
Úskalí v diagnostice srdeční amyloidózy a možnosti terapie
Difficulties in the Diagnosis of Cardiac Amyloidosis and Treatment Options: Case Report
Amyloidosis is a disease characterized by extracellular deposition of amyloid, an insoluble pathological protein. Clinical manifestation is based on the type of amyloid, and to define the specific type of amyloid is crucial to determining prognosis and optimal therapy. Echocardiography may be the first method to highlight the diagnosis of cardiac amyloidosis, but this is not a specific method. Typical echocardiographic findings in cardiac amyloidosis are myocardial enlargement, atrial dilatation, decreased longitudinal contraction with relatively long preserved systolic function of the left ventricle and left ventricular diastolic dysfunction with restrictive type of filling. Magnetic resonance gives a somewhat specific image of late gadolinium enhancement, but cannot distinguish individual types of amyloid. The biopsy can be falsely negative. In recent years, methods of nuclear medicine have become more important, especially in case of the transthyretin form of amyloidosis. Our case report shows a 72-year-old male, in whom 99mTc-DPD scintigraphy revealed the senile form of transthyretin amyloidosis. The therapeutic possibilities of transthyretin form of cardiac amyloidosis are currently being explored.
Keywords:
Biopsy – scintigraphy – magnetic resonance – echocardiography – Amyloidosis – doxycycline – tauroursodeoxycholic acid – 99mTc-Diphosphono-Propanodicarboxylic Acid
Authors:
Renáta Závodná 1; Kamil Zeman 1; Martin Pleva 2; Milan Kamínek 3
Authors‘ workplace:
Interní oddělení Nemocnice ve Frýdku-Místku
1; Oddělení intervenční radiologie, Magnetická rezonanceKomplexního kardiovaskulárního centra, Nemocnice Podlesí, Třinec
2; Klinika nukleární medicíny LF UP a FN Olomouc
3
Published in:
Vnitř Lék 2020; 66(1): 71-77
Category:
Case Report
Overview
Amyloidóza je onemocnění charakterizované extracelulárním ukládáním amyloidu, nerozpustné patologické bílkoviny. Klinická manifestace se odvíjí od typu amyloidu a určení správného typu amyloidu je zcela zásadní pro stanovení prognózy a optimální terapie. Echokardiografie nás může jako první upozornit na tuto diagnózu, nejedná se však o specifickou metodu. Typickým echokardiografickým nálezem u srdeční amyloidózy je zbytnělý myokard, dilatace síní, snížení longitudinální kontrakce při relativně dlouho zachovalé systolické funkci levé komory a diastolická dysfunkce levé komory s restriktivním typem plnění. Magnetická rezonance poskytuje specifický obraz difuzního pozdního sycení myokardu gadoliniovou kontrastní látkou, avšak nedokáže rozlišit jednotlivé typy amyloidu. Biopsie může být falešně negativní. V posledních letech nabývají na diagnostickém významu také metody nukleární medicíny. Naše kazuistika poukazuje na případ 72letého muže, u kterého 99mTc-DPD scintigrafie odhalila transthyretinovou formu srdeční amyloidózy. Terapeutické možnosti transthyretinové formy srdeční amyloidózy jsou v současné době stále předmětem zkoumání.
Klíčová slova:
amyloidóza – Biopsie – doxycyklin – echokardiografie – magnetická rezonance – tauroursodeoxycholová kyselina – 99mTc-dihydroxypropandifosfonová kyselina – scintigrafie
Úvod
Amyloidóza je onemocnění charakterizované extracelulárním ukládáním amyloidu, nerozpustné patologické bílkoviny. Onemocnění může být lokalizované nebo systémové. Klinická manifestace se odvíjí od typu amyloidu. Systémová amyloidóza může být fatální a vést k rychlému úmrtí (1). V současné době jsou známy 2 hlavní typy srdeční amyloidózy, které se liší původem a terapeutickými možnostmi, a to AL-amyloidóza a transthyretinová amyloidóza (ATTR), kterou rozdělujeme na 2 subtypy – hereditární (mATTR) a senilní (wtATTR neboli wildtype) (2, 3, 8, 9). U AL-amyloidózy jsou prekurzorem patologické bílkoviny volné lehké řetězce imunoglobulinu produkované klonální populací plazmatických buněk. Nejúčinnější léčebnou metodou je zde vysokodávkovaná chemoterapie s autologní transplantací buněk kostní dřeně (22). Naproti tomu u mATTR amyloidózy se jedná o mutaci genu kódujícího prekurzorový protein – transthyretin (TTR), dříve nazývaný prealbumin (8, 23). Terapeutické možnosti ATTR jsou v současné době předmětem zkoumání. Při diagnostice srdeční amyloidózy vycházíme z anamnézy, klinického obrazu, elektrokardiografie (EKG), echokardiografie, biochemických ukazatelů a magnetické rezonance (MRI). Definitivní diagnózu může stanovit biopsie, ta však může být falešně negativní v případě odběru nepostižené tkáně (22). V odlišení mutantní od senilní formy ATTR nám může pomoci genetické vyšetření (23). V posledních letech nabývají na významu v diagnostice tohoto onemocnění také metody nukleární medicíny, jak prezentuje naše kazuistika.
Popis případu
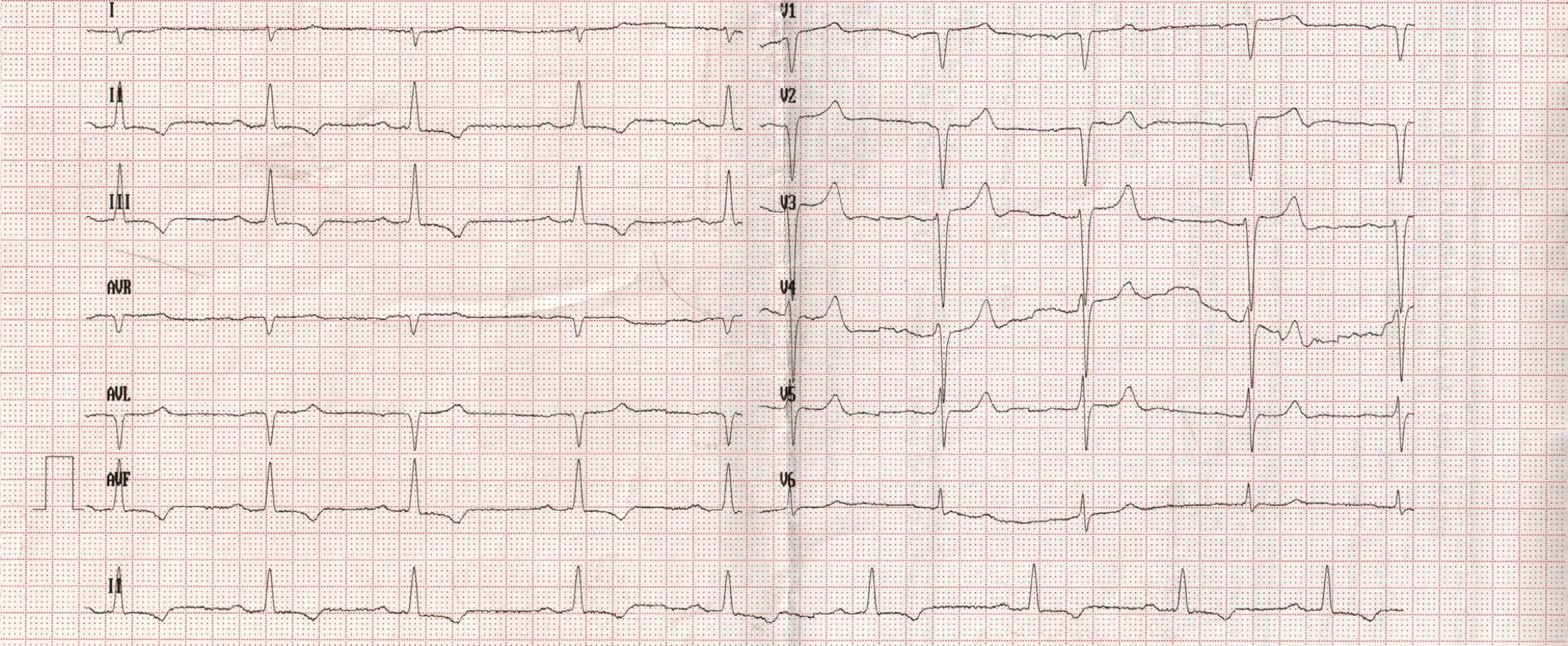
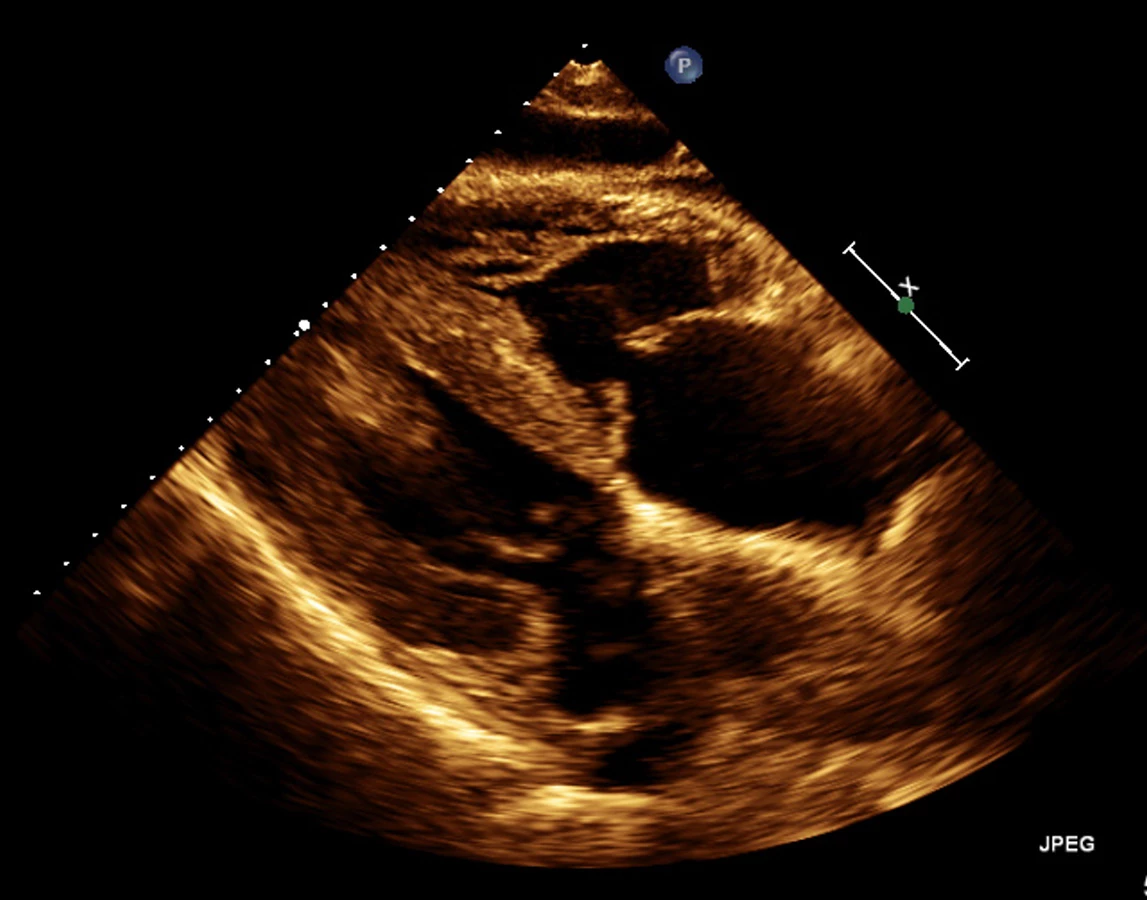
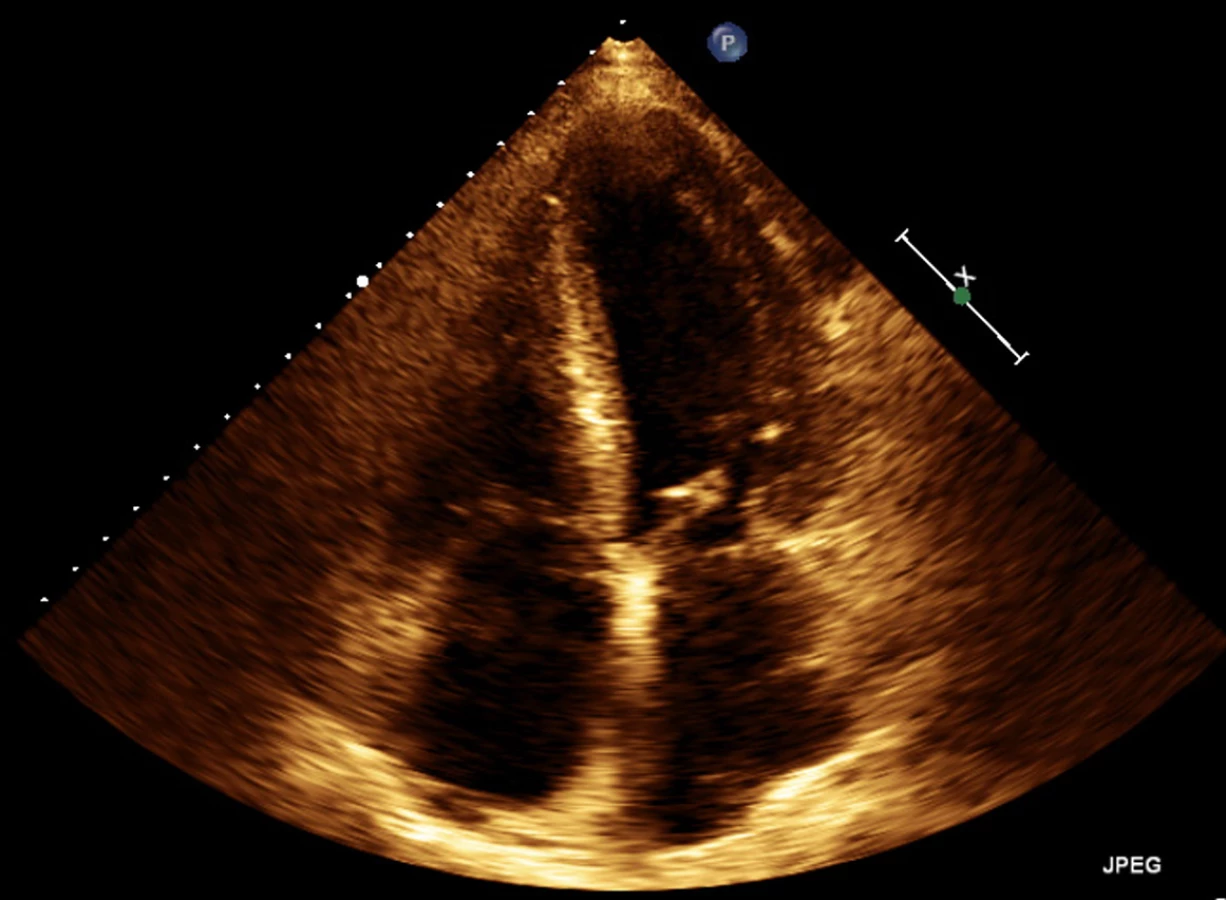
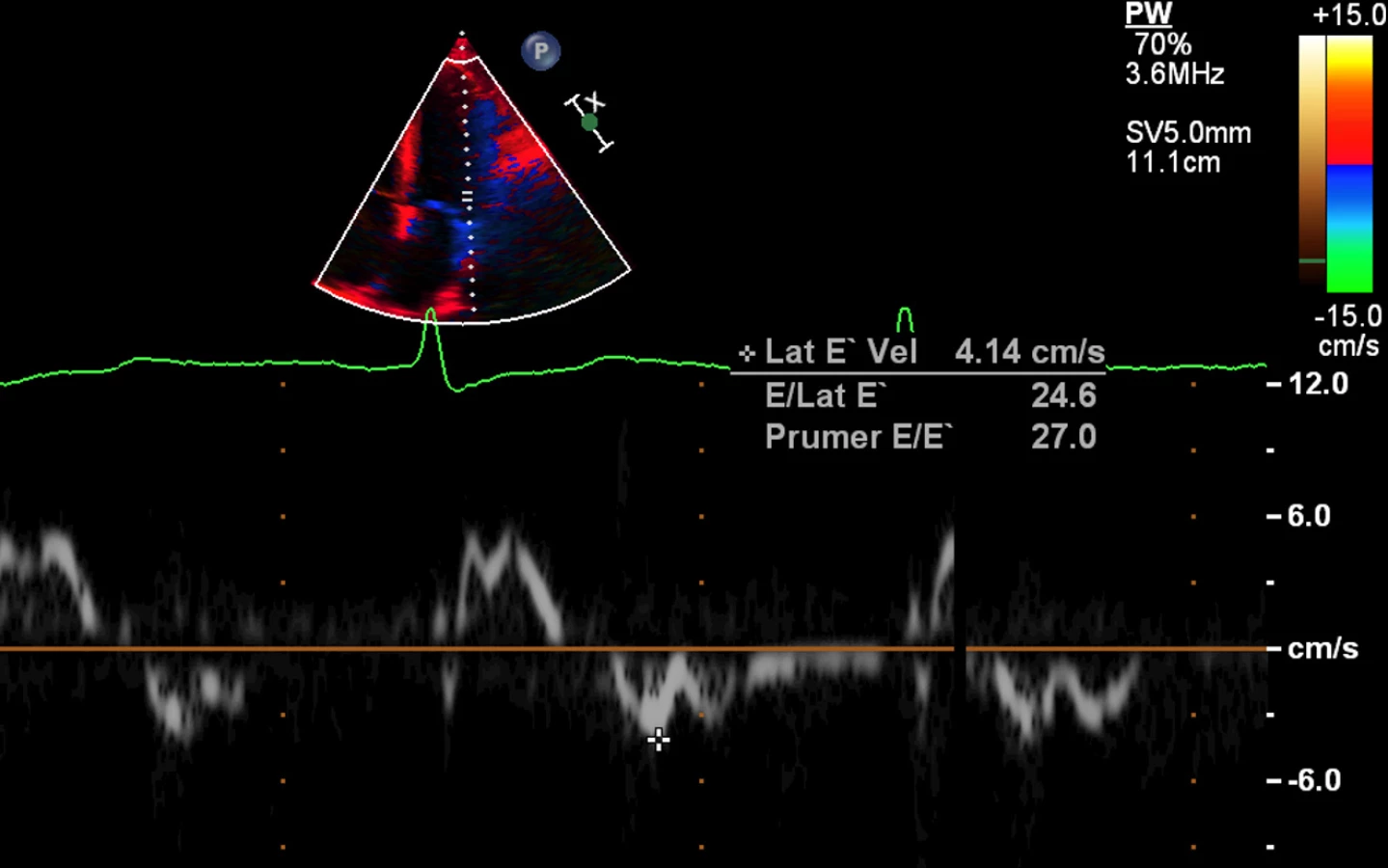
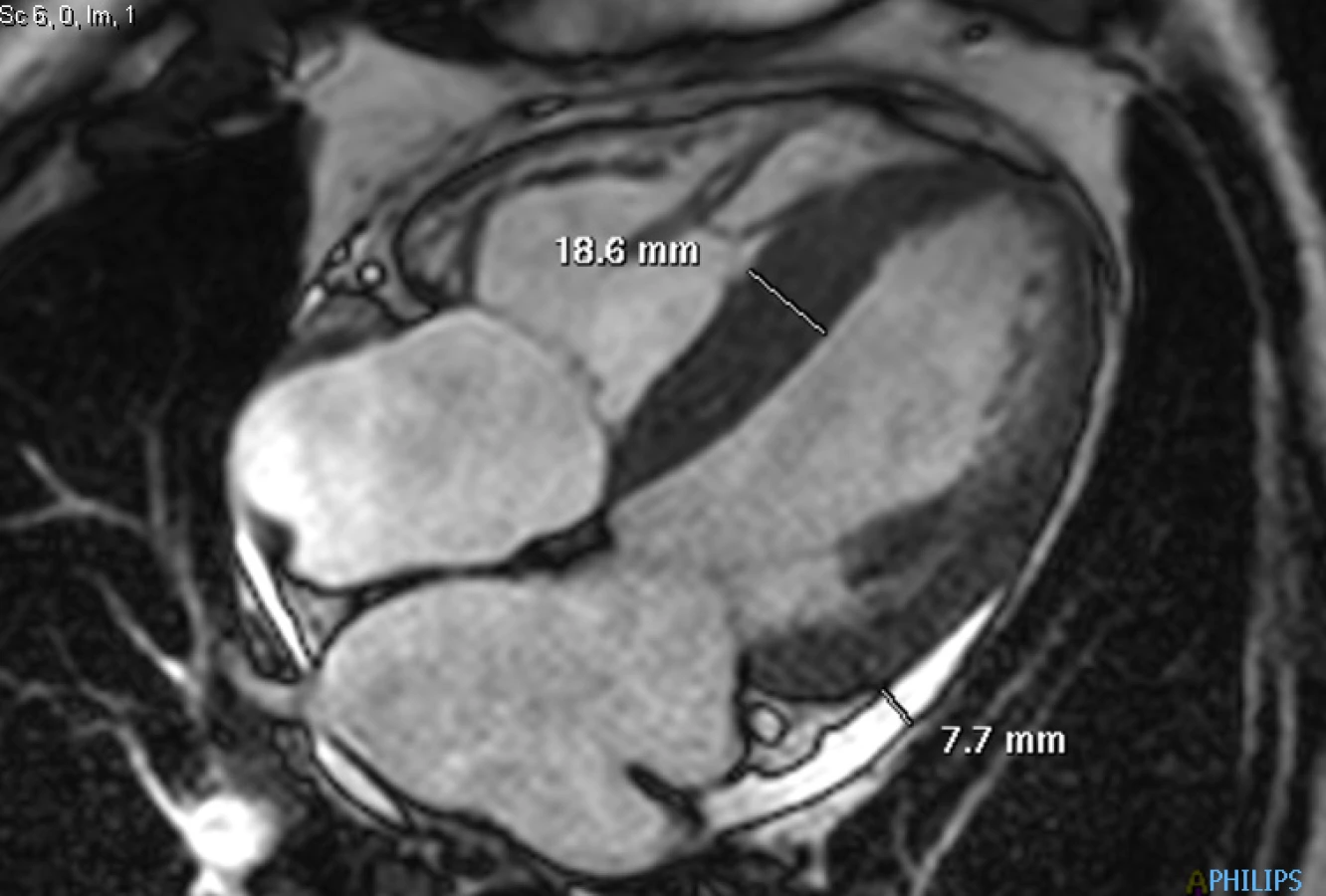
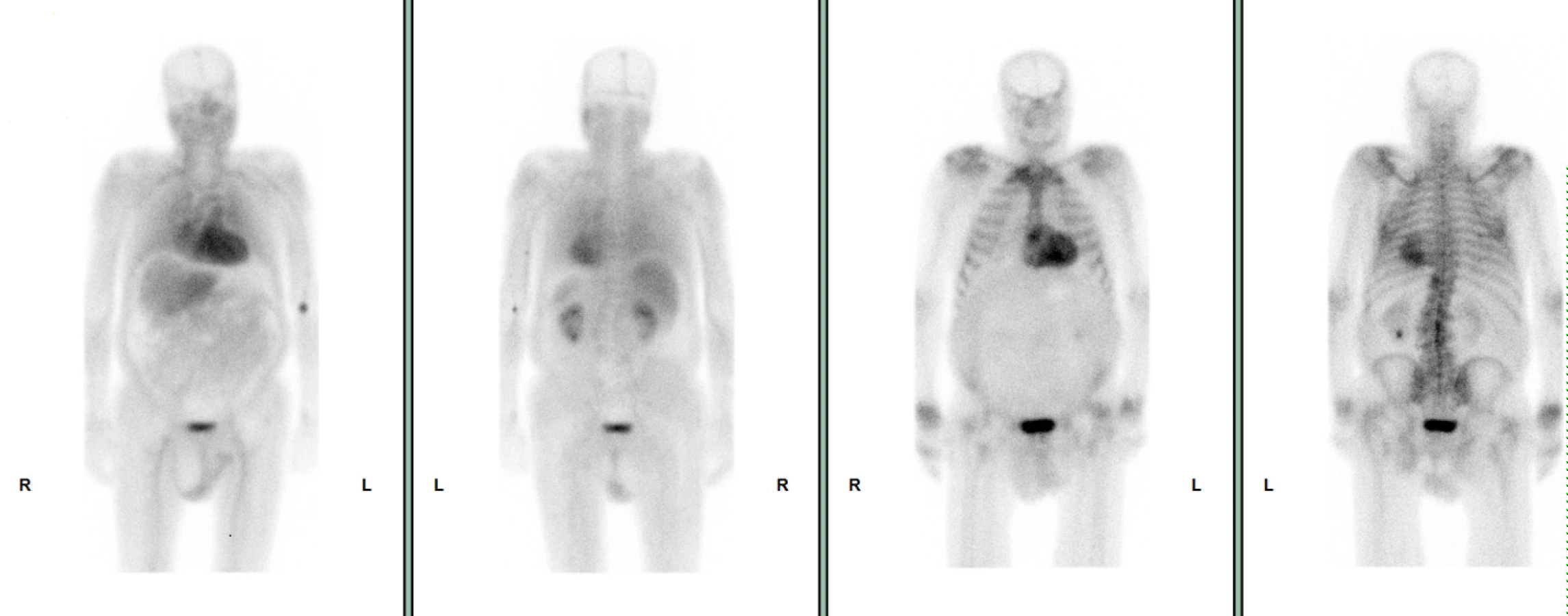
72letý pacient, hypertonik s chronickou obstrukční plicní nemocí, s anamnézou plicní hypertenze, po elektrické kardioverzi v minulosti pro symptomatický atypický flutter síní s nově nasazeným amiodaronem a warfarinem, byl odeslán praktickým lékařem do naší nemocnice pro zhoršující se dušnost, nárůst otoků dolních končetin, zvětšení objemu břicha a skrota. Praktický lékař pro trvající sinusový rytmus recentně vysadil amiodaron i warfarin, ponechal pacienta na antiagregační terapii a frekvenční kontrole betablokátorem, pro sklon k hypotenzi redukoval stávající antihypertenzní medikaci. RTG srdce a plic prokazoval zvýraznění plicních hilů s redistribucí cévní kresby v rámci kongesce, laboratorně zjištěna lehce snížená hodnota glomerulární filtrace (46,7 ml/min), sérový kreatinin 131 μmol/l, elevace kardiomarkerů troponinu I (91 ng/l, referenční mez 0–60 ng/l) a NT-proBNP – natriuretický peptid typu B (10 854 ng/l, referenční mez 0–125 ng/l). Na EKG jsme pozorovali sinusový rytmus, repolarizační změny nad spodní stěnou starého data, bez známek hypertrofie myokardu levé komory (Obr. 1). Sonografie břicha odhalila zvětšená hyperechogenní játra bez známek portální hypertenze, ascitickou tekutinu v dutině břišní a solitární cholecystolitiázu. Za hospitalizace byla provedena jak odlehčovací, tak diagnostická punkce ascitu s negativním výsledkem cytologického i kultivačního vyšetření, biochemicky stanovena hladina bílkoviny v ascitu 42,4 g/l (hodnota svědčící pro transsudativní ascites). Echokardiografie prokázala lehce sníženou systolickou funkci pravé i levé komory (LK) s ejekční frakcí LK 45–50 %, hypertrofii myokardu s restriktivním typem plnění LK, aortální stenózu do střední významnosti, plicní hypertenzi s odhadovaným systolickým tlakem v plicnici 50–55 mm Hg, známky hyperhydratace s velkým ascitem, bez průkazu fluidotoraxu oboustranně (Obr. 2–4). S ohledem na klinický obraz a echokardiografický nález bylo vysloveno podezření na infiltrativní onemocnění myokardu. V diferenciálně diagnostickém procesu byla provedena elektroforéza bílkovin séra se zvýšenou zónou β2 -globulinů a dále přítomna hyperimunoglobulinemie IgA. Imunofixací bílkovin séra a moči neprokázán M protein (neboli paraprotein) – diagnóza mnohočetného myelomu proto málo pravděpodobná. Za hospitalizace nastavena terapie srdečního selhání a v druhé době doplněna magnetická rezonance srdce, která v nativním obraze verifikuje hypertrofii stěn LK s maximem v oblasti mezikomorového septa (18,6 mm) se stopovým perikardiálním výpotkem (Obr. 5.1) a dále prokazuje difuzní pozdní sycení myokardu obou komor gadoliniovou kontrastní látkou (late gadolinium enhancement – LGE) – nález vysoce suspektní pro srdeční amyloidózu (Obr. 5.2). S výsledkem MRI byl pacient rehospitalizován ke komplexnímu došetření suspektního infiltrativního onemocnění. Na interním oddělení naší nemocnice odebrány sérové volné lehké řetězce κ (128,9 mg/l) a poměr κ/λ (2,6 mg/l), který zvýšen (referenční mez 0,3–1,6 mg/l, v případě renální insuficience až do 3,1 mg/l) (16), provedená biopsie z rekta neprokázala přítomnost amyloidu. Pacient následně objednán k dalšímu došetření na hematoonkologickou kliniku, kde provedená imunofixace vzorku séra a moči neprokázala přítomnost monoklonálního imunoglobulinu a průtoková cytometrie vzorku periferní krve a kostní dřeně neprokázala klonální plazmatické buňky. Hladina sérových volných lehkých řetězců κ činila 137 mg/l, λ 54 mg/l a stanovený poměr κ/λ (2,54 mg/l) byl opět nesignifikantně zvýšen v rámci renální insuficience (hodnota kreatininu 174 μmol/l). Hladina polyklonálního IgG činila 23,2 g/l, IgA 6,3 g/l a IgM 0,37 g/l. Biopsie z paraumbilikálního tuku s následujícím speciálním vyšetřením cíleným na průkaz amyloidu neprokázala jeho přítomnost. Pacient propuštěn z hematoonkologické kliniky se závěrem: o amyloidózu se s největší pravděpodobností nejedná, příčinou polyklonálního zmnožení IgG a IgA je chronické interní onemocnění. Pro naše trvající podezření na postižení srdce amyloidózou byl pacient odeslán na kliniku nukleární medicíny, kde za použití 99mTc-DPD (DiphosphonoPropanodicarboxylic Acid) scintigrafie bylo prokázáno zvýšené vychytávání 99mTc-DPD myokardem, které svědčí pro ATTR (Obr. 6). S výsledkem DPD skenu konzultován specialista zabývající se problematikou transthyretinové srdeční amyloidózy a pacientovi po jeho předchozím souhlasu byla nasazena off-label léčba ATTR (doxycyklin + kyselina tauroursodeoxycholová).

Diskuze
Prvním projevem amyloidózy srdce bývá diastolické srdeční selhání. Fibrilace síní může být přítomna několik let před tím, než vůbec začneme pomýšlet na toto onemocnění. V osobní anamnéze často nacházíme diagnózu syndromu karpálního tunelu, případně stenózu páteřního kanálu při infiltraci ligamentum flavum, a to především u wtATTR. Periferní a autonomní neuropatie nebývá v případě wtATTR tak obvyklá, naopak u mATTR ji nacházíme velmi často (23). U AL formy bývá patognomická makroglosie, periorbitální purpura, proteinurie či ortostatická hypotenze. Varovným příznakem je hypotenze u pacienta s předchozí anamnézou arteriální hypertenze a intolerance léků používaných běžně v terapii srdečního selhání (inhibitory angiotenzin-konvertujícího enzymu – ACEi a betablokátory) (3). Na EKG můžeme vidět nízkou voltáž (60% u AL, 40% u wtATTR), obraz pseudoinfarktu zvláště v prekordiálních svodech, nespecifické repolarizační změny, převodní poruchy typu atrioventrikulárních blokád prvního či vyšších stupňů, raménkové blokády. Vzácností nejsou ani srdeční arytmie typu fibrilace či flutteru síní (zejména u ATTR) (3, 4, 23). Echokardiograficky prokazujeme zbytnělý myokard levé komory s relativně typickou zrnitou strukturou (22). Ztluštění myokardu při absenci jiné příčiny hypertrofie LK by v nás vždy mělo vzbudit podezření na možnost tohoto infiltrativního onemocnění. V případě AL formy se jedná především o symetrickou hypertrofii stěn myokardu, naproti tomu u ATTR je hypertrofie častěji asymetrická a tloušťka mezikomorového septa bývá větší (okolo 18 mm) (3). Dalšími typickými echokardiografickými nálezy jsou dilatace síní, ztluštění mezisíňového septa a cípů chlopní, zvýšený tlak v pravé síni s dilatací dolní duté žíly či známky plicní hypertenze. Tkáňová dopplerometrie odhalí diastolickou dysfunkci LK s restriktivním typem plnění, nápadné může být zřetelné snížení longitudinální kontrakce LK i přes zachovalou ejekční frakci, případně perikardiální výpotek (4). Vyšetření hladin volných lehkých řetězců imunoglobulinu v séru κ a λ, resp. jejich poměr κ ku λ bývá v případě AL-amyloidózy abnormální (zvýšený) a jeho senzitivita činí 91%, v kombinaci s imunofixací až 99% (5). Imunofixace odhalí přítomnost M proteinu (3, 19, 21). Stanovení kardiálních markerů, a to především troponinu T/I a BNP/NT-proBNP, má vliv především v prognostické stratifikaci a v monitoraci léčby (6). Vyšší hladiny NT-proBNP nacházíme především u AL formy (2, 3, 22). Vyšetření magnetickou rezonancí prokáže globální subendokardiální či transmurální LGE s poměrně vysokou senzitivitou i specifitou (7). K průkazu časných forem srdeční amyloidózy a v rámci diferenciální diagnostiky etiologie hypertrofie LK může do budoucna napomoci standardně prováděné T1 mapování s výpočtem extracelulárního objemu (ECV) myokardu LK. Pro srdeční amyloidózu je typické výrazné zvýšení nativního T1 relaxačního času myokardu a ECV. Jejich zvýšení umožňuje detekovat i časné fáze srdeční amyloidózy u pacientů, u kterých ještě nedošlo k rozvoji hypertrofie LK a u kterých není přítomné LGE (17). Je nutné zdůraznit, že MRI nedokáže rozlišit jednotlivé typy amyloidu a právě identifikace typu amyloidu je zásadní pro stanovení prognózy a výběru vhodné terapie. Standardně se při diagnostice amyloidózy provádí biopsie z rekta, paraumbilikálního tuku a EMB – endomyokardiální biopsie (20, 22), která je založena na barvení tkáně Kongo červení a na imunohistochemických nebo proteomických metodách (19, 21, 23). Nutno podotknout, že nestačí jen průkaz amyloidu, leč je nutná přesná specifikace jeho typu. V případě provedení biopsie z paraumbilikálního tuku je senzitivita 70–90% u AL formy, nicméně jen 45% u mATTR a 15% u wtATTR. EMB je invazivní, nicméně relativně bezpečná metoda s rizikem komplikací 1% v centrech, kde je rutinně prováděna (4, 20). Postižení srdce se vyskytuje stejně často u AL i u ATTR formy, avšak u AL-amyloidózy vede často k rychlé progresi kardiální dysfunkce, zatímco u ATTR formy jsou příznaky vyjádřeny mírněji a k tak rychlé progresi nedochází. U ATTR dochází spíše k rozvoji diastolické dysfunkce, případně k AV blokádě, a to především u senilní formy (9, 23). Diagnóza mATTR může být jednoduchá v případě průkazu specifické mutace TTR genu, zatímco odlišení wtATTR od AL-amyloidózy může býti problematické zvláště u koincidentující wtATTR a monoklonální gamapatie nejistého významu (10). Hereditární ATTR je způsobena více než 100 mutacemi v genu pro transthyretin. Dědičnost je autozomálně dominantního typu (8, 23). Pinney et al ve své práci z roku 2013 poukazují na to, že v odlišení wtATTR od AL formy nám může pomoci hladina NT-proBNP a stáří pacienta v době stanovení diagnózy. Hladina NT-proBNP >183pmol/l a zároveň věk pod 70 let znamenaly vyšší pravděpodobnost AL typu (10). Velká prospektivní studie TRACS (Transthyretin Amyloid Cardiac Study) z roku 2012 porovnávala mortalitu a morbiditu u celkem 29 pacientů s mATTR (fenotyp V122I) a wtATTR. Ve skupině mATTR byla zaznamenána jak vyšší mortalita, tak i vyšší počet hospitalizací z kardiovaskulární příčiny. Nejčastější příčiny úmrtí byly srdeční selhání, náhlá srdeční smrt a sepse (11).
Bylo zjištěno, že některá radiofarmaka (polyfosfáty) používaná v kostní scintigrafii mají schopnost vázat se na srdeční amyloid, a to s různou ochotou. Nejvyšší afinitu vykazuje DPD. Přesný mechanismus vazby polyfosfátů na amyloid není znám, nicméně za zásadní se považuje role vápníku v amyloidovém depozitu a jeho vysoká afinita k radiofarmaku. TTR fibrily na rozdíl od AL fibril vážou vápník s velkou ochotou. Díky tomu je 99mTc-DPD scintigrafie vysoce senzitivní v diagnóze ATTR amyloidózy a umožňuje odlišit transthyretinovou a AL-amyloidózu se 100% spolehlivostí (12). Na vysokou senzitivitu 99mTc-DPD scintigrafie v diagnostice ATTR poukazuje také studie F. Javier de Haro-del Moral z roku 2012, kde negativní výsledek scintigrafie byl pozorován nejen v AL skupině, ale také ve skupině kontrolních zdravých jedinců a jejich příbuzných první linie, u kterých byla prokázána pouze mutace TTR genu. Z uvedeného vyplývá, že pro pozitivní test je nutná přítomnost amyloidu v myokardu (13).
Podpůrná terapie srdeční amyloidózy je zaměřena na redukci symptomů, udržení euvolemie a především u starších pacientů minimalizaci polypragmazie (8). Nízký srdeční výdej způsobený progresí systolické dysfunkce LK může způsobit symptomatickou hypotenzi a intoleranci léků používaných běžně v léčbě srdečního selhání (ACEi, blokátory receptorů pro angiotenzin II, betablokátory). Jsou-li tyto léky zapotřebí, je nutná jejich titrace s velkou opatrností. Za vysoce efektivní se považuje kombinace kličkových diuretik s antagonisty aldosteronu. Blokátory kalciových kanálů vykazují zvýšenou afinitu k amyloidovým fibrilám, proto je jejich použití kontraindikováno. Zvýšená afinita byla pozorována také v případě digoxinu (3, 4). U pacientů s ATTR, především typu wild-type, jak už bylo řečeno výše, dochází často k poruchám převodního systému myokardu. Pokročilé formy AV blokád, případně symptomatické bradykardie, mohou vést až k nutnosti implantace trvalé kardiostimulace. Nicméně implantace kardiostimulátoru u pacientů se srdeční amyloidózu zhoršuje prognózu, neboť pravokomorová elektroda může zhoršit trikuspidální regurgitaci a tím způsobit další progresi srdečního selhání (8). Jelikož amyloidové fibrily vznikají agregací patologických monomerů původně tetramerního transthyretinu, cílem farmakologické léčby je v současné době jak snížit tvorbu TTR, tak zajistit stabilizaci TTR tetrameru a v neposlední řadě způsobit degradaci již vytvořeného amyloidu ve tkáních. Z blokátorů TTR syntézy jsou známy 2 látky (siRNA – small interfering RNA a ASO – antisense oligonucleotide), které vazbou na messengerovou RNA způsobují její degradaci a tím brání hepatocytům v tvorbě transthyretinu. Pozitivní účinek ASO se projevil zejména v léčbě familiární amyloidové polyneuropatie (FAP), kdy zabránil další progresi neuropatie. Ze skupiny stabilizátorů TTR jsou to Diflunisal, Tafamidis, Tolcapone a AG10. Diflunisal v randomizované studii signifikantně zpomalil progresi neuropatie u pacientů s FAP (3). Tafamidis se ve srovnání s placebem výrazně podílel na snížení celkové mortality a počtu hospitalizací z kardiovaskulární příčiny a v testované populaci nedošlo k dalšímu zhoršení funkční kapacity i kvality života (18). Hlavním zástupcem 3. skupiny léčiv je kombinace doxycyklinu s tauroursodeoxycholovou kyselinou (TUDCA). Na myších modelech tato kombinační léčba vedla k signifikantnímu snížení stupně infiltrace tkáně amyloidem (3, 8). Tato léčba nezpůsobila u testované populace žádné nežádoucí účinky či zhoršení stávající kardiomyopatie či neuropatie (3, 4, 15). Je nutno podotknout, že v současnosti neexistuje žádný lék schválený v této indikaci a použití TUDCA v kombinaci s Doxycyklinem představuje off-label indikaci.
Závěr
Transthyretinová forma srdeční amyloidózy je v současné době stále poddiagnostikovanou příčinou srdečního selhání. Echokardiografie je základní vyšetřovací metoda, která nás může jako první na tuto diagnózu upozornit. Magnetická rezonance prokazuje typický obraz pozdního sycení myokardu gadoliniovou kontrastní látkou a pomocí T1 mapování je schopna detekovat i časnější formy tohoto onemocnění. Za zásadní se považuje odlišit ATTR od AL formy, neboť určení správného typu amyloidu je důležité pro stanovení prognózy a optimální terapie. 99mTc-DPD scintigrafie se zdá býti vysoce senzitivní v diagnostice transthyretinové formy. Negativní hematologické a bioptické vyšetření diagnózu srdeční amyloidózy nevylučuje, a proto bychom měli být kritičtí k závěrům vyšetřovacích metod a v případě jakýchkoli pochybností pátrat po pravé příčině onemocnění.
KORESPONDENČNÍ ADRESA AUTORA:
MUDr. Renáta Závodná, Ph.D.,
Interní oddělení Nemocnice ve Frýdku-Místku,
p.o. El. Krásnohorské 321,
738 01 Frýdek-Místek
Cit. zkr: Vnitř Lék 2020; 66(E-1): e34–e40
Článek přijat redakcí: 6. 1. 2019
Článek přijat k publikaci: 6. 2. 2019
Sources
1. Glaudemans A, Slart R, Zeebregts C, et al. Nuclear imaging in cardiac amyloidosis. Eur J Nucl Med Mol Imaging 2009; 36 : 702–714.
2. Pinney J., Whelan C, Petrie A, et al. Senile Systemic Amyloidosis: Clinical features at presentation and outcome, J Am Heart Assoc. 2013; 2: e000098. .
3. Donnely J, Hanna M. Cardiac amyloidosis: An update on diagnosis and treatment, Cleveland clinic Journal of Medicine 2017; 84(suppl. 3): 12–26.
4. Halatchev I, Zheng J, et al. Wild-type transthyretin cardiac amyloidosis, previsouly known as senile cardiac amyloidosis: clinical presentation, diagnosis, management and emerging therapis. J Thorac Dis 2018; 10 : 2034–2045.
5. Palladini G, Russo P, Bosoni T, et al. Identification of amyloidogenic light chains requires the combination of serum-free light chain assay with immunofixation of serum and urine. Clinical chemistry 2009; 55 : 3.
6. Kumar S, Dispenzieri A, Lacy M, et al. Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements, J Clin Oncol 2012; 30 : 989–995
7. Vogelsberg H, Mahrholdt H, Deluigi C, et al. Cardiovascular Magnetic Resonance in Clinically Suspected Cardiac Amyloidosis. J Am Coll Cardiol 2008; 51 : 1022–1030.
8. Castano A, Drachman B, Judge D, et al. Natural history and therapy of TTR-cardiac amyloidosis: emerging disease-modifying therapis from organ transplantation to stabilizier and silencer drugs. Heart Fail Rev 2015; 20 : 163–178.
9. Ng B, Connors LH, Davidoff R, et al. Senile systemic amyloidosis presenting with heart failure: a comparison with light chainassociated amyloidosis. Arch Intern Med 2005; 165 : 1425–1429.
10. Pinney J, Whelan C, Petrie A, et al. Senile Systemic Amyloidosis: Clinical features at presentation and outcome. J Am Heart Assoc 2013; 2: e000098.
11. Ruberg FL. et al. Prospective evaluation of the morbidity and mortality of wild-type and V122I mutant transthyretin amyloid cardiomyopathy: the transthyretin amyloidosis cardiac study (TRACS). Am Heart J 2012; 164 : 222–228.
12. Perugini E, Guidalotti PL, Salvi F, et al. Noninvasive etiologic diagnosis of cardiac amyloidosis using 99mTc-3,3-diphosphono-1,2-propanodicarboxylicacid scintigraphy. J Am Coll Cardiol 2005; 46 : 1076–1084.
13. Javier de Haro-del Moral F, Sánchez-Lajusticia A, Gómez-Bueno M, et al. Role of cardiac scintigraphy with 99mTc-DPD in the differentiation of cardiac amyloidosis subtype. Rev Esp Cardiol 2012; 65 : 440–446.
14. Kristen AV, et al. Prophylactic implantation of cardioverter-defibrillator in patients with severe cardiac amyloidosis and high risk for sudden cardiac death. Heart Rhythm 2008; 5 : 235–240.
15. Obici L, et al. Doxycycline plus tauroursodeoxycholic acid for transthyretin amyloidosis: a phase II study. Amyloid 2012; 19(Suppl. 1): 34–36.
16. Hutchison CA, Plant T, Drayson M, et al. Serum free light chain measurement aids the diagnosis of myeloma in patients with severe renal failure. BMC Nephrology. Dostupné z DOI: .
17. Pleva M, Borová J, Plevová I, et al. Význam zobrazení srdce pomocí magnetické rezonance v diagnostice hypertrofické kardiomyopatie, část II. Vnitř Lék 2017; 63 : 249–253.
18. Mathew S, Maurer MD, Jeffrey H, et al. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. N Engl J Med 2018; 379 : 1007–1016.
19. Flodrova P, Flodr P, Pika T, et al. Cardiac amyloidosis: from clinical suspicion to morphological diagnosis. Pathology (Sydney) 2018; 50 : 261–268.
20. Aiglová R, Táborský M, Lazárová M, et al. Srdeční transthyretinová amyloidóza – častější, než jsme si mysleli? Cor et Vasa 2017; 59 : 476.
21. Pika T, Heřmanová Z, Flodrová P, et al. Laboratorní aspekty systémové AA amyloidózy. Klinická biochemie a metabolismus 2017; 25 : 56–58.
22. Kováčik F, Táborský M, Hutyra M, et al. Srdeční amyloidóza – kazuistika. Intervenční a akutní kardiologie 2016; 15 : 145–147.
23. Kufová Z, Pika T, Jelínek T, et al. Hereditární amyloidózy – etiologie, klinický obraz a léčba. Transfuze a hematologie dnes 2015; 21 : 184–192.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2020 Issue 1
Most read in this issue
- Sarkopenická obezita – aktuální přehled problematiky
- Chronický stres, psychická nepohoda a deprese zvyšují četnost infekčních, autoimunitních, ale i maligních nemocí
- Odešel velký člověk a lékař prof. MUDr. Vítězslav Kolek, DrSc., FCCP
- Autoimunitní hepatitida