezření na poruchu funkce krevních destiček by mělo vést zjištění těchto skutečností: zvýšený sklon ke krvácení v anamnéze, pozitivní rodinná anamnéza, vyloučení hlubší trombocytopenie a prodloužení krvácivosti s jinak normálním koagulačním nálezem.
Ke klinicky nejčastějším hereditárním trombocytopatiím zařazujeme všechny uvedené níže.
Poruchy trombocytárních glykoproteinů
Bernardův-Soulierův syndrom (BSS)
Jedná se o vrozenou krvácivou poruchu s převážně autosomálně recesivní dědičností. K onemocnění dochází v důsledku snížené exprese glykoproteinu Ib/IX/V (receptor pro vWF) na povrchu trombocytů, s následnou poruchou adheze krevních destiček v místech cévního poškození.
V klinickém obrazu dominuje slizniční krvácení, méně časté je poúrazové krvácení, hematurie a krvácení do CNS (4 %).
Téměř u všech pacientů nacházíme trombocytopenii se záchytem obrovských trombocytů (někdy až do velikosti lymfocytů). V koagulačním nálezu je prodloužení krvácivosti a výrazně snížená agregační odpověď po podání ristocetinu, kterou nelze korigovat přidáním normální plazmy (na rozdíl od vWD). Pomocí průtokové cytometrie se prokáže defektní exprese GPIb/IX/V.
Antifibrinolytická terapie (např. PAMBA) je vhodná při slizničním krvácení. U části pacientů vede podání desamino-D-argininu-vazopresinu (DDAVP) ke zkrácení krvácivosti, a lze ho použít u menších krvácení. Před operačními zákroky a při život ohrožujícím krvácení je obecně lékem volby aplikace trombocytárních náplavů, ale tato terapie může indukovat tvorbu antitrombocytárních protilátek. Získané formy BSS, hlavně autoimunitního původu jsou velice raritní, byly popsány u lymfoproliferativních a jaterních nemocí, případně při chromosomálních poruchách.
Glantzmannova trombastenie (GT)
Jedná se o raritní krvácivé onemocnění s autosomálně recesivní dědičností vznikající v důsledku deficientní exprese GPIIb-IIIa, což vede k poruše agregace trombocytů. Z krvácivých projevů jsou nejčastější menoragie, purpura a gingivální krvácení. Typický laboratorní nález zahrnuje prodloužení krvácivosti, poruchu retrakce koagula, sníženou agregační odpověď po podání adenozindifosfátu (ADP), adrenalinu, kolagenu a trombinu. Agregace po ristocetinu a velikost trombocytů jsou normální. K zástavě a v prevenci většího krvácení je nutná aplikace trombocytů. Klinická odpověď na podávání antifibrinolytik a DDAVP je variabilní. U menstruačního krvácení se osvědčila aplikace progesteronu a hormonální antikoncepce.
Trombocytární typ (pseudo) von Willebrandovy choroby (PvWD)
Je to raritní porucha s většinou autosomálně dominantní dědičností vznikající v důsledku kvalitativních poruch GPIb. Jejich následkem je zvýšená afinita velkých multimerů vWF k trombocytům s jejich následnou deplecí v plazmě. Laboratorní a fenotypový nález je podobný vWD typu 2B s lehkou trombocytopenií, prodloužením krvácivosti, poklesem plazmatických hladin vWF a FVIII, zvýšenou senzitivitou k ristocetinu a deficitem velkých multimerů vWF v plazmě.
Léčba PvWD je komplikovaná rizikem prohloubení trombocytopenie po aplikaci preparátu obsahujících vWF, proto je při krvácení doporučováno podávat malé dávky kryoprecipitátu (dostatečný hemostatický efekt, bez indukce trombocytopenie) nebo náplavy trombocytů.
Poruchy granul trombocytů
Heterogenní skupina nemocí, kterou podle typu postižených granul dělíme na:
- poruchy denzních granul (Dense Granule Deficiency – DGD: defekt trombocytárních σ-granul/ σ-granul deficiency)
- poruchy α-granul (syndrom šedých trombocytů/Gray Platelet Syndrome – GPS)
- kombinované poruchy denzních a α-granul (defekt trombocytárních α-granul a σ-granul)
Klinicky jsou charakterizovány různě intenzivním krvácením (většinou mírného stupně) slizničního původu a krvácením po úrazech nebo operacích.
V laboratorním nálezu je prodloužení krvácivosti, poruchy trombocytárních funkcí, případně trombocytopenie s morfologickými změnami trombocytů (např. GPS).
Pacienti s DGD většinou dobře odpovídají na terapii DDAVP a antifibrinolytiky (např. Pamba). Náplavy trombocytů jsou vyhrazeny jenom pro velká krvácení. Terapeutická odpověď nemocných s GPS na DDAVP je horší a podávání kortikoidů je většinou bez efektu.
Poruchy signální transdukce
Do této skupiny trombocytopatií patří poruchy povrchových receptorů trombocytů (kolagenový a ADP receptor) a poruchy intratrombocytárního přenosu informací mezi trombocytární membránou a efektorovým enzymem (defekty G-proteinů, fosfolipázy C, metabolizmu kyseliny arachidonové). Krvácivé projevy jsou většinou mírného stupně. K zástavě a prevenci krvácení lze použít DDAVP, trombocytární náplavy (pro nebezpečí aloimunizace indikovat uváženě), antifibrinolytika, případně krátkodobě i kortikoidy (např. prednison 25–50 mg p. o. 3–4 dny).
Poruchy trombocytární prokoagulační aktivity
Scottův syndrom – nejznámější onemocnění z této skupiny trombocytopatií. Patofyziologicky je charakterizován poruchou transportu fosfatidylserinu z vnitřní na vnější stranu povrchové membrány trombocytů, v důsledku které dochází k nedostatečné vazbě FVa a FXa na trombocytární fosfolipidy a následné insuficientní generaci trombinu. Klinickým projevem poruchy je krvácivá diatéza mírného stupně.
Získané trombocytopatie
Poruchy destičkových funkcí vznikají i druhotně v průběhu řady chorobných stavů. Patogeneze těchto poruch hemostázy není vždy jednoznačně objasněna a na jejím vzniku se často podílí postižení i dalších mechanizmů hemostázy.
Téměř pravidelně bývají kvalitativní odchylky krevních destiček u myeloproliferativních onemocnění. Ty mohou být příčinou krvácivého stavu nebo jej prohloubit při současné trombocytopenii. Také u chronických ledvinných onemocnění s uremií jsou krvácivé projevy připisovány funkčním poruchám krevních destiček. Úprava odchylek destičkových funkcí po dialýze ukazuje na jejich plazmatický původ. Podobně paraprotein ve vyšších koncentracích u monoklonálních gamapatií může interferovat s destičkovými funkcemi. Krvácivé projevy v této skupině nemocí však bývají především v důsledku trombocytopenie při infiltraci kostní dřeně. Poruchy hemostázy s krvácivými projevy mohou vyvolat i léky inhibující destičkové funkce. Patří k nim především nesteroidní protizánětlivé léky (např. kyselina acetylsalicylová, indometacin, ibuprofen).
U získaných destičkových poruch léčba základního procesu vede také k úpravě poměrů hemostázy. V případě krvácivých komplikací nebo při přípravě k operačnímu výkonu u trombocytopatií podáváme převody krevních destiček případně antifibrinolytika.
Koagulopatie
Pojmem koagulopatie označujeme krvácivé stavy zapříčiněné poruchou hladin nebo poruchou funkce koagulačních faktorů. Do této skupiny krvácivých stavů patří vrozené i získané deficity koagulačních faktorů, které mohou být z hlediska počtu postižených faktorů izolované, nebo kombinované.
Hemofilie
Hemofilie jsou recesivně dědičné krvácivé stavy charakterizované deficitem koagulačního faktoru VIII (hemofilie A) nebo koagulačního faktoru IX (hemofilie B). Obě onemocnění jsou si geneticky, biologicky a klinicky podobná, dochází k nim v důsledku heterogenních defektů genů pro FVIII a FIX.
Výskyt onemocnění je u hemofilie A 1/10 000 a u hemofilie B 1/60 000 nově narozených dětí.
Asi u 30 % pacientů dochází k onemocnění v důsledku spontánních (de novo) mutací s negativní rodinnou anamnézou.
Z laboratorních testů je typické prodloužení aktivovaného parciálního tromboplastinovéhu času (Activated Parcial Tromboplastine Time – APTT), s jeho korekcí po inkubaci pacientské a normální plazmy. Hloubku deficitu FVIII a FIX, resp. tíži onemocnění zjistíme přímým stanovením hladiny koagulačních faktorů (koagulační aktivity a antigenní hladiny).
Součástí diagnostiky hemofilií je i DNA analýza genů pro FVIII a FIX, důležitá nejenom pro objasnění rozsahu a lokalizace genetické poruchy, ale i pro stanovení přenašečství a prenatální diagnostiku plodů rizikových matek.
Hlavním klinickým projevem onemocnění je krvácení, jehož rozsah a intenzita jsou závislé na hladině FVIII:C, resp. FIX:C. Podle tíže deficitu koagulačních faktorů rozdělujeme hemofilie do 3 klinických forem (tab. 3). U hemofilických pacientů představuje 90 % krvácivých epizod krvácení do kloubů a svalů. Kolenní a loketní klouby jsou vzhledem ke své relativní nestabilitě a kombinované rotačně-úhlové zátěži postiženy nejčastěji. Intraartikulární krvácení s následnou autolýzou erytrocytů vede k depozici železa v synovii a chondrocytech kloubní chrupavky. Následkem recidivujících krvácení dochází k synoviální hypertrofii, která zvyšuje náchylnost kloubu k dalšímu krvácení (vzniká tzv. cílový kloub – locus minoris resistentiae). Tento bludný kruh vede k dalšímu poškozování kloubního aparátu, k vzestupu koncentrace hydrolytických enzymů, destrukci synovie, chrupavek a kostí s ireverzibilním funkčním a anatomickým poškozením. Klinickou známkou krvácení je bolest následovaná otokem a fixací kloubu ve flexním postavení. Intramuskulární krvácení je druhým nejčastějším krvácením u těžkých forem hemofilie (30 % krvácivých epizod). Krvácení do velkých svalů se obvykle vstřebává bez komplikací, ale malé krvácení do uzavřených interfasciálních prostorů může vést ke kompresi vitálních struktur s distální ischemií, gangrénou, flexní kontrakturou a neuropatií (např. m. iliopsoas). U těžkých forem hemofilie je poměrně časté krvácení do urogenitálního traktu, hlavně makroskopická hematurie. Probíhá většinou benigně. Při krvácení do této oblasti je nutno se vyvarovat aplikaci antifibrinolytik pro vysoké riziko obstrukce močovodů koagulem. Gastrointestinální krvácení postihuje 10–15 % pacientů s těžkou formou hemofilie, přičemž jeho frekvence stoupá u nemocných s chronickou hepatitidou a cirhózou v důsledku portální hypertenze. K život ohrožujícímu krvácení do CNS dochází u 2,8–13,8 % pacientů, z toho 30 % krvácení je smrtelných a v 50 % vede k dlouhodobému neurologickému postižení.
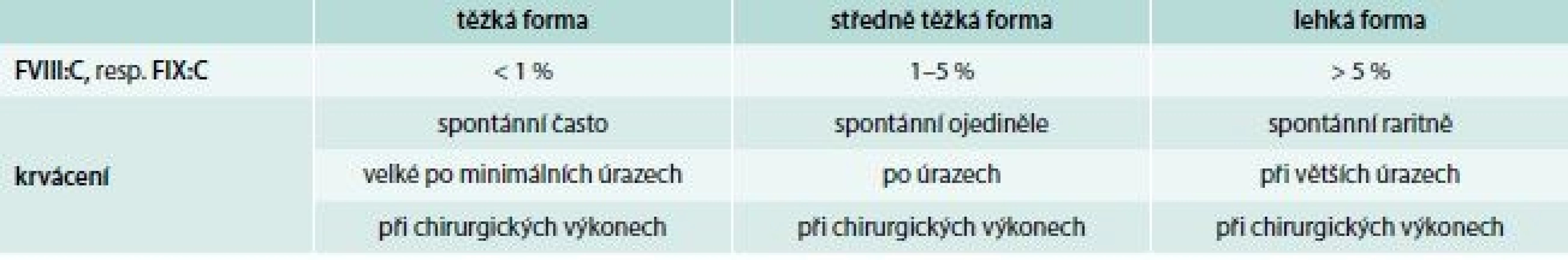
Hlavním cílem terapie a prevence krvácení u pacientů s hemofilií A a B je promptní a dostatečná intravenózní substituce chybějících koagulačních faktorů. Časná léčba podaná už při prvních známkách krvácení může omezit rozsah krvácení a zabrání následnému tkáňovému poškození. To vedlo k zavedení tzv. domácí terapie u pacientů s těžkou formou onemocnění, při které si pacient sám (případně člen rodiny) aplikuje koncentráty FVIII nebo FIX. V současnosti jsou lékem volby koncentráty koagulačních faktorů s dostatečnou virovou inaktivací, vyrobené z lidské plazmy, případně rekombinantní technikou. Jejich dávkování je závislé na hladině deficitního faktoru u pacienta, cílové hladině, kterou chceme dosáhnout, a typu krvácení. Pro výpočet potřebné aplikační dávky platí: podání 1 U koagulačního faktoru/kg hmotnosti pacienta vede ke zvýšení plazmatické hladiny o 2 % u FVIII a o 1 % u FIX. V současnosti se do klinické praxe dostávají rekombinantní preparáty s prodlouženým poločasem. Jejích používání v profylaktických režimech snižuje časnost intravenózních aplikací chybějících faktorů u pacienta.
von Willebrandova choroba
Patří mezi nejčastější dědičné krvácivé nemoci s prevalencí okolo 100 případů/milion obyvatel.
Příčinou onemocnění je deficit von Willbrandova faktoru (vWF), který fyziologicky zajišťuje adhezi trombocytů na místa cévního poškození a současně stabilizuje plazmatickou hladinu koagulačního faktoru VIII v cirkulaci.
Deficit vWF, tedy von Willebrandovu chorobu (von Willebrand Disease – vWD) rozdělujeme do 3 základních skupin (tab. 4), přičemž typ 1 a typ 3 představují kvantitativní defekt (parciální, resp. totální deficit vWF) a typ 2 kvalitativní poruchu struktury a funkce vWF.

V klinickém obrazu dominují krvácivé projevy mírného až středně těžkého stupně s lokalizací do sliznic a menoragie (tab. 5).
![Frekvence krvácení (%) podle lokalizace u vWD a těžké formy hemofilie. Upraveno podle [10,11]](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image_pdf/3190f5cdafaf620f06e50bde420c37a1.jpeg)
V léčbě pacientů s vWD lze použít 4 terapeutické postupy v závislosti na podtypu onemocnění a závažnosti krvácení: indukce vyplavení zásob koagulačních faktorů, substituce koagulačních faktorů, zmírnění krvácení antifibrinolytickou léčbou a snížení rizika krvácení pomocí estrogenů u žen.
Desmopresin (DDAVP) je syntetický analog vazopresinu s reziduální presorickou aktivitou < 1 % přirozeného hormonu. Aplikace DDAVP vede k rychlému vyplavení FVIII a vWF ze zásobních granul u zdravých jedinců a pacientů s vWD typu 1. Lze ho podávat intravenózně, subkutánně a intranazálně. Opakované aplikace, hlavně v intervalech do 24 hod, vedou k poklesu efektu terapie z důvodu vyčerpání zásob FVIII a vWF. U vWD typu 2 je terapie DDAVP většinou neefektivní, navíc pro riziko transientní trombocytopenie je u vWD typu 2B kontraindikována.
Substituční terapie je léčbou první volby u vWD typu 3 a u ostatních typů při selhání nebo kontraindikaci DDAVP. Zahrnuje aplikaci koncentrátů z plazmy bohatých na vWF a FVIII. Dávkování substituční terapie je závislé na tíži onemocnění a rozsahu krvácení.
Antifibrinolytika (kyselina tranexamová, kyselina ε-aminokapronová) jsou alternativní terapie pro menší krvácení, hlavně v orofaryngeální oblasti. Lze je podávat samostatně nebo v kombinaci s jinou terapií. Obecně jsou antifibrinolytika kontraindikována při krvácení do močového traktu.
Estrogeny zvyšují plazmatické hladiny vWF, ale jejich účinek je tak variabilní a nepředvídatelný, že v terapii krvácivých epizod u vWD se skoro nepoužívají. Jedině nasazení hormonální antikoncepce může vést k redukci hypermenoragií u žen s vWD, včetně typu 1.
Deficity ostatních koagulačních faktorů
Dysfibrinogenemie
Strukturální poruchy fibrinogenu vedoucí k defektní tvorbě fibrinu. Mohou být vrozeného původu (autosomálně dominantní nebo kodominantní dědičnost), nebo sekundární, hlavně při jaterním postižení. U 40–55 % pacientů je průběh onemocnění asymptomatický. U 25–50 % dochází ke krvácení, které je většinou lehkého stupně, hlavně při chirurgických zákrocích. Méně časté je spontánní krvácení typu epistaxe, hypermenoragie a podkožních hematomů. U 10–20 % pacientů je zvýšený sklon k trombotickým komplikacím (venózní a arteriální trombózy, spontánní potraty).
V koagulačním nálezu je prodloužení v APTT, protrombinového času (Prothrombin Time – PT) a trombinovém času (Thrombin Time – TT), který je nejcitlivějším testem pro dysfibrinogenemii. Patologický je i reptilázový test (RT) – na rozdíl od prodloužení TT při přítomnosti heparinu, při němž RT je normální. Hladina fibrinogenu je nesměrodatná, může byt zvýšená, normální, nebo snížená.
Terapie pacientů s dysfibrinogenemií je závislá na jejich klinické symptomatologii. Při sklonu ke krvácení (pozitivní osobní anamnéza) je v předoperační přípravě indikována substituce fibrinogenu (kryoprecipitát, koncentráty fibrinogenu) v dávce zajišťující dostatečnou hemostaticky účinnou hladinu (1 g/l). Antifibrinolytika jsou vhodná ke zmírnění běžných krvácivých epizod, ale neměla by se podávat pacientům se sklonem k trombózám, u kterých se řídíme obecnými principy terapie a prevence trombofilních stavů.
Pojmem hypofibrinogenemie a afibrinogenemie označujeme kvantitativní poruchy fibrinogenu. Jsou spojeny s celoživotním sklonem ke krvácení různé intenzity, přičemž podobně jako u dysfibrinogenemie je bezpečnou hladinou fibrinogenu > 1 g/l.
Deficit koagulačního faktoru II
Rozlišujeme 2 typy deficitních poruch koagulačního faktoru II (protrombin, FII) – hypoprotrombinemie a dysprotrombinemie. Jedná se o raritní onemocnění s autosomální dědičností, přičemž pacienti s heterozygotním postižením (FII > 50 %) jsou většinou bez klinické symptomatologie. Homozygotní, resp. kombinované heterozygotní postižení (FII 2–25 %) je spojeno s krvácivými projevy typu epistaxe, hypermenoragie, s porodním, peroperačním a poúrazovým krvácením. Krvácení do kloubu je raritní. V laboratorním nálezu je prodloužení PT a APTT s nálezem snížené hladiny FII při specifickém funkčním nebo imunologickém vyšetření. V diferenciální diagnostice je nutné vyloučit deficit FV a FX, případně nedostatek vitaminu K a dikumarolovou intoxikaci (kombinované deficity vitamin K dependentních faktorů). Substituční terapie je indikována při větších krvácivých příhodách a před chirurgickým zákrokem. Lze podat plazmu 10–20 ml/kg váhy s následnou udržovací dávkou 3 ml/kg po 24 hod nebo koncentrát protrombinového komplexu.
Deficit koagulačního faktoru V
Deficit koagulačního faktoru V (proakcelerin, FV) je raritní, autosomálně recesivně dědičný deficit s klinickými projevy jenom u homozygotních pacientů (u asymptomatických heterozygotů je plazmatická hladina FV okolo 50 %).
V klinickém obrazu dominují krvácivé projevy různého stupně, od slizničního, podkožního, peroperačního krvácení, zvýšeného menstruačního krvácení až po krvácení do CNS. Intenzita a rozsah krvácení bývají obvykle menší než u deficitu FVII, ale i u těchto pacientů může dojít k život ohrožujícímu krvácení.
Z diagnostického hlediska je typické: prodloužení APTT a zejména PT, u třetiny nemocných je patologická i doba krvácivosti.
Léčba nemoci je substituční, přičemž komerční koncentrát FV není dostupný. Lékem volby je plazma.
Deficit koagulačního faktoru VII
Z hlediska etiologie lze deficity koagulačního faktoru VII (FVII) rozdělit na vrozené (vzácné autosomálně recesivně dědičné onemocnění s hemoragickou symptomatologií u homozygotních forem a symptomatickým průběhem u heterozygotů) a získané, přičemž nejčastější příčinou získaného deficitu FVII bývají jaterní onemocnění, intoxikace dikumaroly a malabsorpce vitaminu K.
Ke krvácení dochází typicky u pacientů s hladinou FVII < 1 %. Rozsah a typ krvácení je podobný jako u hemofilií, včetně krvácení do kloubů a svalů. Běžně dochází i k slizničním krvácením, hypermenoragii, hematurii, krvácení do GIT, případně do retroperitonea a CNS (novorozenci). Při hladině FVII okolo 10 % je krvácení jenom mírného stupně. Navzdory hemoragické povaze nemoci, ke komplikacím průběhu onemocnění patří i tromboembolické příhody.
Z diagnostického hlediska je typické prodloužení PT při většinou normálním APTT. Diagnózu potvrdí až průkaz deficitu FVII.
Léčbou volby je substituce deficitního FVII. V terapii lze použít koncentráty FVII, koncentráty protrombinového komplexu, plazmu nebo rekombinantní aktivovaný FVIIa.
Vzhledem ke krátkému poločasu FVII je nutné, v případě potřeby, následnou dávku aplikovat v intervalu 2–6 hod. Za bezpečnou hladinu FVII potřebnou k zástavě většiny krvácení je považována hladina 15–20 %.
Deficit faktoru X
Výskyt vrozené autosomálně dědičné formy deficitu faktoru X (FX) je ojedinělý. Získané formy byly zachyceny u nemocných s amyloidózou, při respiračních infekcích a akutní myeloidní leukemii.
Rozsah krvácení u pacientů s hladinou < 1 % je podobný jako u hemofilií, přičemž dominuje krvácení do sliznic, měkkých tkání, ale nezřídka i kloubů. Při hladině > 10 % je riziko krvácení malé, je však třeba pamatovat na zvýšené riziko při úrazech a operacích.
V koagulačním nálezu je prodloužení APTT a PT při normální době krvácivosti.
Terapie je substituční, lze podat plazmu i koncentráty protrombinového komplexu. Cílový vzestup hladiny FX na 10–40 % zajistí dostatečný hemostatický efekt.
Deficit koagulačního faktoru XI
Deficit koagulačního faktoru XI (FXI) představuje raritní koagulační deficit s autosomálně recesivní dědičností a zvýšeným výskytem u aškenázských (středo - a východoevropských) Židů.
Krvácení je většinou mírné, nejčastěji v souvislosti s extrakcí zubů, úrazem, nebo chirurgickým zákrokem. Zvýšené menstruační krvácení se vyskytuje u většiny žen s deficitem FXI. Petechie, podkožní hematomy a krvácení do kloubů nepatří do klinického obrazu nedostatku FXI. Riziko krvácení nekoreluje s hladinou FXI u pacienta. Deficit FXI je laboratorně charakterizován prodloužením APTT při normální krvácivosti, PT a TT. Definitivní diagnózu stanovíme až vyšetřením hladiny FXI pomocí specifických funkčních testů.
Lékem volby je substituční terapie pomocí koncentrátů FXI, nebo plazmou. Dosažení hladiny 30–45 % FXI je většinou dostatečné k zástavě krvácení nebo k přípravě před operačním zákrokem. Poločas substituovaného FXI je okolo 52 hod. Aplikace vysoce čištěných koncentrátů FXI může být spojena se vznikem trombotických komplikací (10 % pacientů) typu DIC a arteriálních trombóz. K rizikovým faktorům patří vyšší věk a dávka přesahující 40 U/kg hmotnosti. U menších krvácení, nebo v kombinaci se substituční terapií jsou indikována antifibrinolytika, respektive DDAVP u pacientů s lehčím deficitem FXI.
Deficit faktorů kontaktní fáze – faktor XII, prekalikrein a vysokomolekulární kininogen
Deficity faktorů kontaktní fáze – faktor XII (FXII), prekalikrein (PK) a vysokomolekulární kininogen (High Molecular Weight Kininogen – HMWK) – nejsou spojeny se zvýšeným krvácením. O klinických projevech těchto defektů se vedou spory, někteří autoři považují hluboké deficity těchto faktorů za trombofilní (první popsaný pacient s deficitem FXII zemřel na embolii plic), současně se předpokládá výskyt častějších spontánních potratů.
Laboratorně u těžkých defektů FXII, PK a HMWK nacházíme extrémně prodloužené APTT.
Substituční terapie není nutná, ale při chirurgickém zákroku je doporučovaná preventivní antikoagulační terapie. V případě trombózy nebo embolie plic se podává běžná antikoagulační léčba.
Deficit koagulačního faktoru XIII
Z hlediska etiologie poklesu koagulačního faktoru XIII (FXIII) dělíme onemocnění na vrozené a získané formy.
Vrozené formy deficitu FXIII jsou velice vzácná onemocnění s autosomálně recesivní dědičností a krvácivými projevy až při hladině FXIII < 1 %. Proto ke krvácení dochází jenom u pacientů s homozygotní formou postižení.
Získané deficity FXIII byly popsány u pacientů s Henochovou-Schönleinovou purpurou, Crohnovou nemocí, ulcerózní kolitidou a u jaterních onemocnění.
Ke krvácení dochází často už po narození – do měkkých tkání, kloubů, podkoží a CNS (až u 25 % pacientů). Typické bývá krvácení ze špatně se hojícího pupečníku. Často dochází k tvorbě posthemoragických pseudocyst a kombinací peroperačního krvácení s pozdním pooperačním krvácením (až s několikadenním odstupem). S deficitem FXIII bývá spojeno i horší hojení ran, oligospermie a sklon k spontánním potratům.
Laboratorní nález včetně APTT, PT, krvácivosti a počtu trombocytů je normální.
Rozpustnost koagula v 5M močovině je nejpoužívanější screeningový test k detekci těžkého deficitu FXIII, ale přesnou diagnózu lze stanovit až kvantitativním stanovením hladiny FXIII.
Vzhledem k tomu, že komerční koncentrát FXIII není dostupný, je lékem volby jak v prevenci, tak i k zástavě krvácení aplikace plazmy nebo kryoprecipitátu. K zástavě krvácení většinou stačí aplikovat 2–3 ml plazmy/kg hmotnosti. Většinou není nutné podávat následné dávky plazmy, protože poločas FXIII je 9–19 dnů.
Vrozené kombinované deficity koagulačních faktorů
Vrozené kombinované deficity koagulačních faktorů (Familial Multiple Coagulation Factor Deficiencies – FMFD syndromes) představují skupinu raritních onemocnění, která dle typu genetického defektu rozdělujeme na skupinu s náhodným kombinovaným defektem více faktorů, s genetickým defektem pro každý faktor a na skupinu s jedním genetickým defektem spojeným s poklesem více faktorů.
Z hlediska kombinace deficitních faktorů rozlišujeme FMFDI (FV a FVIII), FMFD II (FVIII a FIX), FMFDIII (FII, FVII, FIX a FX), FMFDIV (FVII a FVIII), FMFDV (FVIII, FIX a FXI) a FMFDVI (FIX a FXI).
V klinickém obrazu dominují krvácivé projevy, většinou lehkého až středně těžkého stupně, a to v závislosti na hloubce a kombinaci koagulačního defektu. Při stanovování diagnózy je nutné u těchto pacientů vyloučit přítomnost cirkulujícího antikoagulans, jaterního onemocnění a intoxikaci dikumaroly.
Laboratorní nález a terapie jsou závislé na typu kombinace deficitů koagulačních faktorů. V léčbě lze použít jak plazmu, tak i komerční koncentráty koagulačních faktorů.
Specifické inhibitory koagulačních faktorů
Pojmem specifický inhibitor koagulačního faktoru označujeme přítomnost protilátky proti danému koagulačnímu faktoru inaktivující jeho prokoagulační aktivitu. Teoreticky může být protilátka namířena proti kterémukoliv koagulačnímu faktoru, ale v klinické praxi se prakticky setkáváme jenom s případy inhibitorů FVIII, velice ojediněle FIX a zcela raritně s inhibitory ostatních koagulačních faktorů.
Inhibitor FVIII/FIX se vyskytuje u 4 klinických stavů: u pacientů s hemofilií A, v poporodním období, u pacientů s autoimunitním, resp. nádorovým onemocněním a idiopaticky, bez primárního postižení.
Inhibitor FVIII/FIX u pacientů s hemofilií
Vzniká jako imunitní odpověď na terapii FVIII/FIX obsaženém v krevních derivátech (aloprotilátka – imunoglobuliny převážně třídy IgG). Postihuje 5–10 % pacientů s hemofilií A, ale v 95 % vzniká u nemocných s těžkou a středně těžkou formou onemocnění (hladina FVIII/FIX < 5 %). Incidence inhibitoru je závislá na typu genetického postižení pacienta (výskyt inhibitoru u 70–75 % pacientů s velkými deplecemi a nonsense mutacemi v A3 oblasti genu pro FVIII) a na vnějších podmínkách daných způsobem, časem a dávkou, v jaké byl FVIII/FIX aplikován, a dále také na imunogenitě substitučního přípravku. Titr inhibitoru ≤ 10 jednotek Bethesda (Bethesda unit – BU) představuje ředění plazmy, při kterém dojde k 50% redukci normální aktivity FVIII, tento titr označujeme jako nízký, a naopak > 10 BU jako vysoký. Pacienty s nízkým titrem inhibitoru rozdělujeme podle změny hladiny inhibitoru po podání FVIII/FIX do 2 skupin:
- „low responders (LR)“ – podání FVIII/FIX nevede k významnému vzestupu titru inhibitoru (25 % pacientů – vzestup titru ≤ 5 BU)
- „high responders (HR)“ – podání FVIII/FIX je doprovázeno markantním vzestupem titru inhibitoru (75 % pacientů).
Klinickým korelátem vzniku inhibitoru u pacienta trpícího hemofilií A nebo B je zkrácení poločasu účinnosti terapie nebo perzistence krvácení i při dostatečné substituční terapii. V laboratorních testech dochází k prodloužení už patologického APTT, přičemž inkubace normální plazmy s plazmou pacienta s inhibitorem nekoriguje prodloužení v APTT.
Terapeutická strategie hemofilických pacientů s inhibitorem FVIII/FIX je závislá na tíži onemocnění, titru inhibitoru a odpovědi pacienta na aplikaci FVIII/FIX (LR vs HR). Malé krvácení lze zastavit kompresí, případně v kombinaci s antifibrinolytiky. Zástavu větších krvácení můžeme docílit dvěma způsoby – zvednutím hladiny FVIII nebo aplikací přípravku s „by-pass“ aktivitou vůči FVIII/FIX.
Substituční terapie FVIII/FIX je vhodná u pacientů s nízkým titrem inhibitoru a s anamnestickým průkazem, že jsou LR. Dávky FVIII/FIX potřebné k dosažení hemostaticky účinné hladiny jsou z důvodu potřeby neutralizace inhibitoru podstatně vyšší než u hemofilických neinhibitorových pacientů (např. u LR s inhibitorem < 5 BU jsou 2–3krát vyšší). Substituční terapie krevními deriváty obsahující lidský FVIII/FIX není vhodná pro HR a nemocné s vysokým titrem inhibitoru. U těchto nemocných je lékem volby aplikace přípravku s „by-pass“ aktivitou vůči FVIII/FIX – rekombinantní aktivovaný FVIIa (rFVIIa, NovoSeven®), nebo koncentráty aktivovaného protrombinového komplexu (Activated Prothrombin Complex Concentrates – APCCs, Factor Eight Inhibitor Bypassing Activity – FEIBA).
Velkou změnou v terapii pacientů s inhibitorem bude zavedení emicizumabu (Hemlibra) do klinické praxe. Emicizumab je bispecifická protilátka namířená proti faktorům IXa a X. Je navržena tak, aby k sobě přiblížila faktor IXa a faktor X, a obnovila tak proces srážení krve u osob s hemofilií A. Je určená k profylaktické léčbě, přičemž se aplikuje formou podkožní injekce 1krát týdně. Závažnou komplikací této léčby je zvýšený výskyt trombotických mikroangiopatii a trombotických příhod, hlavně při kombinaci s vyššími dávkami APCC.
Efekt imunosupresivní terapie je u hemofilických pacientů menší než u nehemofilických nemocných s inhibitorem.
Speciální terapeutický přístup představuje tzv. navozování imunitní tolerance (IT), což znamená dlouhodobé (12–24 měsíců), časté a pravidelné aplikovaní FVIII pacientům s inhibitorem FVIIII. Cílem IT je výrazné snížení až odstranění inhibitoru. Úspěšnost této léčby je vysoká, ale je cenově nákladná. Nejlepších výsledků bylo dosaženo u pacientů s titrem inhibitoru < 10 BU při pravidelné denní dávce > 100 U/kg tělesné hmotnosti. Navození IT je léčebným postupem volby u všech, hlavně dětských pacientů s čerstvým záchytem inhibitoru.
Získané inhibitory FVIII
Získaný inhibitor FVIII (pacienti bez hemofilie A) patří mezi raritní onemocnění s roční incidencí 1/1 milion obyvatel a s 22% mortalitou. V 50–55 % připadů vzniká idiopaticky, v 8–15 % při jiném autoimunitním nebo nádorovém onemocnění a v 13,5 % v poporodním období. Výskyt je bez pohlavní predilekce převážně ve věku nad 60 let. Na rozdíl od hemofilie A je inhibitor typu autoprotilátky a u 38 % nemocných může dojít k spontánnímu vymizení inhibitoru.
V klinickém obrazu dominuje kožní, slizniční a intramuskulární krvácení. Krvácení do GIT, urogenitálního traktu a CNS je poměrně časté. Ke krvácení do kloubů nedochází.
Laboratorní nález je podobný inhibitorovým pacientům s hemofilií A: výrazné prodloužení APTT nekorigovatelné normální plazmou, výrazně snížená hladina FVIII a pozitivní průkaz specifického inhibitoru.
Terapie pacientů se získaným inhibitorem FVIII zahrnuje léčbu krvácení a eliminaci autoprotilátky.
Terapie krvácivých epizod je podobná léčbě nemocných s hemofilií A – rF VIIa, APCCs. Současně je nutné se vyvarovat intramuskulární aplikaci léků a obecně preparátů s antiagregačním efektem (např. NSAID).
S cílem snížit až eliminovat inhibitor FVIII je doporučována kombinovaná imunosupresivní terapie složená z kortikoidů a cyklofosfamidu. V případě rezistence na daný postup lze místo cyklofosfamidu podat cyklosporin A nebo podání anti CD20. U pacientů s vysokým titrem inhibitoru, krvácením a nedostatečnou odpovědí na předchozí terapii lze do terapeutického postupu zařadit plazmaferézy nebo extrakorporální imunoadsorpci.
MUDr. Peter Salaj
Ústav hematologie a krevní transfuze, Praha
Doručeno do redakce 31. 1. 2018
Přijato po recenzi 17. 3. 2018
Sources
- Salaj P. Poruchy hemostázy. Maxdorf Jessenius: Praha 2017. ISBN 978–80–7345–513–2.
- Alving BM. How I treat heparin-Induced thrombocytopenia and thrombosis. Blood 2003; 101(1): 31–37. Dostupné z DOI: <http://dx.doi.org/10.1182/blood-2002–04–1089>.
- Cetkovský J. Intenzivní péče v hematologii. Galén: Praha 2004. ISBN 80–7262–255–2.
- Colman RW, Hirsh J, Marder VJ et al. Hemostasis and Thrombosis: Basic Principles and Clinical Practice. 4th ed. Lippincott Williams Wilkins: 2000. ISBN 978–0781714556.
- Mitchell Lewis S, Bain BJ, Bates I. Dacie and Lewis Practical Haematology. 10th ed. Churchill Livingstone Elsevier: 2006. ISBN 978–0443066603.
- Furlan M, Robles R, Solenthaler M et al. Acquired deficiency of von Willebrand factor-cleaving protease in a patient with thrombotic thrombocytopenic purpura. Blood 1998; 91(8): 2839–2846.
- George JN, Woolf SH, Raskob GE et al. Idiopathic Thrombocytopenic Purpura: A Practice Guideline Developed by Explicit Methods for The American Society of Hematology. Blood 1996; 88(1): 3–40.
- Sadler JE, Mannucci PM, Berntorp E et al. Impact, diagnosis and treatment of von Willebrand disease. Thromb Haemost 2000; 84(2): 160–174.
- Tsai HM, Lian EC. Antibodies to von Willebrand factor-cleaving protease in acute thrombotic thrombocytopenic purpura. N Engl J Med 1998; 339(22): 1585–1594.
- Silwer J. von Willebrand‘s disease in Sweden. Acta Paediatr Scand 1973; 238 (Suppl): 1–159.
- Lak M, Peyvandi F, Mannucci PM. Clinical manifestations and complications of childbirth and replacement therapy in 385 Iranian patients with type 3 von Willebrand disease. Br J Haematol 2000; 111(4): 1236–1239
Labels
Diabetology Endocrinology Haematology Internal medicineArticle was published in
Internal Medicine

2018 Issue 5
- Impact of Prophylaxis on Reducing the Risk of Neurological Problems Associated with Bleeding in Hemophilia A
- Minimum and Optimal Factor Levels in Physically Active Hemophiliacs
- The Importance of Hydration in Wound Healing
- Cost Effectiveness of FVIII Substitution Versus Non-Factor Therapy for Hemophilia A
- Vascular Disease in the Gradually Aging Population of Hemophiliacs: An Underestimated Problem?
Most read in this issue
- Autoimunitní hemolytická anémie
- Diferenciální diagnostika anémií
- Vrozené a získané krvácivé stavy
- Hemoglobinopatie